
Abstract
Novel 2-aminopyrimidine derivatives were prepared from acyclic starting materials, benzylidene acetones and ammonium thiocyanates, via 5 steps, including ring closure, aromatization, S-methylation, oxidation to methylsulfonyl compounds, and formation of guanidines with suitable amines. The prepared compounds differ from each other by the substitutions of their amino group and of their phenyl ring. The 2-aminopyrimidines were tested by use of microplate assays for their in vitro activities against a causative organism of sleeping sickness, Trypanosoma brucei rhodesiense, as well as against a causative organism of malaria, Plasmodium falciparum NF54. Their cytotoxic properties were determined with L-6 cells (rat skeletal myoblasts). Some of the compounds exhibited quite good antitrypanosomal activity, and others showed excellent antiplasmodial activity. The influence of the structural modifications on these activities is discussed.
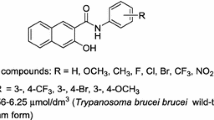
Graphic abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the past two decades, over two billion of the world’s poorest people have been affected by neglected tropical diseases (NTDs). One of the 11 major NTDs studied is human African trypanosomiasis (HAT) [1]. HAT or sleeping sickness is caused by protozoa of the genus Trypanosoma like Trypanosoma brucei gambiense (Tbg) and Trypanosoma brucei rhodesiense (Tbr). The vector is the tsetse fly. Only one drug, melarsoprol, is available for the late-stage Tbr infection treatment [2]. This toxic arsenic compound causes severe side effects including a deadly encephalopathy in more than 5% of the patients [3]. Therefore it is an urgent need to develop new efficient antitrypanosomal compounds with less side effects.
In 2018 malaria globally affected 228 million people and caused 405,000 deaths [4]. The emergence and spread of resistance in Plasmodium falciparum malaria to artemisinin combination therapies in the Greater Mekong subregion poses a major threat to malaria control and elimination [5]. Since the last defense line, the artemisinines, might fall possibly, there is a great demand for antiplasmodial compounds with alternative mechanism of action.
Substituted pyrimidines were described early as antiplasmodial compounds [6,7,8,9,10,11,12]. Some 2-aminopyrimidines were reported to be active in low micromolar to submicromolar concentration [13]. We prepared new methyl-aryl-substituted 2-aminopyrimidines including compounds bearing partial structures of chloroquine which were connected to the nitrogen in ring position 2.
Results and discussion
Starting from benzylidene acetones 1a–1d pyrimidine-thiones 2a–2d were prepared by reaction with ammonium thiocyanate in refluxing benzene/cyclohexanol [14]. Aromatization of the heterocyclic ring took place in boiling xylene in the presence of sulfur giving compounds 3a–3d. Subsequently the SH group was methylated using methyl iodide in chloroform yielding 4a–4d. The next step was an oxidation to the methylsulfonyl compounds 5a–5d with m-chloroperbenzoic acid in dichloromethane. The final formation of the target compounds 6–10 took place in dioxane or tetrahydrofuran in the presence of the various amines under microwave irradiation at 120 °C or under reflux. Structural modifications were restricted to the amino substituent including amino, a (pyrrolidin-1-yl) and (4-methylpiperazin-1-yl) groups of compounds 6–8. Moreover, partial structures of chloroquine were used as substituents for compounds 9 and 10. The chiral aliphatic amine moiety was connected with the pyrimidine core giving compounds 9 as racemates. In a further step, the quinoline residue was attached. Similar compounds, bearing an additional ester function have already been investigated [15]. Further variations concerned the substitution pattern of a phenyl ring (Scheme 1).

Scheme 1. Syntheses of compounds 2–10. Reagents and conditions: (i) benzene, cyclohexanol, water separator, reflux, 6 h, (ii) S, xylene, reflux, overnight, (iii) CH3I, CHCl3, r.t., overnight to 3 days, (iv) m-chloroperbenzoic acid, CH2Cl2, 0–20 °C, 2 h, (v) NH3 conc. or amine, dioxane or THF, 85 °C, reflux overnight or microwave 120 °C 2–13 h. For the aromatization from 2a–2d to 3a–3d we observed the disappearance of the proton signal at 4.9 ppm of the CH group attached to the aromatic moiety in 1H NMR spectra as well as the shift of the signal of the olefinic proton from 4.7 ppm to 7.3 ppm. Furthermore, the signals of the NH protons at 8.8 and 9.5 ppm disappeared and a new signal was observed for the SH group at 13.5 ppm. In 13C spectra, the signal of the carbon attached to the aromatic moiety shifted from 54 to 165 ppm due to aromatization. S-Methylation to compounds 4a–4d caused appearance of an additional signal at 2.6 ppm for the methylthio group in 1H NMR spectra and at 13.8 ppm in 13C spectra. The subsequent oxidation to the methylsulfonyl group in 5a–5d shifted the signal of the attached methyl group from 2.6 ppm to 3.4 ppm in 1H NMR spectra and from 13.8 ppm to 39 ppm in 13C spectra. The replacement of the methylsulfonyl group of compunds 5a–5d by amino substituents shifted the signal for the C-2 2–6 ppm to lower frequencies. Moreover, we observed long-range couplings from protons of the amino substituent to C-2 in HMBC spectra of compounds 6–10 which confirmed the attachment of the amino groups to this ring position.
All 2-aminopyrimidine derivatives 6–10 were tested for their antiplasmodial activities against P. falciparum NF54 and for their antitrypanosomal potencies against Trypanosoma brucei rhodesiense STIB 200 as well as for their cytotoxicity against rat skeletal myoblasts (L-6 cells) in microplate assays. The results are presented in Table 1.

Structures of reference compounds
A series of 4-alkyl-6-(hetero)arylpyrimidin-2-amines was reported to possess promising antiplasmodial activity (IC50 = 0.115–3.96 µM); however, the significance of the results was not sustained by cytotoxicity data [13]. Our 2-amino compounds 6a–6d, 7a–7d, and 8a–8d were completely inactive (IC50 = 11.2–270 µM) against P. falciparum NF54. The by far lower activity of our 6-methyl compound 6b (IC50 = 139 µM) compared to its 6-isopropyl analogue (IC50 = 3.96 µM) [13] may be explained by the use of different strains test methods. The substitution of the amino group with the side chain of chloroquine improved the antiplasmodial activity (9a–9c: IC50 = 2.62–73.0 µM) only slightly. Moreover, the selectivity indexes of compounds 6–9 (SIPN = 1.08–7.84) were very low. High activity against P. falciparum NF54 (IC50 = 0.04–0.14 µM) and good selectivity (SIPN = 81–220) was observed for the 2-aminopyrimidines 10a–10d, which exhibit a 4-aminoquinoline partial structure linked to their amino nitrogen. The most active compound 10d was additionally tested against the multiresistant K1 strain of P. falciparum and showed slightly decreased activity (IC50 = 0.14 µM) but is more active than chloroquine (IC50 = 0.27 µM) [16] against this strain.
Most of the new compounds exhibited weak or negligible antitrypanosomal activity (IC50 = 6.20–214 µM) or low selectivity (SIT = 0.68–5.78) or both of them. However, moderate activity (IC50 = 1.90, 2.40 µM) and quite good selectivity (SIT = 17.4, 25.5) were observed for compounds 8b, 8c with 4-methylpiperazinyl substitution. The most promising antitrypanosomal compounds 9b and 9c (IC50 = 0.41, 1.03 µM; SIT = 30.7, 30.9) feature the side chain of chloroquine.
Conclusion
Several new methyl-aryl-substituted 2-aminopyrimidines with differing amino and phenyl substitution have been prepared. The antitrypanosomal and antiplasmodial activities of the new compounds were determined. The most active antitrypanosomal compounds (IC50 = 0.41, 1.03 µM) exhibited the same side chain as chloroquine. Compounds possessing the 7-chloroquinoline partial structure of chloroquine showed excellent activity against P. falciparum NF54 (IC50 = 0.04–0.14 µM). The most active compound was additionally tested against the multiresistant K1 strain of P. falciparum and showed twice the activity of chloroquine against this strain. Therefore, this compound could be a lead for further optimization.
