
Abstract
The diarylheptanoid curcumin is the yellow coloring agent accumulated in the rhizome of the common spice turmeric (Curcuma longa L.). It has gathered a lot of pharmaceutical interest over the last decades due to some positive effects on human health. However, the use of curcumin as a drug is prevented by its low bioavailability and solubility in water. Interestingly, piperine, the pungent constituent of household pepper (Piper nigrum L.) is able to increase the bioavailability of curcumin up to 20-fold without any known adverse effects. The mechanism responsible for this piperine-based increase of curcumin bioavailability is, however, not fully understood. In a recent publication, a quantum chemical study suggested the formation of a molecular complex between curcumin and piperine being responsible for this effect. The present work now revealed that indeed a 1:1 complex formation can be observed in NMR titration experiments and by mass spectrometry, but the complex strength is rather low (K ~ 1.5 dm3/mol). Furthermore, it is shown that the presence of piperine does not increase the water solubility of curcumin, which makes it rather improbable that such a complex is the main reason for an enhanced curcumin bioavailability.
Graphic abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
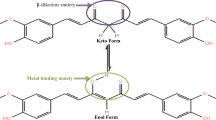
The diarylheptanoid curcumin (1), the yellow coloring agent of the rhizome of turmeric (Curcuma longa L.), is probably best known from its use in commercial curry mixtures. Over the last decades, a large variety of positive pharmacological effects have been attributed to curcumin, although the validity of these findings in vivo is currently debated [1]. Furthermore, curcumin is safe to consume even at high doses (ca. 10 g orally) [2]. Unfortunately, curcumin is hardly bioavailable, which can be attributed to the fact that it is rapidly metabolized and eliminated from the system [2]. Furthermore, it is poorly soluble in water and hence, not very well absorbed by the human body [3, 4]. One interesting way to mitigate these problems is the addition of piperine (2). This is the pungent constituent of household pepper (Piper nigrum L.) and is able to increase the bioavailability of curcumin up to 20-fold with no known adverse effects [5,6,7]. Structures of curcumin (1) and piperine (2) are shown in Fig. 1.

Molecular structures of curcumin (1) and piperine (2). There are two tautomeric forms of curcumin (1a and 1b), although in solution the enol form 1b is predominantly present [8]. Numbering of positions is in accordance with those given in Tables S1 and S2
However, the mechanism of how piperine is able to increase the bioavailability of curcumin is not fully understood, although many different hypotheses exist. First, piperine is a known inhibitor of hepatic and intestinal glucuronidation, which is a major metabolic pathway for orally applied curcumin [9]. Second, piperine is reported to decrease the secretion of hydrochloric acid in the stomach, increase the blood supply to the gastrointestinal tract and increase the permeability of intestinal epithelial cells [10] as well as stimulate γ-glutamyl transpeptidase, a protein which transports various amino acids across cellular membranes [5, 11].
Recently, a quantum chemical study suggested that piperine features appropriate electronic properties to form an intercalation complex with curcumin through enolic intermolecular hydrogen bonding and π − π interactions between these two molecules. This complex may facilitate the transport of curcumin through the metabolic pathways and transport through the blood–brain barrier [6]. The present work now investigates the properties of such complex formation utilizing NMR titrations, mass spectrometry, and solubility measurements.
Results and discussion
NMR titrations and mass spectrometry of the complex
For this study, commercially available curcumin (1) and piperine (2) were used. NMR measurements were performed using methanol-d4 as solvent due to the low solubility of curcumin in water as well as in buffered aqueous solvents with defined pH values. Methanol-d4 is one of the few common ‘protic’ NMR solvents and thus probably resembles aqueous conditions the most, although solubility of both compounds is rather moderate. Using aqueous solvents with an addition of non-‘protic’ polar solvents, e.g. DMSO-d6, an amount of up to 70% of DMSO is necessary to enable reasonable solubility of curcumin [8].
Figures 2 and S1 show the results from an NMR titration (Job's method) of curcumin and piperine performed in methanol-d4 to identify the resulting complex and to determine its stoichiometry. The maximum of the curve resides at a molar fraction of 0.5 indicating a 1:1 complex [12]. However, the differences in the chemical shift determined within the titration are very small, they only appeared on the third digit after the comma on the parts per million scale (Figs. 3 and S2). This indicates a rather weak interaction between the molecules and therefore the presence of a weak complex. Only very small dipole–dipole interactions, hardly visible in a NOESY measurement of the complex, support such a weak interaction (Fig. S3).

