
Abstract
The microsomal protein cytochrome b 5 , which is located in the membrane of the endoplasmic reticulum, has been shown to modulate many reactions catalyzed by cytochrome P450 (CYP) enzymes. We investigated the influence of exposure to the anticancer drug ellipticine and to two environmental carcinogens, benzo[a]pyrene (BaP) and 1-phenylazo-2-naphthol (Sudan I), on the expression of cytochrome b 5 in livers of rats, both at the mRNA and protein levels. We also studied the effects of these compounds on their own metabolism and the formation of DNA adducts generated by their activation metabolite(s) in vitro. The relative amounts of cytochrome b 5 mRNA, measured by real-time polymerase chain reaction analysis, were induced by the test compounds up to 11.7-fold in rat livers. Western blotting using antibodies raised against cytochrome b 5 showed that protein expression was induced by up to sevenfold in livers of treated rats. Microsomes isolated from livers of exposed rats catalyzed the oxidation of ellipticine, BaP, and Sudan I and the formation of DNA adducts generated by their reactive metabolite(s) more effectively than hepatic microsomes isolated from control rats. All test compounds are known to induce CYP1A1. This induction is one of the reasons responsible for increased oxidation of these xenobiotics by microsomes. However, induction of cytochrome b 5 can also contribute to their enhanced metabolism.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cytochromes b 5 are heme proteins which are capable of accepting and transferring a single electron [1]. One cytochrome b 5 protein, which is located in the membrane of endoplasmic reticulum (microsomal cytochrome b 5 ), is involved in fatty acid desaturation, cholesterol and plasmalogen biosynthesis, as well as in various hydroxylation reactions catalyzed by cytochrome P450 (CYP) enzymes [1–7]. The endoplasmic reticulum (microsomal) cytochrome b 5 is an integral membrane protein located on the outer surface of this cell compartment [3–5, 8] and consists of two domains, the larger soluble N-terminal heme-binding core and the smaller hydrophobic C-terminal tail, which anchors the protein to the membrane. A 15-amino acid flexible linker connects these two domains providing the heme domain with sufficient mobility to bind different redox partners, whereas the protein remains in the membrane. It has been postulated that the linker, consisting of at least sevenamino acids, is necessary for a productive interaction with CYP enzymes [9].
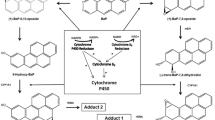
Cytochrome b 5 can accept an electron from either NADH:cytochrome b 5 reductase or NADPH:cytochrome P450 (CYP) oxidoreductase (POR) [4, 7, 10] and then reduced cytochrome b 5 transfers this electron to CYPs or other enzymes (Fig. 1). The role of microsomal cytochrome b 5 in the catalytic function of CYPs has not yet been fully understood. Cytochrome b 5 has been shown to be able to stimulate, inhibit or to have no effect on CYP-mediated reactions (reviewed in [2, 6, 7, 10]. One hypothesis trying to explain the influence of cytochrome b 5 on CYP reactions suggests a role of cytochrome b 5 in the direct transfer of the second electron to the CYP enzyme, which is considered to be the rate-limiting step in the catalytic cycle of the CYP monooxygenase reaction [10]. The electron transfer from reduced cytochrome b 5 to CYP is faster than the input of electrons from POR [7, 11]. Another possible mechanism of the cytochrome b 5 action is the formation of a complex between cytochrome b 5 and CYP, which can receive two electrons from POR in a single step, one for the reduction of CYP and the other for the reduction of cytochrome b 5 [7]. While CYP without cytochrome b 5 has to undergo two separate interactions with POR to complete one catalytic cycle, in the presence of cytochrome b 5 only one single interaction complex of CYP and cytochrome b 5 with POR is sufficient; cytochrome b 5 provides the second electron to CYP promptly after oxygen binding. Interaction of cytochrome b 5 with CYP may also induce conformational changes in CYP proteins leading to the breakdown of the oxygenated hemoprotein complex with substrates to products. This hypothesis is based on findings showing that not only the holoprotein of cytochrome b 5 , but also its apo-form (devoid of heme) which is not capable of electron transfer, can contribute to stimulation effects [6, 7, 10, 12].