Experimental
Melting points were obtained on a digital melting point apparatus Electrothermal IA 9200. IR spectra: Bruker Alpha Platinum ATR FT-IR spectrometer (KBr discs). NMR spectra: Varian Inova 400 (300 K) 5 mm tubes, spectra were acquired in CDCl3 containing 0.03% TMS. Chemical shifts were recorded in parts per million (ppm), for 1H spectra TMS (0.00 ppm) was used as internal standard and for 13C spectra the central peak of the CDCl3 peak was used as the internal reference (77.0 ppm). Some spectra were acquired in DMSO-d6. Here the proton signal at 2.49 ppm served as internal reference as well as the central peak of the DMSO-d6 signal at 39.7 ppm. Abbreviations: aromatic H, ArH; aromatic C, ArC, quaternary aromatic C, ArCq. Signal multiplicities are abbreviated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; q, quartet; br, broad. Coupling constants (J) are reported in Hertz (Hz). 1H and 13C resonances were assigned using 1H, 1H- and 1H, 13C-correlation spectra. 1H and 13C resonances are numbered as given in the formulae. Assignments marked with an asterisk are interchangeable. HRMS: GCT-Premier, Waters (EI, 70 eV). Materials: column chromatography (CC): silica gel 60 (Merck 70–230 mesh, pore-diameter 0.6 nm), aluminium oxide (Alox) basic (Fluka for chromatography, 0.05–0.15 mm, Brockmann activity I, basic); Alox neutral 90 (Merck, 0.063–0.2 mm, activity I, neutral); thin-layer chromatography (TLC): TLC plates (Merck, silica gel 60 F254 0.2 mm, 200 × 200 mm); TLC plates (Merck, Alox 60 F254 neutral, 200 × 200 mm); the substances were detected in UV light at 254 nm. Unless otherwise stated silica gel was used for separations (CC, TLC). Microwave-assisted reactions were carried out in a CEM Discover/Explorer system in sealed 10 cm3 standard vessels with temperature control. Syntheses of compounds 2a, 2b, and 2d were described previously [14]. Compund 3a was prepared following a reported procedure [17]. Its melting point (201–205 °C) corresponded well with the reported one (202–204 °C) [18]. The synthesis of 4a was already described and the melting point (214 °C) corresponded well with the reported one (213 °C) [19].
6-Methyl-4-(4-methylphenyl)-3,4-dihydropyrimidine-2(1H)-thione (2c, C12H14N2S)
The reaction of 21.27 g of 1c (132.76 mmol) with 8.26 g of ammonium thiocyanate (108.5 mmol) was carried out in 400 cm3 of toluene in the presence of 4.12 g of cyclohexanol (41.16 mmol). The mixture was heated for 18 h at 160 °C oil bath temperature using a water separator filled with molecular sieves 4 Å. After cooling to r.t., the orange precipitate was collected by filtration, washed with ether and ethanol. Then it was dissolved in a mixture of hot ethanol/isopropanol (3:1), treated with charcoal and filtered. The filtrate was concentrated in vacuo. Thereafter it was allowed to stand overnight at r.t. to complete crystallization. The product was collected by filtration, washed with ethanol, and dried. Yield: 11.85 g of 2c (41%) as white crystals. Rf = 0.84 (benzene:CHCl3:EtOH = 4:4:1); m.p.: 207 °C; 1H NMR (DMSO-d6, 400 MHz):δ = 1.69 (s, 3H, CH3), 2.26 (s, 3H, ArCH3), 4.68 (d, J = 1.8 Hz, 1H, H-5), 4.85 (s, 1H, H-4), 7.10 (d, J = 8.1 Hz, 2H, ArH), 7.15 (d, J = 7.7 Hz, 2H, ArH), 8.76 (s, 1H, H-3), 9.52 (s, 1H, H-1) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 17.87 (CH3), 20.90 (ArCH3), 54.75 (C-4), 99.49 (C-5), 126.43, 129.24 (ArC), 130.78 (C-6), 136.73, 142.05 (ArCq), 174.18 (C-2) ppm; IR (KBr): \(\overline{v}\) = 3184, 2987, 1706, 1569, 1489, 1323, 1197, 1180, 844, 819, 790 cm−1; HRMS (EI +): m/z calcd C12H14N2S [M+] 218.0878, found 218.0874.
Preparation of pyrimidine-2-thiols 3b–3d
The aromatization of dihydropyrimidine-2(1H)-thiones 2b, 2c, and 2d took place overnight in refluxing xylene in the presence of sulfur. The solution was then allowed to cool to r.t.. A precipitate was formed which was collected by filtration. The solid was stirred with 1 N NaOH and filtered. The filtrate was acidified with 2 N HCl. The precipitate was collected by filtration, washed with water and recrystallized.
4-(4-Chlorophenyl)-6-methylpyrimidine-2-thiol (3b, C11H9ClN2S)
Reaction of 3.99 g of 2b (16.7 mmol) with 3.2 g of sulfur (0.1 mol) in 24 cm3 of xylene yielded after crystallization from a mixture of ethanol and propan-2-ol 1.75 g of 3b (44%) as orange powder. Rf = 0.58 (CH2Cl2:CH3OH = 10:1); m.p.: 249 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 2.34 (s, 3H, CH3), 7.37 (s, 1H, H-5), 7.59 (d, J = 8.7 Hz, 2H, ArH), 8.13 (d, J = 8.6 Hz, 2H, ArH), 13.72 (br, s, 1H, SH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 18.82 (CH3), 106.08 (C-5), 129.23, 129.84 (ArC), 134.31, 137.25 (ArCq), 159.37 (C-6), 164.36 (C-4), 181.34 (C-2) ppm; IR (KBr): \(\overline{v}\) = 3441, 2920, 2360, 1611, 1577, 1552, 1457, 1280, 1230, 1169, 1090, 977, 809 cm−1; HRMS (EI +): m/z calcd. (C11H9ClN2S) [M+] 236.0175, found 236.0162.
4-Methyl-6-(4-methylphenyl)pyrimidine-2-thiol (3c, C12H12N2S)
Reaction of 11.85 g of 2c (54.28 mmol) with 2.09 g of sulfur (65.2 mmol) in 80 cm3 of xylene yielded after crystallization from ethanol 4.35 g of 3c (37%) as yellow powder. Rf = 0.57 (CH2Cl2:CH3OH = 10:1); m.p.: 249 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 2.33 (s, 3H, CH3), 2.36 (s, 3H, ArCH3), 7.32–7.34 (m, 3H, H-5, ArH), 8.03 (d, J = 8.1 Hz, 2H, ArH), 13.60 (br, s, 1H, SH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 18.75 (CH3), 21.26 (ArCH3), 105.78 (C-5), 128.05, 129.74 (ArC), 132.69, 142.58 (ArCq), 158.67 (C-4), 165.39 (C-6), 181.27 (C-2) ppm; IR (KBr): \(\overline{v}\) = 2827, 1612, 1577, 1553, 1453, 1336, 1288, 1238, 1198, 1173, 976, 811 cm−1; HRMS (EI +): m/z calcd. (C12H12N2S) [M+] 216.0721, found 216.0712.
4-(4-Methoxyphenyl)-6-methylpyrimidine-2-thiol (3d, C12H12N2OS)
Reaction of 6.29 g of 2d (26.86 mmol) with 1.03 g of sulfur (32.23 mmol) in 40 cm3 of xylene yielded after crystallization with ethanol 2.02 g of 3d (32%) as buff powder. Rf = 0.48 (CH2Cl2:CH3OH = 10:1); m.p.: 243–245 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 2.32 (s, 3H, CH3), 3.83 (s, 3H, OCH3), 7.06 (d, J = 8.8 Hz, 2H, ArH), 7.30 (s, 1H, H-5), 8.11 (d, J = 8.8 Hz, 2H, ArH), 13.50 (br, s, 1H, SH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 18.71 (CH3), 55.69 (OCH3), 105.26 (C-5), 114.53 (ArC), 127.62 (ArCq), 130.03 (ArC), 158.22 (C-6), 162.81 (ArCq), 164.92 (C-4), 181.09 (C-2) ppm; IR (KBr): \(\overline{v}\) = 2836, 1619, 1605, 1581, 1553, 1456, 1340, 1257, 1242, 1203, 1168, 1027, 976, 818 cm−1; HRMS (EI +): m/z calcd. (C12H12N2OS) [M+] 232.0670, found 232.0655.
Preparation of (methylsulfanyl)pyrimidine hydroiodides 4b–4d
To a stirred suspension of 3b, 3c, or 3d in CHCl3 methyliodide (CH3I) was added dropwise. Stirring was continued at r.t. for up to 3 days. Then the formed solid was collected by filtration. The filtrate was evaporated to dryness and the residue was crystallized by treatment with ethyl acetate to give a second portion of the product. The solids were combined and dried.