Job’s Plot of curcumin (1) and piperine (2) determined by chemical shift changes of the signal from protons in positions 1 and 7, recorded in methanol-d4. Comparable Job’s Plot of protons in position 3 of piperine is shown in Fig. S1

Observed 1H NMR signals (doublets) of protons in position 1 and 7 of curcumin (1) from representative spectra of the Job’s Plot methanol-d4. Note the extremely small shift differences between the measurements. Molar ratio of 1/2: a: 10/0; b 9/1; c: 7/3; d: 5/5; e: 3/7; f: 1/9; g: 0/10. Comparable 1H NMR signal shifts of other protons in curcumin and piperine (2) are shown in Fig. S2
The presence of the complex was proven using mass spectrometric measurements, whereas a small peak of the 1:1 complex was detectable next to the overwhelming peaks of the two pure substances when measuring a mixture of curcumin and piperine. Such peak of the course cannot be detected, when only one of the compounds is present. Further MS/MS investigations of this peak utilizing a more concentrated stock solution showed some dissociation into curcumin (1) and piperine (2) even at no collision energy. This indicates that the complex is not formed in the gas phase, but in solution prior to evaporation and charging by addition of Na+. Figure 4 shows the MS/MS spectrum of the 1:1 complex peak at 6 eV collision energy. These results indicate and confirm a weak interaction between the two molecules in the formed intermolecular complex.

MS–MS spectrum of a curcumin–piperine 1:1 complex at 6 eV collision energy. Note the dissociation into two fragments with the m/z of curcumin (1) and piperine (2). Relevant calculated m/z values are: curcumin: [M+Na]+ 391.11576; [M]+ 368.12599; piperine: [M+Na]+ 308.12620; [M]+ 285.13649; 1:1 curcumin–piperine complex: [M+Na]+ 676.25225. Figure is edited for better readability, the unmodified figure can be found in Fig. S4
The determination of the complex formation constant (K) proved to be rather difficult due to the weak binding and relatively low solubility of curcumin (1) and piperine (2) in methanol-d4. The curcumin (1, host) concentration for the NMR titration was set to 1.3 mM which is at the lower end of observable concentrations in NMR. The piperine (2, guest) stock solution was set at a concentration of 245 mM, which is close to the solubility threshold for piperine (2) in methanol-d4. The thereby obtainable concentration difference is still far from a complete titration. Hence, a Rose–Drago Plot was chosen to interpret visually the obtained data (Fig. 5) [13]. It is clearly visible that the lines cross each other between 1/K = 0.4 and 0.6 mol/dm3, which corresponds to a K value between 1.1 and 2.0 dm3/mol. This signifies a weak complexation and corresponds well with ‘simple’ π − π interactions between the aromatic moieties of the two compounds [13, 14].

Rose–Drago Plot of protons in position 1 and 7 of curcumin (1) to determine the binding constant K of the curcumin–piperine complex in methanol-d4. 1/K can be determined at the meeting point of all lines, Δ0 represents the shift difference between bound and free host molecules. As this is unknown, it is varied arbitrarily. The method was originally developed for photometric measurements and later adapted for NMR measurements [13]. Comparable Rose–Drago Plots of protons in the two methoxy-groups of curcumin are shown in Fig. S5
Solubility experiments
Since water is the main solvent in human cells, measurements at aqueous conditions were performed. As solubility problems prohibit sensible (NMR) titration experiments in water, we decided to observe a possible influence of piperine on the solubility of curcumin. A comprehensive literature survey revealed that no definitive value has been reported so far. Reported values range between 0.6 µg/cm3 and 7.4 µg/cm3, in which the latter was determined after treatment with heat [15]. Jagannathan et al. [16] explored the solution behavior of curcumin in water and reported its ability to form stable aggregates of various sizes in water. The resulting yellow suspension/solution remains stable for more than a month.
Since the removal of these aggregates by centrifugation failed, we chose sterile filtering through a hydrophobic 0.2 µm PTFE filter. This resulted in a clear, non-yellow solution with a high reproducibility. While this method may not remove very small aggregates, we mainly focused on changes in curcumin (1) solubility in the presence of piperine (2) and not determining the absolute solubility of 1. After the filtration step, the solution was diluted 1:1 with DMSO which offers subsequently many advantages. On one hand, the calibration for determining the curcumin concentration can be done in this solvent mixture. Establishing this calibration curve in methanol caused some errors due to solvent effects (Fig. S6). On the other hand, DMSO stabilizes the solution and changes in temperature does not lead to re-aggregation and precipitation of curcumin. If any aggregates pass the sterile filter, the greatly improved solubility in the DMSO/water solution should dissolve them so that they do not interfere with the fluorescence measurements. Furthermore, the fluorescence emission strength slightly increased with the addition of DMSO through solvent effects [17].
To accelerate the dissolution of curcumin in water, the samples were heated to 90 °C for 1 h and to reach the equilibrium the samples were then stirred at RT for further 72 h. While this does not ensure a saturated solution, reproducible results have been obtained with this method that are in accordance with published values.