Electron transfer between components of the mixed-function oxidase system (adopted from [4])
It has become clear from these investigations that the expression levels of cytochrome b 5 are crucial for the efficiency of several CYPs to oxidize some xenobiotics. This is also true for the oxidation of the anticancer drug ellipticine and the two environmental carcinogens benzo[a]pyrene (BaP) and 1-phenylazo-2-naphthol (Sudan I). The oxidation of these xenobiotics by CYP1A1 and/or 3A4 which dictates their biological effects can be strongly influenced by cytochrome b 5 [12–19]. However, the expression levels of cytochrome b 5 as well as the regulation of its expression after xenobiotic exposure remains largely unknown. Therefore, in this study we investigated the effect of ellipticine, BaP, and Sudan I on expression of cytochrome b 5 in vivo, both at the transcriptional and translational levels. Rats, shown to mimic the metabolism of these compounds in humans [14–25], were used as the animal model.
Results and discussion
Expression of cytochrome b5 mRNA and protein levels in rat liver after treatment with ellipticine, BaP, and Sudan I
We examined the effect of exposure of rats to three xenobiotics, the anticancer drug ellipticine and the two environmental carcinogens BaP and Sudan I, on the expression of cytochrome b 5 at the mRNA and protein levels in the liver, the major organ responsible for xenobiotic metabolism. We used the real-time polymerase chain reaction (RT-PCR) and Western blotting utilizing antibodies raised against rat cytochrome b 5 , to measure the mRNA and protein expression levels, respectively. As shown in Fig. 2, treatment of rats with ellipticine, BaP, and Sudan I induced expression of hepatic cytochrome b 5 mRNA. The highest induction was seen in the livers of Sudan I-exposed rats, where cytochrome b 5 mRNA expression increased more than 11-fold; mRNA expression was up to 1.7- and 2.7-fold higher in rats after treatment with ellipticine and BaP, respectively (Fig. 2).

Ellipticine-, BaP-, and Sudan I increased expression of cytochrome b 5 mRNA in rat livers. Values represent mean (n = 3); SDs were less than 10 %. Numbers (F) represent the fold increase over the control in the amounts of mRNA caused by the treatment of rats with ellipticine, BaP, and Sudan I. **P < 0.01; *** P < 0.001 (Student’s t test), significantly different from controls
In addition to the induction of cytochrome b 5 mRNA, treatment of rats to the test compounds resulted in an up to sevenfold increase in cytochrome b 5 protein levels. This increase in cytochrome b5 protein levels was similar for all compounds tested; ellipticine and BaP both caused a fivefold increase, while Sudan I elevated the level of this protein by sevenfold (Fig. 3).

Induction of cytochrome b 5 protein in livers of untreated (control) rats or rats treated with 40 mg/kg b.w. ellipticine, 125 mg/kg b.w. BaP, and 60 mg/kg b.w. Sudan I determined by Western blotting. Insert: Representative immunoblots of hepatic microsomal cytochrome b 5 (cyt b5) stained with antibody against rat cytochrome b 5 ; imunoblots of glyceraldehyde phosphate dehydrogenase (GAPDH) that was used as loading control. Control, microsomal fractions of control (untreated) rats. Values represent mean (n = 3); SDs were less than 10 %. Numbers (F) represent the fold increase over the control in the amounts of protein caused by the treatment of rats with ellipticine, BaP and Sudan I. ***P < 0.001 (Student’s t test), significantly different from control
The differences in cytochrome b 5 mRNA and protein induction by individual test xenobiotics might be caused by several phenomena, i.e. a route of administration of test compounds, a dosage scheme and/or different induction potency of these xenobiotics. Since ellipticine, BaP, and Sudan I are strong inducers of CYP1A1, we here used the route of administration and the dosage schemes that were analogous to those utilized in studies evaluating the induction of CYP1A1 mRNA and protein of this enzyme. Under these conditions, the ~34- and ~40-fold increases in expression levels of CYP1A1 protein were due to ellipticine and BaP, respectively [13, 26], whereas higher potency to induce this enzyme was caused by Sudan I; an ~80-fold increase in CYP1A1 protein expression levels was found [27]. These results indicate that the tendency for induction of CYP1A1 by the compounds examined was similar to that of cytochrome b 5 found in the present work (see Figs. 2 and 3).