4-(4-Chlorophenyl)-6-methyl-2-(methylsulfanyl)pyrimidine hydroiodide (4b, C12H12ClIN2S)
Reaction of 1.65 g of 3b (6.9 mmol) in 50 cm3 of CHCl3 with 2.48 g of CH3I (17.4 mmol) yielded overnight 1.948 g of 4b (75%) as yellow-orange precipitate. Rf = 0.60 (CH2Cl2:CH3OH = 10:1); m.p.: 193 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 2.46 (s, 3H, CH3), 2.57 (s, 3H, SCH3), 7.60 (d, J = 8.6 Hz, 2H, ArH), 7.70 (s, 1H, H-5), 8.20 (d, J = 8.6 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 13.81 (SCH3), 23.96 (CH3), 112.00 (C-5), 129.04, 129.29 (ArC), 134.85, 136.32 (ArCq), 161.71 (C-4), 168.66 (C-6), 171.25 (C-2) ppm; IR (KBr): \(\overline{v}\) = 2862, 1610, 1577, 1552, 1455, 1335, 1279, 1229, 1168, 1089, 1010, 977, 818, 808 cm−1; HRMS (EI +): m/z calcd. (C11H8ClN2S) [M+-H-CH3I] 235.0097, found 235.0092.
4-Methyl-6-(4-methylphenyl)-2-(methylsulfanyl)pyrimidine hydroiodide (4c, C13H15IN2S)
Reaction of 4.21 g of 3c (19.46 mmol) in 250 cm3 of CHCl3 with 6.63 g of CH3I (46.71 mmol) yielded after 3 days 5.73 g of 4c (82%) as brown solid. Rf = 0.81 (CH2Cl2:MeOH = 10:1); m.p.: 208–209 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 2.34 (s, 3H, ArCH3), 2.43 (s, 3H, CH3), 2.56 (s, 3H, SCH3), 7.31 (d, J = 8.1 Hz, 2H, ArH), 7.66 (s, 1H, H-5), 8.06 (d, J = 8.1 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 13.79 (SCH3), 21.27 (ArCH3), 23.55 (CH3), 111.68 (C-5), 127.31, 129.82 (ArC), 132.99, 141.77 (ArCq), 163.21 (C-6), 167.73 (C-4), 170.49 (C-2) ppm; IR (KBr): \(\overline{v}\) = 2732, 1620, 1578, 1429, 1349, 1298, 1273, 1206, 1190, 834 cm−1; HRMS (EI +): m/z calcd. (C13H14N2S) [M+-HI] 230.0878, found 230.0877.
4-(4-Methoxyphenyl)-6-methyl-2-(methylsulfanyl)pyrimidine hydroiodide (4d, C13H15lIN2OS)
Reaction of 1.88 g of 4d (8.1 mmol) in 120 cm3 of CHCl3 with 2.75 g of CH3I yielded after 2 days 2.65 g of 4d (87%) as yellow solid. Rf = 0.76 (CH2Cl2:MeOH = 10:1); m.p.: 213 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 2.43 (s, 3H, CH3), 2.57 (s, 3H, SCH3), 3.81 (s, 3H, OCH3), 7.05 (d, J = 8.8 Hz, 2H, ArH), 7.66 (s, 1H, H-5), 8.15 (d, J = 8.8 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 13.79 (SCH3), 23.17 (CH3), 55.75 (OCH3), 111.19 (C-5), 114.66 (ArC), 127.78 (ArCq), 129.33 (ArC), 162.45 (ArCq), 163.21 (C-4), 166.90 (C-6), 169.85 (C-2) ppm; IR (KBr): \(\overline{v}\) = 2698, 1602, 1560, 1399, 1348, 1262, 1212, 1178, 1022, 834 cm−1; HRMS (EI +): m/z calcd. (C13H14N2OS) [M+-HI] 246.0827, found 246.0812.
Preparation of (methanesulfonyl)pyrimidines 5a–5d
The methylthio-pyrimidine hydroiodides 4a, 4b, 4c, or 4d were dissolved in CH2Cl2. The resulting solution was cooled down to 0 °C and 3-chloroperoxybenzoic acid (77%) was added slowly with stirring and cooling. The color of the solution turned to cerise, the ice bath was removed and the reaction mixture was stirred for 2 h at r.t.. During the reaction the color of the solution turned to violet due to the oxidation of iodide to iodine. The organic layer was washed once with saturated NaHCO3 solution, once with an aqueous Na2S2O3 solution, and finally with brine. Then it was dried over anhydrous Na2SO4 and filtered. The solvent was evaporated in vacuo giving white solids which were recrystallized with a small amount of ethyl acetate giving colorless needles.
2-(Methanesulfonyl)-4-methyl-6-phenylpyrimidine (5a, C12H12N2O2S)
Reaction of 1.124 g of 4a (3.27 mmol) in 120 cm3 of CH2Cl2 with 2.452 g of 3-chloroperoxybenzoic acid (10.94 mmol) yielded 751 mg of 5a (92%). Rf = 0.75 (CH2Cl2:CH3OH = 10:1); m.p.: 176 °C; 1H NMR (CDCl3, 400 MHz): δ = 2.73 (s, 3H, CH3), 3.44 (s, 3H, SO2CH3), 7.52–7.58 (m, 3H, ArH), 7.75 (s, 1H, H-5), 8.14 (d, J = 6.6 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 24.38 (CH3), 39.01 (SO2CH3), 118.31 (C-5), 127.41, 129.09, 132.01 (ArC), 134.63 (ArCq), 165.18 (C-6), 165.90 (C-2), 170.36 (C-4) ppm; IR (KBr): \(\overline{v}\) = 3015, 1590, 1515, 1499, 1426, 1353, 1296, 1223, 1141, 974, 965, 883, 793, 765, 751, 697 cm−1; HRMS (EI +): m/z calcd. (C12H12N2O2S) [M+] 248.0620, found 248.0623.
4-(4-Chlorophenyl)-2-(methanesulfonyl)-6-methylpyrimidine (5b, C12H11ClN2O2S)
Reaction of 378 mg of 4b (1.00 mmol) in 36 cm3 of CH2Cl2 with 1 g of 3-chloroperoxybenzoic acid (4.46 mmol) yielded 246 mg of 5b (87%). Rf = 0.75 (CH2Cl2:CH3OH = 10:1); m.p.: = 161 °C; 1H NMR (CDCl3, 400 MHz): δ = 2.71 (s, 3H, CH3), 3.42 (s, 3H, SO2CH3), 7.49 (d, J = 8.6 Hz, 2H, ArH), 7.73 (s, 1H, H-5), 8.08 (d, J = 8.6 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 24.41 (CH3), 39.02 (SO2CH3), 118.12 (C-5), 128.72, 129.39 (ArC), 133.06, 138.42 (ArCq), 164.02 (C-4), 165.94 (C-2), 170.61 (C-6) ppm; IR (KBr): \(\overline{v}\) = 3013, 1591, 1575, 1513, 1494, 1444, 1398, 1356, 1324, 1301, 1228, 1137, 1094, 1012, 968, 840, 754 cm−1; HRMS (EI +): m/z calcd. (C12H11ClN2O2S) [M+] 282.0230, found 282.0224.
2-(Methanesulfonyl)-4-methyl-6-(4-methylphenyl)pyrimidine (5c, C13H14N2O2S)
Reaction of 5.61 g of 4c (15.66 mmol) in 575 cm3 of CH2Cl2 with 11.77 g of 3-chloroperoxybenzoic acid (52.52 mmol) yielded 2.996 g of 5c (73%). Rf = 0.44 (cyclohexane:ethyl acetate = 1:3); m.p.: 146–147 °C; 1H NMR (CDCl3, 400 MHz): δ = 2.43 (s, 3H, ArCH3), 2.69 (s, 3H, CH3), 3.42 (s, 3H, SO2CH3), 7.32 (d, J = 8.1 Hz, 2H, ArH), 7.71 (s, 1H, H-5), 8.03 (d, J = 8.1 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 21.46 (ArCH3), 24.34 (CH3), 38.99 (SO2CH3), 117.84 (C-5), 127.34, 129.82 (ArC), 131.83, 142.75 (ArCq), 165.11 (C-6), 165.82 (C-2), 170.08 (C-4) ppm; IR (KBr): \(\overline{v}\) = 3013, 1587, 1521, 1504, 1444, 1355, 1304, 1138, 966, 828, 746 cm−1; HRMS (EI +): m/z calcd. (C13H14N2O2S) [M+] 262.0776, found 262.0766.