The undertaken solubility experiments showed that the solubility of curcumin alone is in the expected range (2.01 ± 0.28 µg/cm3) but drops significantly in the presence of piperine to 0.52 ± 0.1 µg/cm3 (Fig. 6). This finding could be an indication that piperine causes an effect on the size of curcumin aggregates in water, but it is much more likely that piperine competes against curcumin for ‘soluble places’ in water, since piperine itself is also sparingly soluble in water [18].

Solubility of curcumin (1) in water with and without the presence of piperine (2; saturated solution)
Conclusion
The 1:1 complex between piperine (2) and curcumin (1), which was earlier predicted based on quantum chemical calculations, could now be observed by spectroscopic and spectrometric methods. However, this complex is relatively weak and has a low binding constant in methanol-d4. Furthermore, it was shown that piperine does not have any influence to increase the solubility of curcumin in water. The observed complex between the two natural products can therefore not explain the reported bioavailability enhancement by improved solubility. Other hypotheses that do not rely on a direct molecular interaction between the two molecules seem therefore more plausible [9,10,11, 19, 20].
Experimental
All solvents and reagent-grade chemicals were purchased from commercial suppliers (Fluka, Aldrich, Arcos and TCI). The structures of curcumin (1) and piperine (2) were confirmed with NMR measurements (Tables S1 and S2) and proven to be free of relevant metal ion contents via elemental analysis (Table S3). Purchased curcumin was a mixture of three curcuminoids, hence curcumin was purified for titration experiments according to the procedure described in the following section. NMR spectra were recorded on a Bruker AVIII 600 spectrometer at 600.25 MHz (spectra (Figs. 3 and S2), Job’s Plots (Figs. 2 and S1), Rose–Drago Plots (Figs. 5 and S5), shift assignment (Tables S1 and S2), as well as a Bruker Ascend 700 spectrometer at 700.40 MHz (NOESY spectrum, Fig. S3). For a single measurement in an NMR titration experiment (Job’s Plot and Rose–Drago Plot) 32 scans with 64 k data points and a spectral width of 20 ppm were accumulated. All spectra were recorded at 300 K and processed with Topspin 3.5 software with 128 k data points and without window function.
Chemical shifts (δ) are reported in ppm: for 1H relative to residual non-deuterated solvent signals in methanol (δH = 3.31 ppm) and for 13C relative to solvent signals (methanol-d4, δC = 49.0 ppm). This allows to avoid an addition of possibly interfering reference compounds. Coupling patterns are designated as s(inglet), d(oublet), t(riplet), and m(ultiplet).
Purification of curcumin
An amount of 55 mg curcumin was dissolved in EtOAc, 1.0 g silica gel 60 (0.063–0.2 mm) was added and the resulting suspension was slowly dried to a fine powder under reduced pressure. The adsorbed curcumin on silica gel was placed on top of a packed column of 8 g silica gel. The chromatography was performed with a gradient from 1% EtOH in CHCl3 to 2% EtOH in CHCl3. Fractions of pure curcumin were identified via TLC (2% EtOH in CHCl3) and combined. The solvent was removed under reduced pressure and 22 mg (40% yield of total mass; 52% yield of theoretical curcumin content) of pure curcumin was obtained. Purity was verified via 1H NMR [21].
General considerations
For all NMR measurements, curcumin (1) was indicated as the “host molecule” and piperine (2) as the “guest molecule”. The curcumin doublet at 7.58 ppm was evaluated in all measurements. It is well separated from all other curcumin and piperine peaks and is composed of two protons (positions 1 and 7) which are located close to the important β-diketone moiety.
Determination of stoichiometry via NMR spectroscopy
Stock solutions of curcumin (1; 5.9 mM in methanol-d4) and piperine (2; 5.9 mM in methanol-d4) were prepared. The NMR samples were created by pipetting the necessary amounts to create different curcumin/piperine ratios directly into the NMR tubes. The solutions were then thoroughly homogenized by turning the NMR tubes upside down and back up for several minutes before the samples were measured on the 600 MHz spectrometer. The results were evaluated following the procedure described by Hirose [12].
Determination of stoichiometry via mass spectrometry
The stock solution was prepared by dissolving 2.7 mg curcumin and 2.1 mg piperine (1 eq.) in 1.0 cm3 of solvent (49.5% methanol/49.5% acetonitrile/1.0% water) and diluted 1:10 000 (two times 10 mm3 sample in 990 mm3 solvent). Samples were measured on a Bruker maXis ESI-Qq-oaRTOF mass spectrometer. To measure the complex itself, a higher concentrated sample was prepared by addition of 100 mm3 of the 1:100 diluted stock solution to the 1:10,000 diluted stock solution resulting in a dilution of 1:1000.