The effect of the treatment of rats with ellipticine, BaP, and Sudan I on hepatic microsomal oxidation of these compounds and formation of DNA adducts by their reactive metabolite(s)
Using microsomes isolated from the livers of untreated and treated rats, we analyzed the oxidation of the test compounds to their metabolites by HPLC. As shown in Fig. 4, conversion of each of the test compounds to their metabolites by hepatic microsomes was changed by treatment of rats with the examined xenobiotic, thereby indicating that the test compound can influence their own metabolism. The main metabolites formed after the microsomal oxidation of ellipticine are 9-hydroxy-, 12-hydroxy-, 13-hydroxy-, 7-hydroxyellipticine, and ellipticine N 2-oxide (Fig. 4A) which is in line with previous studies [28]. Microsomal incubation with Sudan I resulted in the formation of 1-(4-hydroxyphenylazo)-2-naphthol (4′-OH-Sudan I), 1-(phenylazo)-naphthalene-2,6-diol (6-OH-Sudan I), and 1-(4-hydroxyphenylazo)-naphthalene-2,6-diol (4′,6-diOH-Sudan I) (Fig. 4C) which is in accordance with studies published previously [20]. Whereas pretreatment of the rats with ellipticine and Sudan I both resulted in an increase of the metabolites formed by these test compounds in microsomal incubations, the changes in the metabolic profile of BaP were more complex (Fig. 4b). The main BaP metabolites structurally identified previously in hepatic microsomal incubation [18] are: BaP-4,5-dihydrodiol, BaP-7,8-dihydrodiol, BaP-9,10-dihydrodiol, BaP-1,6-dione, BaP-3,6-dione, BaP-3-ol, and BaP-9-ol. In addition one metabolite (assigned metabolite Mx) has been found which structure has not yet been identified. The pattern of individual BaP metabolites formed by control microsomes was different from that generated by microsomes isolated from BaP-pretreated rats. While the formation of BaP-7,8-dihydrodiol and BaP-3-ol significantly increased in hepatic microsomal fraction isolated from BaP-pretreated rats, a significant decrease was found for the metabolites BaP-1,6-dione, BaP-3,6-dione, BaP-9-ol, and Mx (Fig. 4b). In this context it is noteworthy that a previous study showed that the formation of BaP-7,8-dihydrodiol and BaP-3-ol in an enzyme system containing rat CYP1A1 was increased by cytochrome b 5 [25]. Therefore, the elevated amounts of these BaP metabolites formed by liver microsomes of BaP-pretreated rats (compare Fig. 4b) might therefore be attributable to the BaP-mediated induction of cytochrome b 5 .