2-(Methanesulfonyl)-4-(4-methoxyphenyl)-6-methylpyrimidine (5d, C13H14N2O3S)
Reaction of 2.512 g of 4d (6.72 mmol) in 250 cm3 of CH2Cl2 with 5.04 g of 3-chloroperoxybenzoic acid (22.49 mmol) yielded 1.32 g of 5d (71%). Rf = 0.75 (CH2Cl2:CH3OH = 10:1); m.p.: 135–136 °C; 1H NMR (CDCl3, 400 MHz): δ = 2.66 (s, 3H, CH3), 3.41 (s, 3H, SO2CH3), 3.88 (s, 3H, OCH3), 7.00 (d, J = 8.8 Hz, 2H, ArH), 7.65 (s, 1H, H-5), 8.10 (d, J = 8.8 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 24.29 (CH3), 38.96 (SO2CH3), 55.42 (OCH3), 114.40 (ArC), 117.14 (C-5), 126.89 (ArCq), 129.12 (ArC), 162.82 (ArCq), 164.61 (C-4), 165.72 (C-2), 169.75 (C-6) ppm; IR (KBr): \(\overline{v}\) = 2932, 1587, 1524, 1417, 1354, 1298, 1281, 1261, 1223, 1189, 1140, 1021, 840, 750 cm−1; HRMS (EI +): m/z calcd. (C13H14N2O3S) [M+] 278.0725, found 278.0727.
Preparation of pyrimidin-2-amines 6a–6d
The pyrimidin-2-amines were prepared similar to a reported procedure [20]. Compounds 5a, 5b, 5c, or 5d were suspended in dioxane and concentrated aqueous NH3 was added. The reaction mixture was subjected to microwave irradiation at 120 °C. The solvents were evaporated in vacuo to dryness. Water was added and the mixture was extracted 5 times with CH2Cl2. The combined organic layers were washed once with water, dried over anhydrous Na2SO4, and filtered. The solvent was evaporated in vacuo giving a crystalline residue. The crude products were purified by means of sublimation at reduced pressure yielding the products as white needles.
4-Methyl-6-phenylpyrimidin-2-amine (6a, C11H11N3)
Reaction of 200 mg of 5a (0.81 mmol) with 1.34 cm3 of concentrated aqueous NH3 (18 mmol) in 4.8 cm3 of dioxane for 2 h yielded 66 mg of 6a (44%). The melting point (169–170 °C) corresponded well with the reported one (169–171 °C) [21]. Rf = 0.63 (CH2Cl2:CH3OH = 9:1); 1H NMR (DMSO-d6, 400 MHz): δ = 2.29 (s, 3H, CH3), 6.58 (s, 2H, NH2), 7.02 (s, 1H, H-5), 7.45–7.47 (m, 3H, ArH), 8.02–8.05 (m, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 23.91 (CH3), 105.38 (C-5), 126.86, 128.80, 130.43 (ArC), 137.46 (ArCq), 163.75 (C-6), 163.90 (C-2), 168.39 (C-4) ppm; IR (KBr): \(\overline{v}\) = 3323, 3194, 1637, 1601, 1579, 1551, 1464, 1380, 1353, 1227, 770, 708 cm−1; HRMS (EI +): m/z calcd. (C11H11N3) [M+] 185.0953, found 185.0942.
4-(4-Chlorophenyl)-6-methylpyrimidin-2-amine (6b, C11H10ClN3)
Reaction of 215 mg of 5b (0.76 mmol) with 1.27 cm3 of concentrated aqueous NH3 (17 mmol) in 4.6 cm3 of dioxane for 3 h yielded 50 mg of 6b (30%). The melting point (198 °C) was similar to the reported one (204 °C) [22]. Rf = 0.47 (CH2Cl2:CH3OH = 20:1); 1H NMR (DMSO-d6, 400 MHz): δ = 2.28 (s, 3H, CH3), 6.62 (s, 2H, NH2), 7.03 (s, 1H, H-5), 7.52 (d, J = 8.4 Hz, 2H, ArH), 8.05 (d, J = 8.8 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 23.91 (CH3), 105.27 (C-5), 128.62, 128.86 (ArC), 135.19, 136.26 (ArCq), 162.42 (C-4), 163.86 (C-2), 168.68 (C-6) ppm; IR (KBr): \(\overline{v}\) = 3306, 3162, 1639, 1595, 1572, 1551, 1492, 1463, 1089, 808 cm−1; HRMS (EI +): m/z calcd. (C11H10ClN3) [M+] 219.0563, found 219.0553.
4-Methyl-6-(4-methylphenyl)pyrimidin-2-amine (6c, C12H13N3)
Reaction of 200 mg of 5c (0.76 mmol) with 1.27 cm3 (16.70 mmol) of concentrated aqueous NH3 in 4.6 cm3 of dioxane for 3 h yielded 100 mg of 6c (66%). The melting point (148–149 °C) corresponded well with the reported one (149–150 °C) [22, 22]. Rf = 0.54 (CH2Cl2:CH3OH = 9:1); 1H NMR (DMSO-d6, 400 MHz): δ = 2.27 (s, 3H, CH3), 2.33 (s, 3H, ArCH3), 6.53 (s, 2H, NH2), 6.98 (s, 1H, H-5), 7.26 (d, J = 8.1 Hz, 2H, ArH), 7.94 (d, J = 8.4 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 21.10 (ArCH3), 23.89 (CH3), 105.02 (C-5), 126.79, 129.40 (ArC), 134.65, 140.17 (ArCq), 163.67 (C-6), 163.86 (C-2), 168.19 (C-4) ppm; IR (KBr): \(\overline{v}\) = 3361, 3186, 1638, 1576, 1550, 1514, 1462, 1379, 1351, 809, 799 cm−1; HRMS (EI +): m/z calcd. (C12H13N3) [M+] 199.1109, found 199.1097.
4-(4-Methoxyphenyl)-6-methylpyrimidin-2-amine (6d, C12H13N3O)
Reaction of 213 mg of 5d (0.77 mmol) with 1.27 cm3 of concentrated aqueous NH3 (16.9 mmol) in 4.6 cm3 of dioxane for 3 h yielded 55 mg of 6d (33%). The melting point (202 °C) corresponded well with the reported one (204 °C) [24]. Rf = 0.74 (CH2Cl2:CH3OH = 9:1); 1H NMR (DMSO-d6, 400 MHz): δ = 2.26 (s, 3H, CH3), 3.80 (s, 3H, OCH3), 6.47 (s, 2H, NH2), 6.96 (s, 1H, H-5), 7.01 (d, J = 9.2 Hz, 2H, ArH), 8.01 (d, J = 8.8 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 23.90 (CH3), 55.46 (OCH3), 104.56 (C-5), 114.13, 128.39 (ArC), 129.71, 161.25 (ArCq), 163.33 (C-4), 163.79 (C-2), 167.98 (C-6) ppm; IR (KBr): \(\overline{v}\) = 3310, 3176, 1636, 1608, 1579, 1545, 1514, 1463, 1417, 1382, 1351, 1249, 1233, 1182, 1033, 820 cm−1; HRMS (EI +): m/z calcd. (C12H13N3O) [M+] 215.1059, found 215.1045.
Preparation of (pyrrolidin-1-yl)pyrimidines 7a–7d
The compounds 5a, 5b, 5c, or 5d were dissolved in dry THF and pyrrolidine was added. The reaction mixture was refluxed at 85 °C overnight or subjected to microwave irradiation. Water was added and the mixture was extracted five times with diethyl ether. The combined organic layers were washed neutral with water, dried over anhydrous Na2SO4, and filtered. The solvent was evaporated in vacuo giving pure compounds 7a–7d as white to yellowish needles. For analytical purposes they were recrystallized giving white needles.
4-Methyl-6-phenyl-2-(pyrrolidin-1-yl)pyrimidine (7a, C15H17N)
Method 1: Reaction of 218 mg of 5a (0.88 mmol) in 45 cm3 of dry THF with 157 mg of pyrrolidine (2.21 mmol) yielded 200 mg of 7a (95%). Method 2: Reaction of 200 mg of 5a (0.81 mmol) in 4 cm3 of dry THF with 172 mg of pyrrolidine (2.42 mmol) yielded after 1 h microwave irradiation at 100 °C 148 mg of 7a (76%). Rf = 0.84 (CH2Cl2:CH3OH = 10:1); m.p.: 88 °C (ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ = 1.90 (t, J = 6.6 Hz, 4H, (CH2)2), 2.33 (s, 3H, CH3), 3.60 (br, s, 4H, N(CH2)2), 6.72 (s, 1H, H-5), 7.34–7.36 (m, 3H, ArH), 7.97–7.99 (m, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 24.45 (CH3), 25.51 ((CH2)2), 46.63 (N(CH2)2), 104.33 (C-5), 126.93, 128.46, 129.98 (ArC), 138.10 (ArCq), 160.70 (C-2), 163.95 (C-6), 167.69 (C-4) ppm; IR (KBr): \(\overline{v}\) = 2971, 2928, 2857, 1587, 1555, 1512, 1482, 1458, 1374, 1336, 1223, 1183, 771, 693 cm−1; HRMS (EI +): m/z calcd. (C15H17N3) [M+] 239.1422, found 239.1421.