Determination of the binding constant via NMR
A 1.3 mM host stock solution of curcumin (1) in methanol-d4 was prepared. The guest stock solution consists of 245 mM piperine (2) dissolved in the (curcumin) host stock solution. The NMR tube was then filled with 600 mm3 of host stock solution. After the NMR measurement, the guest stock solution is added to the NMR tube, which is then measured again. This procedure was repeated until the desired concentration ratio of host to guest was reached. Furthermore, 600 mm3 of guest stock solution was added to another NMR tube and measured. The results were then analyzed using the Rose–Drago method adapted for NMR measurements [12, 13].
Water solubility
The calibration curve (Fig. S6) was established with five standard solutions of curcumin (1) dissolved in 50% DMSO/water within the concentration range of 0.45–1.45 μg/cm3. Four samples of 5 mg curcumin in 10 cm3 distilled water and four samples of 5 mg curcumin and 3.9 mg piperine (2; 1.0 eq.) in 10 cm3 distilled water were heated in a round bottom flask to 90 °C for 1 h and then stirred at RT for 72 h. The samples were then filtered (hydrophobic 0.2 μm PTFE sterile syringe filter; the first 1.0 cm3 was discarded). An amount of 1.5 cm3 of the filtered clear solution was diluted with 1.5 cm3 DMSO, left cooled down to RT and then measured in the fluorescence spectrometer (440 nm excitation wavelength, 515 nm emission wavelength, 10 nm excitation slit width, 10 nm emission slit width). The concentration was calculated using the previously established calibration curve.
References
Nelson KM, Dahli JL, Bisson J, Graham GF, Walters MA (2017) J Med Chem 60:1620
Vareed SK, Kakarala M, Ruffin MT, Crowell JA, Normolle DP, Djuric Z, Brenner DE (2008) Cancer Epidemiol Biomarkers Prev 17:1411
Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB (2007) Mol Pharm 4:807
Goel A, Kunnumakkara AB, Aggarwal BB (2008) Biochem Pharmacol 75:787
Dudhatra GB, Mody SK, Awale MM, Patel HB, Modi CM, Kumar A, Kamani DR, Chauhan BN (2012) Sci World J 2012:1
Patil VM, Das S, Balasubramanian K (2016) J Phys Chem A 120:3643
Shoba G, Joy D, Joseph T, Majeed M, Rajendran R, Srinivas P (1998) Planta Medica 64:353
Payton F, Sandusky P, Alworth WL (2007) J Nat Prod 70:143
Moorthi C, Senthil Kumar C, Kathiresan K (2014) Int J Pharm Pharm Sci 6:901
Khajuria A, Thusu N, Zutshi U (2002) Phytomedicine 9:224
Johri RK, Thusu N, Khajuria A, Zutshi U (1992) Biochem Pharmacol 43:1401
Hirose K (2001) J Incl Phenom Macrocycl Chem 39:193
Wachter HN, Fried V (1974) J Chem Educ 51:798
Rasool AA, Hussain AA, Dittert LW (1991) J Pharm Sci 80:387
Kurien BT, Singh A, Matsumoto H, Scofield RH (2007) Assay Drug Dev Technol 5:567
Jagannathan R, Abraham PM, Poddar P (2012) J Phys Chem B 116:14533
van Duuren BL (1961) J Org Chem 26:2954
Padalkar KV, Gaikar VG (2008) Sep Sci Technol 43:3097
Zeng X, Cai D, Zeng Q, Chen Z, Zhong G, Zhuo J, Gan H, Huang X, Zhao Z, Yao N, Huang D, Zhang C, Sun D, Chen Y (2017) Biopharm Drug Dispos 38:3
Pathania V, Gupta AP, Singh B (2006) J Liq Chromatogr Relat Technol 29:877
Reen RK, Jamwal DS, Taneja SC, Koul JL, Dubey RK, Wiebel FJ, Singh J (1993) Biochem Pharmacol 46:229
Acknowledgements
Open access funding provided by University of Vienna. The authors would like to acknowledge Susanne Felsinger (NMR Centre, University of Vienna) for recording the NMR spectra and Peter Unteregger (Mass Spectrometry Centre, University of Vienna) for recording the mass spectra, Johannes Theiner (Microanalysis Service, University of Vienna) for performing the elemental analysis and Aranka Komlodi (Institute of Organic Chemistry, University of Vienna) who performed preliminary experiments during her bachelor’s thesis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Traxler, F., Schinnerl, J. & Brecker, L. Spectroscopic studies on the molecular interactions of curcumin and piperine. Monatsh Chem 151, 325–330 (2020). https://doi.org/10.1007/s00706-020-02563-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-020-02563-z