Oxidation of ellipticine (a), BaP (b), and Sudan I (c) in microsomes isolated from livers of untreated (control) rats or rats pretreated with 40 mg/kg b.w. ellipticine, 125 mg/kg b.w. BaP, and 60 mg/kg b.w. Sudan I. Values represent mean ± SD from three parallel measurements. *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test), significantly different from control
In further experiments, DNA adduct formation by ellipticine, BaP, and Sudan I incubated with microsomes isolated from the livers of untreated and pretreated rats was compared in vitro. Results with ellipticine and Sudan I have been previously published [13, 27, 29]. Ellipticine was activated by these microsomes to form four DNA adducts (data not shown), two of them were identical to those formed by 13-hydroxyellipticine and 12-hydroxyellipticine. Additionally, two minor ellipticine-derived DNA adducts were observed which have been also formed in vivo [22, 24, 30–32] and in vitro in several enzymatic systems including CYP enzymes and peroxidases [21, 23, 31–33], but the low adduct levels have prevented their structural characterization. As shown in Fig. 5 ellipticine-DNA adduct levels generated by hepatic microsomes isolated from ellipticine-pretreated rats were twofold higher than those formed by microsomes isolated from untreated rats [13].

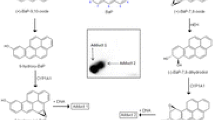
DNA adduct formation by ellipticine (a), BaP (b), and Sudan I (c) activated with microsomes isolated from livers of untreated (control) rats or rats pretreated with 40 mg/kg b.w. ellipticine, 125 mg/kg b.w. BaP, and 60 mg/kg b.w. Sudan I. a Adduct data has been published previously [13]. b Insert Autoradiographic profile of BaP-DNA adducts formed by hepatic microsomes of BaP-pretreated rats incubated with BaP and NADPH. The arrows show adduct 1 formed from 9-hydroxy-BaP-4,5-epoxide with deoxyguanosine in DNA [18] and dG-N 2-BPDE adduct (adduct 2). c Adduct data has been published previously [26]. For all panels, values represent mean total RAL (relative adduct labeling) ± SD (n = 4; duplicate analyses of two independent in vitro incubations). **P < 0.01, ***P < 0.001 (Student’s t test), significantly different from control
BaP was activated by rat hepatic microsomes to form two DNA adducts (Fig. 5b). The DNA adduct pattern obtained by thin-layer chromatrography is shown in the insert of Fig. 5; BaP-derived DNA adducts 1 and 2 were generated by incubation of BaP with DNA and microsomes isolated from livers of both untreated and BaP-pretreated rats. As discussed previously [18, 34], adduct 1 is probably derived from 9-hydroxy-BaP-4,5-epoxide with guanine and adduct 2 formed from BaP-7,8-dihydrodiol-9,10-epoxide with guanine (i.e. 10-(deoxyguanosin-N 2-yl)-7,8,9-trihydroxy-7,8,9,10-tetrahydro-BaP; dG-N 2-BPDE). Pretreatment of rats with BaP resulted in more than 40- and 15-fold higher levels of DNA adducts 1 and 2 in microsomal incubations, respectively (Fig. 5b).
As shown previously [20, 27, 35], hepatic microsomes isolated from both untreated and Sudan I-pretreated rats incubated with Sudan I and DNA in vitro generated one major DNA adduct and two minor adducts (partly overlapping with the major adduct). The major adduct was previously identified by 32P-postlabeling to be the 3′,5′-bisphospho-derivative of the 8-(phenylazo)deoxyguanosine adduct [20, 29]. Microsomes isolated from livers of Sudan I-pretreated rats were tenfold more efficient to form Sudan I-derived DNA adducts than those from untreated rats (compare Fig. 5c).
The increases in oxidation of ellipticine, BaP, and Sudan I and enhanced DNA adduct formation derived from these compounds by hepatic microsomes isolated from pretreated rats might be caused by several phenomena. As shown previously, ellipticine, BaP, and Sudan I act as strong inducers of CYP1A1 in livers of several species including rats [13, 14, 27, 35] and all test compounds are substrates of CYP1A1 [13–15, 18, 23–25, 27, 29, 36, 37]. Therefore, induction of CYP1A1 should be one of the reasons responsible for the elevated oxidation of these compounds and higher DNA adduct levels. This was also postulated by us and others in several former studies [13, 14, 27, 35].