4-(4-Chlorophenyl)-6-methyl-2-(pyrrolidin-1-yl)pyrimidine (7b, C15H16ClN3)
Reaction of 300 mg of 5b (1.06 mmol) in 5 cm3 of dry THF with 225 mg of pyrrolidine (3.16 mmol) yielded after 2 h microwave irradiation at 120 °C 87 mg of 7b (30%). Rf = 0.85 (CH2Cl2:CH3OH = 10:1); m.p.: 87 °C (MeOH); 1H NMR (CDCl3, 400 MHz): δ = 1.97–2.01 (m, 4H, (CH2)2), 2.41 (s, 3H, CH3), 3.65–3.68 (m, 4H, N(CH2)2), 6.76 (s, 1H, H-5), 7.41 (d, J = 8.4 Hz, 2H, ArH), 8.00 (d, J = 8.8 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 24.49 (CH3), 25.52 ((CH2)2), 46.66 (N(CH2)2), 104.04 (C-5), 128.25, 128.68 (ArC), 136.03, 136.56 (ArCq), 160.62 (C-2), 162.70 (C-4), 167.99 (C-6) ppm; IR (KBr): \(\overline{v}\) = 2866, 1595, 1582, 1552, 1513, 1487, 1459, 1376, 1338, 1223, 1090, 1013, 806 cm−1; HRMS (EI +): m/z calcd. (C15H16ClN3) [M+] 273.1033, found 273.1036.
4-Methyl-6-(4-methylphenyl)-2-(pyrrolidin-1-yl)pyrimidine (7c, C16H19N3)
Reaction of 400 mg of 5c (1.52 mmol) in 15 cm3 of dry THF with 651 mg of pyrrolidine (9.15 mmol) yielded 371 mg of 7c (96%). Rf = 0.60 (cyclohexane:ethyl acetate = 1:3); m.p.: 76 °C (ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ = 1.96–1.99 (m, 4H, (CH2)2), 2.38 (s, 3H, ArCH3), 2.39 (s, 3H, CH3), 3.65–3.68 (m, 4H, N(CH2)2), 6.77 (s, 1H, H-5), 7.24 (d, J = 8.1 Hz, 2H, ArH), 7.96 (d, J = 8.1 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 21.34 (ArCH3), 24.53 (CH3), 25.53 ((CH2)2), 46.56 (N(CH2)2), 104.03 (C-5), 126.82, 129.18 (ArC), 135.36, 140.08 (ArCq), 160.88 (C-2), 163.80 (C-6), 167.66 (C-4) ppm; IR (KBr): \(\overline{v}\) = 2944, 2870, 1553, 1508, 1482, 1459, 1375, 1337, 1220, 1182, 1111, 803 cm−1; HRMS (EI +): m/z calcd. (C16H19N3) [M+] 253.1579, found 253.1575.
4-(4-Methoxyphenyl)-6-methyl-2-(pyrrolidin-1-yl)pyrimidine (7d, C16H19N3O)
Reaction of 300 mg of 5d (1.08 mmol) in 10 cm3 of dry THF with 460 mg of pyrrolidine (6.47 mmol) yielded an oil which was purified by means of CC with cyclohexane:ethyl acetate (1:1) as eluent giving 276 mg of 7d (95%). Rf = 0.48 (cyclohexane:ethyl acetate = 1:1); m.p.: 88 °C (ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ = 1.96–1.99 (m, 4H, (CH2)2), 2.39 (s, 3H, CH3), 3.65–3.68 (m, 4H, N(CH2)2), 3.84 (s, 3H, OCH3), 6.74 (s, 1H, H-5), 6.95 (d, J = 8.4 Hz, 2H, ArH), 8.03 (d, J = 8.8 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 24.53 (CH3), 25.52 ((CH2)2), 46.54 (N(CH2)2), 55.26 (OCH3), 103.54 (C-5), 113.76, 128.32 (ArC), 130.62 (ArCq), 160.83 (C-2), 161.21 (ArCq), 163.32 (C-4), 167.52 (C-6) ppm; IR (KBr): \(\overline{v}\) = 2955, 2875, 1607, 1587, 1553, 1512, 1483, 1458, 1379, 1351, 1338, 1249, 1173, 1109, 1033, 839, 808, 789 cm−1; HRMS (EI +): m/z calcd. (C16H19N3O) [M+] 269.1528, found 269.1528.
Preparation of 2-(4-methylpiperazin-1-yl)pyrimidines 8a–8d
The compounds 5a, 5b, 5c, or 5d were dissolved in dry THF and 1-methylpiperazine was added. The reaction mixture was refluxed at 100 °C overnight or subjected to microwave irradiation. Water was added and the mixture was extracted five times with diethyl ether. The combined organic layers were washed neutral with water, dried over anhydrous Na2SO4 and filtered. The solvent was evaporated in vacuo giving a resin which was further purified.
4-Methyl-2-(4-methylpiperazin-1-yl)-6-phenylpyrimidine (8a, C16H20N4)
Reaction of 300 mg of 5a (1.21 mmol) in 12 cm3 of dry THF with 726 mg of 1-methylpiperazine (7.24 mmol) yielded a yellow resin which was purified by means of CC (CH2Cl2:CH3OH = 40:1, Alox neutral) giving 245 mg of 8a (75%) as a yellow oil which crystallized upon cooling. Rf = 0.62 (CH2Cl2:CH3OH = 40:1, Alox neutral); m.p.: 65 °C; 1H NMR (CDCl3, 400 MHz):δ = 2.36 (s, 3H, NCH3), 2.40 (s, 3H, CH3), 2.50–2.53 (m, 4H, H-3′), 3.96–3.98 (m, 4H, H-2′), 6.84 (s, 1H, H-5), 7.44–7.45 (m, 3H, ArH), 8.02–8.04 (m, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 24.50 (CH3), 43.62 (C-2′), 46.21 (NCH3), 55.04 (C-3′), 105.32 (C-5), 126.93, 128.51, 130.11 (ArC), 137.93 (ArCq), 162.00 (C-2), 164.00 (C-6), 168.06 (C-4) ppm; IR (KBr): \(\overline{v}\) = 2924, 2794, 1558, 1493, 1443, 1368, 1345, 1300, 1272, 1223, 1188, 1173, 1073, 1007, 994, 890, 815, 764, 687 cm−1; HRMS (EI +): m/z calcd. (C16H20N4) [M+] 268.1688, found 268.1693.
4-(4-Chlorophenyl)-6-methyl-2-(4-methylpiperazin-1-yl)pyrimidine (8b, C16H19ClN4)
Reaction of 375 mg of 5b (1.33 mmol) in 5 cm3 of dry THF with 787 mg of 1-methylpiperazine (7.86 mmol) yielded after 5 h microwave irradiation at 120 °C a yellow resin. It was purified by means of CC (CH2Cl2: CH3OH = 10:1) giving 235 mg of 8b (58%) as a white amorphous solid. Rf = 0.43 (CH2Cl2:CH3OH = 10:1); 1H NMR (DMSO-d6, 400 MHz):δ = 2.20 (s, 3H, NCH3), 2.33 (s, 3H, CH3), 2.34–2.36 (m, 4H, H-3′), 3.75–3.82 (m, 4H, H-2′), 7.11 (s, 1H, H-5), 7.53 (d, J = 8.4 Hz, 2H, ArH), 8.11 (d, J = 8.4 Hz, 2H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz):δ = 24.36 (CH3), 43.46 (C-2′), 46.06 (NCH3), 54.67 (C-3′), 105.06 (C-5), 128.71, 128.93 (ArC), 135.41, 136.11 (ArCq), 161.59 (C-2), 161.91 (C-4), 168.61 (C-6) ppm; IR (KBr): \(\overline{v}\) = 2930, 2795, 1585, 1556, 1508, 1491, 1447, 1371, 1345, 1303, 1283, 1269, 1093, 1006, 995, 804 cm−1; HRMS (EI +): m/z calcd. (C16H19ClN4) [M+] 302.1298, found 302.1310.
4-Methyl-6-(4-methylphenyl)-2-(4-methylpiperazin-1-yl)pyrimidine (8c, C17H22N4)
Reaction of 400 mg of 5c (1.52 mmol) in 3 cm3 of dry THF with 916 mg of 1-methylpiperazine (9.14 mmol) yielded after 7 h microwave irradiation at 120 °C a resin. It was purified by means of CC (CH2Cl2:CH3OH = 10:1) giving 334 mg of 8c (78%) as an orange oil which crystallized upon cooling. Rf = 0.43 (CH2Cl2:CH3OH = 10:1); m.p.: 73 °C; 1H NMR (CDCl3, 400 MHz): δ = 2.34 (s, 3H, NCH3), 2.38 (s, 3H, CH3), 2.39 (s, 3H, ArCH3), 2.48–2.50 (m, 4H, H-3′), 3.94–3.96 (m, 4H, H-2′), 6.80 (s, 1H, H-5), 7.24 (d, J = 8.1 Hz, 2H, ArH), 7.93 (d, J = 8.1 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 21.33 (ArCH3), 24.46 (CH3), 43.67 (C-2′), 46.25 (NCH3), 55.06 (C-3′), 104.94 (C-5), 126.81, 129.20 (ArC), 135.10, 140.25 (ArCq), 161.99 (C-2), 163.91 (C-6), 167.82 (C-4) ppm; IR (KBr): \(\overline{v}\) = 2930, 2793, 1585, 1570, 1556, 1504, 1447, 1371, 1345, 1300, 1285, 1272, 1008, 804 cm−1; HRMS (EI +): m/z calcd. (C17H22N4) [M+] 282.1844, found 282.1850.
4-(4-Methoxyphenyl)-6-methyl-2-(4-methylpiperazin-1-yl)pyrimidine (8d, C17H22N4O)
Reaction of 250 mg of 5d (0.90 mmol) in 9 cm3 of dry THF with 540 mg of 1-methylpiperazine (5.39 mmol) yielded after 3 days refluxing at 100 °C a residue which was crystallized from ethyl acetate giving 141 mg of 8d (53%) as white needles. Rf = 0.43 (CH2Cl2:CH3OH = 10:1); m.p.: 93 °C; 1H NMR (CDCl3, 400 MHz): δ = 2.34 (s, 3H, NCH3), 2.38 (s, 3H, CH3), 2.48–2.50 (m, 4H, H-3′), 3.84 (s, 3H, OCH3), 3.95 (br, s, 4H, H-2′), 6.77 (s, 1H, H-5), 6.95 (d, J = 8.4 Hz, 2H, ArH), 8.01 (d, J = 8.4 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 24.45 (CH3), 43.67 (C-2′), 46.25 (NCH3), 55.06 (C-3′), 55.24 (OCH3), 104.46 (C-5), 113.78, 128.33 (ArC), 130.34, 161.30 (ArCq), 161.95 (C-2), 163.44 (C-4), 167.67 (C-6) ppm; IR (KBr): \(\overline{v}\)2936, 2788, 1610, 1586, 1570, 1557, 1514, 1492, 1459, 1350, 1301, 1286, 1270, 1258, 1172, 1027, 1009, 813 cm−1; HRMS (EI +): m/z calcd. (C17H22N4O) [M+] 298.1794, found 298.1793.
Preparation of N-[5-(diethylamino)pentan-2-yl]pyrimidin-2-amines 9a–9d
The compounds 5a, 5b, 5c, or 5d were dissolved in dry THF and 2-amino-5-(diethylamino)pentane was added. The reaction mixture was subjected to microwave irradiation. Water was added and the mixture was extracted five times with diethyl ether. The combined organic layers were washed neutral with water, dried over anhydrous Na2SO4 and filtered. The solvent was evaporated in vacuo giving residues which were further purified.
N-[5-(Diethylamino)pentan-2-yl]-4-methyl-6-phenylpyrimidin-2-amine (9a, C20H30N4)
Reaction of 200 mg of 5a (0.81 mmol) in 4 cm3 of dry THF with 766 mg of 2-amino-5-(diethylamino)pentane (4.84 mmol) yielded after 12 h microwave irradiation at 120 °C a residue which was purified by means of CC (CH2Cl2:CH3OH = 40:1, Alox neutral) giving 122 mg of 9a (46%) as yellow oil. Rf = 0.24 (CH2Cl2: CH3OH = 40:1, Alox neutral); 1H NMR (CDCl3, 400 MHz): δ = 1.03 (t, J = 7.1 Hz, 6H, H-2′′), 1.26 (d, J = 6.5 Hz, 3H, H-1′), 1.54–1.65 (m, 4H, H-3′, H-4′), 2.38 (s, 3H, CH3), 2.50–2.55 (m, 2H, H-5′), 2.56 (q, J = 7.1 Hz, 4H, H-1′′), 4.23–4.28 (m, 1H, H-2′), 4.96 (d, J = 8.3 Hz, 1H, NH), 6.84 (s, 1H, H-5), 7.44–7.46 (m, 3H, ArH), 8.01–8.02 (m, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 11.28 (C-2′′), 21.12 (C-1′), 23.13 (C-4′), 24.32 (CH3), 35.11 (C-3′), 46.48 (C-2′), 46.71 (C-1′′), 52.71 (C-5′), 105.75 (C-5), 126.92, 128.55, 130.13 (ArC), 137.85 (ArCq), 162.27 (C-2), 164.44 (C-6), 168.25 (C-4) ppm; IR (KBr): \(\overline{v}\) = 3268, 2965, 1574, 1551, 1495, 1458, 1373, 1346, 1203, 1069, 767, 693 cm−1; HRMS (EI +): m/z calcd. (C20H30N4) [M+] 326.2470, found 326.2471.
4-(4-Chlorophenyl)-N-[5-(diethylamino)pentan-2-yl]-6-methylpyrimidin-2-amine (9b, C20H29ClN4)
Reaction of 246 mg of 5b (0.87 mmol) in 5 cm3 of dry THF with 826 mg of 2-amino-5-(diethylamino)pentane (5.22 mmol) yielded after 7.5 h microwave irradiation at 120 °C a residue which was purified by means of CC (CH2Cl2:CH3OH = 40:1, Alox neutral) giving 196 mg of 9b (62%) as yellow oil. Rf = 0.70 (CH2Cl2:CH3OH = 10:1); 1H NMR (CDCl3, 400 MHz): δ = 1.01 (t, J = 7.2 Hz, 6H, H-2′′), 1.25 (d, J = 6.5 Hz, 3H, H-1′), 1.53–1.62 (m, 4H, H-3′, H-4′), 2.37 (s, 3H, CH3), 2.45–2.48 (m, 2H, H-5′), 2.52 (q, J = 7.2 Hz, 4H, H-1′′), 4.20–4.25 (m, 1H, H-2′), 5.02 (d, J = 8.3 Hz, 1H, NH), 6.79 (s, 1H, H-5), 7.41 (d, J = 8.5 Hz, 2H, ArH), 7.96 (d, J = 8.4 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 11.44 (C-2′′), 21.03 (C-1′), 23.38 (C-4′), 24.29 (CH3), 35.08 (C-3′), 46.53 (C-2′), 46.70 (C-1′′), 52.78 (C-5′), 105.36 (C-5), 128.19, 128.71 (ArC), 136.12, 136.27 (ArCq), 162.21 (C-2), 163.11 (C-4), 168.45 (C-6) ppm; IR (KBr): \(\overline{v}\) = 3268, 2965, 2798, 1573, 1549, 1491, 1456, 1402, 1374, 1342, 1091, 1013, 808 cm−1; HRMS (EI +): m/z calcd. (C20H29ClN4) [M+] 360.2081, found 360.2103.