However, for ellipticine and BaP, induction of cytochrome b 5 also seems to modulate their CYP-mediated metabolism. The results showing an increase in the oxidation of ellipticine to 7-hydroxyellipticine and 9-hydroxyellipticine by pretreatment of rats with this drug (Fig. 4a) are consistent with the fact that their formation is mainly catalyzed by CYP1A1 which is induced by ellipticine treatment [15, 23, 24, 32]. In addition, a twofold increase in the generation of 12-hydroxy- and 13-hydroxyellipticine was observed which, in the absence of cytochrome b 5 , are mainly formed by CYP3A (Fig. 4a). However, under the increased levels of this protein, caused by its induction in rat liver microsomes, the increase in formation of these ellipticine metabolites seems to be mediated mainly by CYP1A1. Namely, a previous study has shown that cytochrome b 5 alters the ellipticine metabolite profile formed by rat CYP1A1, demonstrating that the formation of 12-hydroxyellipticine and 13-hydroxyellipticine by rat CYP1A1 increased significantly by cytochrome b 5 [15]. The increase in ellipticine oxidation by hepatic microsomes from the ellipticine-pretreated rats to 12-hydroxy- and 13-hydroxyellipticine also explains the increase in the levels of ellipticine-derived DNA adducts 1 and 2, because these DNA adducts are generated by 12-hydroxy- (adduct 2) and 13-hydroxyellipticine (adduct 1) [23, 31].
Conclusion
Our study demonstrates that ellipticine, BaP, and Sudan I are capable of inducing cytochrome b 5 in the livers of rats exposed to these xenobiotics. Cytochrome b 5 mRNA expression was 1.7-, 2.7-, and 11-fold higher by treatment rats with ellipticine, BaP, and Sudan I, respectively, while 5-, 5- and 7-times higher levels of cytochrome b 5 protein were caused by the test xenobiotics. Since cytochrome b 5 is important for the oxidation of these xenobiotics by CYP1A1, the induction of both this enzyme and cytochrome b 5 by the test compounds exerts a concerted regulatory control on the CYP1A1-mediated oxidation of these xenobiotics, thereby modulating their own pharmacological and genotoxic potency. However, the mechanism of cytochrome b 5 induction is not yet known. As shown in several previous studies, all test compounds are ligands of the aryl hydrocarbon receptor (AhR) [13, 14, 18, 27, 32, 35, 38–42], the receptor whose activation is essential for the induction of several xenobiotic-metabolizing enzymes. Consequently, ellipticine, BaP, and Sudan I induce enzymes that are regulated by the activation of AhR such as CYP1A1 and NADPH:quinone oxidoreductase (NQO1) [13, 14, 18, 19, 27, 32, 35]. These findings suggest that the mechanism of induction of cytochrome b 5 might be analogous to the induction of these enzymes, namely a mechanism which depends on the activation of AhR. However, this hypothesis needs to be explored in future investigations.
Experimental
Ellipticine, BaP, and Sudan I (1-(phenylazo)-2-hydroxynaphthalene) were from Sigma Chemical Co (St Louis, MO, USA).