N-[5-(Diethylamino)pentan-2-yl]-4-methyl-6-(4-methylphenyl)pyrimidin-2-amine (9c, C21H32N4)
Reaction of 300 mg of 5c (1.14 mmol) in 3 cm3 of dry THF with 1.09 g of 2-amino-5-(diethylamino)pentane (6.86 mmol) yielded after 13 h microwave irradiation at 120 °C a residue which was purified by means of CC (CH2Cl2:CH3OH = 40:1, Alox neutral) giving 313 mg of 9c (81%) as orange oil. Rf = 0.23 (CH2Cl2:CH3OH = 40:1, Alox neutral); 1H NMR (CDCl3, 400 MHz):δ = 1.00 (t, J = 7.1 Hz, 6H, H-2′′), 1.24 (d, J = 6.6 Hz, 3H, H-1′), 1.51–1.60 (m, 4H, H-3′, H-4′), 2.36 (s, 3H, CH3), 2.40 (s, 3H, ArCH3), 2.43–2.46 (m, 2H, H-5′), 2.51 (q, J = 7.1 Hz, 4H, H-1′′), 4.21–4.24 (m, 1H, H-2′), 4.97 (d, J = 8.1 Hz, 1H, NH), 6.80 (s, 1H, H-5), 7.24 (d, J = 8.1 Hz, 2H, ArH), 7.92 (d, J = 8.1 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 11.56 (C-2′′), 21.02 (C-1′), 21.33 (ArCH3), 23.45 (C-4′), 24.24 (CH3), 35.14 (C-3′), 46.50 (C-2′), 46.71 (C-1′′), 52.84 (C-5′), 105.30 (C-5), 126.79, 129.21 (ArC), 135.01, 140.25 (ArCq), 162.21 (C-2), 164.32 (C-6), 167.96 (C-4) ppm; IR (KBr): \(\overline{v}\) = 2965, 1574, 1549, 1510, 1454, 1374, 1345, 1182, 806 cm−1; HRMS (EI +): m/z calcd. (C21H32N4) [M+] 340.2627, found 340.2636.
N-[5-(Diethylamino)pentan-2-yl]-4-(4-methoxyphenyl)-6-methylpyrimidin-2-amine (9d, C21H32N4O)
Reaction of 300 mg of 5d (1.08 mmol) in 3 cm3 of dry THF with 1.02 g of 2-amino-5-(diethylamino)pentane (6.47 mmol) yielded after 6 h microwave irradiation at 120 °C a residue which was purified by means of CC (CH2Cl2:CH3OH = 40:1, Alox neutral). The fractions containing the product were combined, solvents evaporated, and the residue was subjected again to CC (CH2Cl2:CH3OH = 60:1, Alox neutral) giving 250 mg of 9d (65%) as yellow oil. Rf = 0.35 (CH2Cl2:CH3OH = 60:1, Alox neutral); 1H NMR (CDCl3, 400 MHz):δ = 1.00 (t, J = 7.3 Hz, 6H, H-2′′), 1.24 (d, J = 6.6 Hz, 3H, H-1′), 1.50–1.60 (m, 4H, H-3′, H-4′), 2.35 (s, 3H, CH3), 2.43–2.46 (m, 2H, H-5′), 2.51 (q, J = 7.3 Hz, 4H, H-1′′), 3.85 (s, 3H, OCH3), 4.21–4.24 (m, 1H, H-2′), 4.96 (d, J = 8.1 Hz, 1H, NH), 6.77 (s, 1H, H-5), 6.96 (d, J = 8.8 Hz, 2H, ArH), 8.00 (d, J = 8.8 Hz, 2H, ArH) ppm; 13C NMR (CDCl3, 100 MHz): δ = 11.56 (C-2′′), 21.02 (C-1′), 23.46 (C-4′), 24.24 (CH3), 35.15 (C-3′), 46.49 (C-2′), 46.71 (C-1′′), 52.85 (C-5′), 55.26 (OCH3), 104.80 (C-5), 113.80, 128.33 (ArC), 130.25, 161.30 (ArCq), 162.16 (C-2), 163.85 (C-4), 167.81 (C-6) ppm; IR (KBr): \(\overline{v}\) = 2964, 1575, 1547, 1510, 1453, 1374, 1348, 1305, 1294, 1250, 1171, 1033, 810 cm−1; HRMS (EI +): m/z calcd. (C21H32N4O) [M+] 356.2576, found 356.2585.
Preparation of N-[2-[(pyrimidin-2-yl)amino]ethyl]-7-chloroquinolin-4-amines 10a–10d
The compounds 5a, 5b, 5c, or 5d were dissolved in dry THF and N-(2-aminoethyl)-7-chloroquinolin-4-amine was added. The reaction mixture was subjected to microwave irradiation. The solution was transferred into a separatory funnel and water was added. The mixture was extracted fives times. The combined organic layers were washed with water, dried over anhydrous Na2SO4, and filtered. The solvent was evaporated in vacuo giving a residue which was further purified.
7-Chloro-N-[2-[(4-methyl-6-phenylpyrimidin-2-yl)amino]ethyl]quinolin-4-amine (10a, C22H20ClN5)
Reaction of 400 mg of 5a (1.61 mmol) in 8 cm3 of dry THF with 1.43 g of N-(2-aminoethyl)-7-chloroquinolin-4-amine (6.44 mmol) yielded after 7 h microwave irradiation at 120 °C and extraction with CH2Cl2 a residue. This was purified using CC (CH2Cl2:CH3OH = 40:1, Alox neutral) giving 285 mg of 10a (45%) as white foam. Rf = 0.36 (CH2Cl2:CH3OH = 40:1, Alox neutral); 1H NMR (DMSO-d6, 400 MHz): δ = 2.33 (br, s, 3H, CH3), 3.46–3.51 (m, 2H, H-3′), 3.58–3.72 (m, 2H, H-2′), 6.62–6.88 (m, 1H, ArH), 7.07 (s, 1H, H-5), 7.29–7.50 (m, 6H, 2NH, ArH), 7.77 (s, 1H, ArH), 8.04–8.24 (m, 3H, ArH), 8.28–8.42 (m, 1H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 24.04 (CH3), 39.07 (C-2′), 42.45 (C-3′), 98.96 (ArC), 105.61 (C-5), 117.60 (ArCq), 124.10, 124.20, 126.96, 127.73, 128.90, 130.60 (ArC), 133.54, 137.54, 149.29, 150.33 (ArCq), 152.07 (ArC), 162.75 (C-2), 163.53 (C-6), 168.55 (C-4) ppm; IR (KBr): \(\overline{v}\) = 3269, 2953, 1582, 1556, 1429, 1374, 1348, 1299, 1231, 1140, 769 cm−1; HRMS (EI +): m/z calcd. (C22H20ClN5) [M+] 389.1407, found 389.1404.
7-Chloro-N-[2-[[4-(4-chlorophenyl)-6-methylpyrimidin-2-yl]amino]ethyl]quinolin-4-amine (10b, C22H19Cl2N5)
Reaction of 226 mg of 5b (0.8 mmol) in 5 cm3 of dry THF with 714 mg of N-(2-aminoethyl)-7-chloroquinolin-4-amine (3.22 mmol) yielded after 5.5 h microwave irradiation at 120 °C and extraction with tert-butylmethyl ether a residue. This was purified by CC (CH2Cl2:CH3OH = 10:1, Alox neutral) giving 62 mg of 10b (18%) as a white-yellow amorphous solid. Rf = 0.26 (CH2Cl2:CH3OH = 10:1, Alox neutral); 1H NMR (DMSO-d6, 400 MHz): δ = 2.32 (br, s, 3H, CH3), 3.47–3.50 (m, 2H, H-3′), 3.56–3.70 (m, 2H, H-2′), 6.64–6.80 (m, 1H, ArH), 7.09 (s, 1H, H-5), 7.33–7-56 (m, 5H, 2NH, ArH), 7.76 (s, 1H, ArH), 8.08 (d, J = 8.3 Hz, 2H, ArH), 8.14 (br, s, 1H, ArH), 8.34–8.38 (m, 1H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 23.94 (CH3), 38.81 (C-2′), 42.01 (C-3′), 98.98 (ArC), 105.47 (C-5), 117.63 (ArCq), 124.12, 127.72, 128.70, 128.93 (ArC), 133.54, 135.32, 149.29, 150.33 (ArCq), 152.07 (ArC), 162.24 (C-4), 162.70 (C-2), 168.63 (C-6) ppm; IR (KBr): \(\overline{v}\) = 3433, 3268, 2927, 1577, 1556, 1492, 1456, 1371, 1342, 1330, 1239, 1140, 1092, 1013, 808 cm−1; HRMS (EI +): m/z calcd. (C22H19Cl2N5) [M+] 423.1017, found 423.1056.