Animal experiments and isolation of microsomes
All animal experiments were conducted in accordance with the Regulations for the Care and Use of Laboratory Animals (311/1997, Ministry of Agriculture, Czech Republic), which is in compliance with the Declaration of Helsinki. Male Wistar rats (~125–150 g, AnLab, Czech Republic) placed in cages in temperature- and humidity-controlled rooms were acclimatized for 5 days and maintained at 22 °C with a 12 h light/dark period. Standardized diet (ST-1 diet from Velaz, Czech Republic) and water were provided ad libitum. Rats were treated with ellipticine, BaP, and Sudan I as follows: (1) Three 5-week-old male Wistar rats (~125–150 g) were treated i.p. with one dose of 40 mg of ellipticine per kg body weight (b.w.) by intraperitoneal injection as reported previously [13]. Ellipticine was dissolved in sunflower oil/dimethyl sulfoxide (1:1 (v/v), 1 cm3). Three control animals received an equal volume of solvent only [13]. This route of ellipticine administration was used here, because we utilized it in our former study that investigated ellipticine potency to induce CYP1A1 [13]. Rats were killed 48 h after the treatment by cervical dislocation. (2) Three 5-week-old male Wistar rats (~125–150 g) were treated p.o. by gastric gavages with a single dose of 150 mg/kg b.w. BaP dissolved in 1 cm3 sunflower oil as described previously [42]. Animals in the control group were treated with 1 cm3 of sunflower oil only. This route of BaP administration was used, because we utilized it in our former study that investigated potency of BaP to induce CYP1A1 [26]. Rats were killed 24 h after the last treatment by cervical dislocation. (3) Three 5-week-old male Wistar rats (~125–150 g) were injected i.p. with 20 mg/kg b.w. Sudan I dissolved in 1 cm3 maize oil once a day for 3 consecutive days as reported previously [27]. Animals in the control group received the same volume of maize oil on 3 days. This route of Sudan I administration and a dosage scheme were used, because we utilized them in our former study that investigated potency of Sudan I to induce CYP1A1 [27]. Rats were killed 24 h after the last treatment by cervical dislocation. For all treatment groups, livers of the animals were removed immediately after killing, frozen in liquid nitrogen, and stored at −80 °C until isolation of microsomal fractions. Pooled microsomes were prepared from 3 rat livers/group as reported [22, 26, 27]. Microsomal fractions were stored at −80 °C until analysis. Protein concentrations in the microsomal fractions were assessed using the bicinchoninic acid protein assay with bovine serum albumin as a standard [43].
Content of cytochrome b5 mRNA in rat livers
Total RNA was isolated from another aliquot of frozen organs using Trizol Reagent (Invitrogen, Carlsbad, CA, USA) according to the procedure supplied by the manufacturer. The quality of isolated RNA was verified by horizontal agarose gel electrophoresis, RNA quantity was assessed by UV–Vis spectrophotometry on a Carry 300 spectrophotometer (Varian, Palo Alto, CA, USA). RNA samples (1 μg) were reversely transcribed into cDNA using 200 U of reverse transcriptase per sample with random hexamer primers utilizing RevertAid™ First Strand cDNA Synthesis Kit (MBI Fermentas, Vilnius, Lithuania) according to the manufacturer’s instructions. RT-PCR was performed in RotorGene 2000 (Corbett Research, Sydney, Australia) under the following cycling conditions: incubation at 50 °C for 2 min and initial denaturation at 95 °C for 10 min, then 50 cycles of denaturation at 95 °C for 15 s and annealing at 60 °C for 1 min, and elongation for 30 s at 72 °C. Gain was set to 7 and fluorescence was acquired after elongation step. The PCR reaction mixtures (0.02 cm3) contained 0.009 cm3 cDNA diluted 10-times in Milli-Q ultrapure water (Biocel A10, Millipore, Billerica, MA, USA), 0.01 cm3 TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA), and 0.001 cm3 TaqMan Gene Expression Assay Mix (commercially available unlabeled PCR primers and FAM™ dye-labeled probe for rat cytochrome b 5 as the target gene and β-actin as internal reference standard gene). Each sample was analyzed in two parallel aliquots. Negative controls had the same compositions as samples but cDNA was omitted from the mixture. Data were analyzed by the program RotorGene v6 (Corbett Research, Sydney, Australia) and evaluated by comparative cycle threshold (cT) method for relative quantitation of gene expression as described [13].