7-Chloro-N-[2-[[4-methyl-6-(4-methylphenyl)pyrimidin-2-yl]amino]ethyl]quinolin-4-amine (10c, C23H22ClN5)
Reaction of 200 mg of 5c (0.76 mmol) in 4 cm3 of dry THF with 676 mg of N-(2-aminoethyl)-7-chloroquinolin-4-amine (3.05 mmol) yielded after 7 h microwave irradiation at 120 °C and extraction with CH2Cl2 a residue. This was purified by CC (CH2Cl2:CH3OH = 40:1, Alox neutral) giving 133 mg of 10c (43%) as an off-white amorphous solid. Rf = 0.27 (CH2Cl2:CH3OH = 40:1, Alox neutral); 1H NMR (DMSO-d6, 400 MHz): δ = 2.31 (br, s, 3H, CH3), 2.36 (s, 3H, ArCH3), 3.47–3.50 (m, 2H, H-3′), 3.54–3.71 (m, 2H, H-2′), 6.59–6.88 (m, 1H, ArH), 7.04 (s, 1H, H-5), 7.25–7.31 (m, 4H, ArH, NH), 7.45 (t, J = 4.9 Hz, 1H, NH), 7.77 (br, s, 1H, ArH), 7.97–7.99 (m, 2H, ArH), 8.13 (br, s, 1H, ArH), 8.30–8.45 (m, 1H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 21.15 (ArCH3), 24.06 (CH3), 39.25 (C-2′), 42.49 (C-3′), 98.95 (ArC), 105.24 (C-5), 117.60 (ArCq), 124.11, 124.20, 126.89, 127.73, 129.48 (ArC), 133.54, 134.72, 140.39, 149.28, 150.34 (ArCq), 152.06 (ArC), 162.70 (C-2), 163.45 (C-6), 168.14 (C-4) ppm; IR (KBr): \(\overline{v}\) = 3264, 2924, 1582, 1513, 1447, 1431, 1369, 1347, 1330, 1239, 1140, 810, 802 cm−1; HRMS (EI +): m/z calcd. (C23H22ClN5) [M+] 403.1564, found 403.1579.
7-Chloro-N-[2-[[4-(4-methoxyphenyl)-6-methylpyrimidin-2-yl]amino]ethyl]quinolin-4-amine (10d, C23H22ClN5O)
Reaction of 200 mg of 5d (0.72 mmol) in 4 cm3 of dry THF with 637 mg of N-(2-aminoethyl)-7-chloroquinolin-4-amine (2.87 mmol) yielded after 7 h microwave irradiation at 120 °C and extraction with CH2Cl2 a residue. This was purified by CC (CH2Cl2:CH3OH = 60:1, Alox neutral) giving 194 mg of 10d (64%) as a yellowish amorphous solid. Rf = 0.20 (CH2Cl2:CH3OH = 60:1, Alox neutral); 1H NMR (DMSO-d6, 400 MHz): δ = 2.31 (br, s, 3H, CH3), 3.47–3.50 (m, 2H, H-3′), 3.54–3.72 (m 2H, H-2′), 3.82 (s, 3H, OCH3), 6.59–6.87 (m, 1H, ArH), 7.02–7.04 (m, 3H, H-5, ArH), 7.22 (t, J = 5.9 Hz, 1H, NH), 7.27–7.43 (m, 1H, ArH), 7.45 (t, J = 5.1 Hz, 1H, NH), 7.77 (s, 1H, ArH), 8.05 (d, J = 8.1 Hz, 2H, ArH), 8.14 (br, s, 1H, ArH), 8.32–8.44 (m, 1H, ArH) ppm; 13C NMR (DMSO-d6, 100 MHz): δ = 24.18 (CH3), 38.92 (C-2′), 42.58 (C-3′), 55.52 (OCH3), 98.95 (ArC), 104.76 (C-5), 114.22 (ArC), 117.59 (ArCq), 124.11, 124.20, 127.71, 128.49 (ArC), 129.68, 133.55, 149.26, 150.34 (ArCq), 152.06 (ArC), 161.36 (ArCq), 162.65 (C-2), 163.18 (C-4), 168.06 (C-6) ppm; IR (KBr): \(\overline{v}\) = 3265, 2929, 1579, 1559, 1510, 1457, 1437, 1371, 1342, 1331, 1251, 1240, 1178, 1031, 807 cm−1; HRMS (EI +): m/z calcd. (C23H22ClN5O) [M+] 419.1513, found 419.1529.
In vitro assays
The in vitro growth inhibition assay of P. falciparum K1 was performed according to an established procedure [25]. The in vitro growth inhibition assay of P. falciparum NF54 and the in vitro growth inhibition assay of Trypanosoma b. rhodesiense, as well as the assay for the determination of cytotoxicity against L6-cells were performed as described earlier [26].
References
Akinsolu FT, Nemieboka PO, Njuguna DW, Ahadji MN, Dezso D, Varga O (2019) Int J Environ Res Public Health 16:1925
Holota S, Kryshchyshyn A, Derkach H, Trufin Y, Demchuk I, Gzella A, Grellier P, Lesyk R (2019) Bioorg Chem 86:126
Kennedy PGE (2006) Int J Parasitol 36:505
WHO (2019) World Malaria Report 2019. World Health Organization, Geneva
van der Pluijm RW, Imwong M, Chau NH, Hoa NT, Thuy-Nhien NT, Thanh NV, Jittamala P, Hanboonkunupakarn B, Chutasmit K, Saelow C, Runjarern R, Kaewmok W, Tripura R, Peto JT, Yok S, Suon S, Sreng S, Mao S, Oun S, Yen S, Amaratunga C, Lek D, Huy R, Dhorda M, Chotivanich K, Ashley EA, Mukaka M, Waithira N, Cheah PY, Maude RJ, Amato R, Pearson RD, Gonçalves S, Jacob CG, Hamilton WL, Fairhurst RM, Tarning J, Winterberg M, Kwiatkowski DP, Pukrittayakamee S, Hien TT, Day NPJ, Miotto O, White NJ, Dondorp AM (2019) Lancet Infect Dis 201:952
Curd FHS, Rose FL (1946) J Chem Soc 343
Curd FHS, Davis MI, Rose FL (1946) J Chem Soc 351
Curd FHS, Richardson DN, Rose FL (1946) J Chem Soc 378
Curd FHS, Davis MI (1946) J Chem Soc 370
Basford FR, Curd FHS, Rose FL (1946) J Chem Soc 713
Curd FHS, Davis MI (1946) J Chem Soc 720
Hull R, Lovell BJ (1947) J Chem Soc 41
Christian DJ, Bhoi MN, Borad MA, Rajani DP, Rajani SD, Patel HD (2014) World J Pharm Pharm Sci 3:1955
Seebacher W, Faist J, Presser A, Weis R, Saf R, Kaserer T, Temml V, Schuster D, Ortmann S, Otto N, Bauer R (2015) Eur J Med Chem 101:552
Singh K, Kaur H, Chibale K, Balzarini J, Little S, Bharatam PV (2012) Eur J Med Chem 52:82
Hoffelner M, Petritsch M, Ahmad S, Seebacher W, Dolensky J, Hochegger P, Kaiser M, Mäser P, Saf R, Weis R (2019) Monatsh Chem 150:1959
Zigeuner G, Hamberger H, Ecker R (1970) Monatsh Chem 101:881
Gotor V, Brieva R, Foces-Foces MC, Cano FH (1989) Tetrahedron 45:1783
Wendelin W, Schermanz K, Schweiger K, Fuchsgruber A (1983) Monatsh Chem 114:1371
Lu W, Liu Y, Ma H, Zheng J, Tian S, Sun Z, Luo L, Li J, Zhang H, Yang Z-J, Zhang X (2017) ACS Chem Neurosci 8:1980
Goswami S, Jana S, Dey S, Adak AK (2007) Aust J Chem 60:120
Yu Z, Chen F (1990) Yingyong Huaxue 7:54
Yu Z, Chen F (1991) Chem Abstr 114:185428
Chauhan SMS, Junjappa H (1976) Tetrahedron 32:1911
Seebacher W, Wolkinger V, Faist J, Kaiser M, Brun R, Saf R, Bucar F, Gröblacher B, Brantner A, Merino V, Kalia Y, Scapozza L, Perozzo R, Weis R (2015) Bioorg Med Chem Lett 25:1390
Mohsin N-ul-A, Seebacher W, Faist J, Hochegger P, Kaiser M, Mäser P, Saf R, Weis R (2018) Monatsh Chem 149:99
Funding
Open access funding provided by University of Graz.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hoffelner, M., Hassan, U., Seebacher, W. et al. New 2-aminopyrimidine derivatives and their antitrypanosomal and antiplasmodial activities. Monatsh Chem 151, 1375–1385 (2020). https://doi.org/10.1007/s00706-020-02674-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-020-02674-7