Cytochrome b5 protein content in microsomes isolated from rat livers
Microsomes containing 30 μg microsomal proteins were subjected to electrophoresis on the 15 % polyacrylamide gel and applied onto a polyvinylidene fluoride (PVDF) membrane as reported [44]. The membranes were then exposed to specific rabbit polyclonal anti-cytochrome b 5 (1:750, AbCam, MA, USA) antibodies overnight at 4 °C and the antigen–antibody complex was visualized with an alkaline phosphatase-conjugated goat anti-rabbit IgG antibody (1:1428, Sigma-Aldrich, USA) and 5-bromo-4-chloro-3-indolylphosphate/nitrobluetetrazolium as chromogenic substrate. Protein bands were expressed as arbitrary units (AU)/mg protein as described previously [14, 19, 20]. Antibody against glyceraldehyde phosphate dehydrogenase (GAPDH) (1:750, Millipore, MA, USA) was used as loading control as recommended by the antibody producer.
Microsomal incubations to study metabolism of ellipticine, BaP, and Sudan I
Incubation mixtures used to study ellipticine oxidation were performed as described previously [21, 24]. Besides the NADPH-generation system the incubation mixtures contained 0.5 mg protein of pooled hepatic microsomal fraction and 0.01 mmol dm−3 ellipticine (dissolved in 0.005 cm3 methanol) in a final volume of 0.5 cm3. The reaction was initiated by adding ellipticine. In the control incubation, ellipticine or NADPH-generating system was omitted from the incubation mixture. After 20 min (37 °C) the reaction was stopped by adding 0.1 cm3 of 2 mol dm−3 NaOH. The oxidation of ellipticine has been shown to be linear up to 30 min of incubation [23, 28]. After incubation, 0.005 cm3 of 1 mol dm−3 phenacetin in methanol was added as an internal standard and the ellipticine metabolites were extracted twice with ethyl acetate (2 × 1 cm3). Subsequently the solvent was evaporated to dryness, the residues dissolved in 0.025 cm3 methanol and ellipticine metabolites separated by HPLC as described [15, 17, 23, 28].
Incubation mixtures used for studying BaP metabolism by hepatic microsomes contained 100 mmol dm−3 potassium phosphate buffer (pH 7.4), NADPH–generating system (1 mmol dm−3 NADP+, 10 mmol dm−3 D–glucose-6-phosphate, 1 U/cm3 d-glucose-6-phosphate dehydrogenase), 0.5 mg protein of pooled hepatic microsomal fraction and 0.05 mmol dm−3 BaP (dissolved in 0.005 cm3 DMSO) in a final volume of 0.5 cm3. The reaction was initiated by adding 0.05 cm3 of the NADPH-generating system. Control incubations were carried out either without microsomes, or without NADPH-generating system, or without BaP. After incubation (37 °C, 20 min), 0.005 cm3 1 mmol dm−3 phenacetin in methanol was added as an internal standard. BaP metabolism by microsomes has been shown to be linear up to 30 min of incubation [18]. BaP metabolites were extracted twice with ethyl acetate (2 × 1 cm3), solvent evaporated to dryness, residues dissolved in 0.025 cm3 methanol and BaP metabolites separated by HPLC as reported [45]. BaP metabolite peaks were collected and analyzed by NMR and/or mass spectrometry as described recently [18].
Incubation mixtures used to study Sudan I oxidation had a final volume of 0.75 cm3, and consisted of 50 mmol dm−3 potassium phosphate buffer (pH 7.4), 1 mmol dm−3 NADPH, 10 mmol dm−3 d-glucose 6-phosphate, 1 unit cm−3 D-glucose 6-phosphate dehydrogenase, 10 mmol dm−3 MgCl2, 0.5 mg protein of pooled hepatic microsomal fraction, and 0.1 mmol dm−3 Sudan I (dissolved in 0.0075 cm3 methanol). After incubation (37 °C, 20 min), Sudan I metabolites were extracted with ethyl acetate (2 × 1 cm3), the extracts evaporated, the residues dissolved 0.025 cm3 in methanol. Sudan I oxidation by microsomes has been shown to be linear up to 30 min [20]. HPLC analysis was performed as described previously [20].
Determination of DNA adduct formation by ellipticine, BaP, and Sudan I in vitro by 32P-postlabeling
Incubation mixtures used to assess DNA adduct formation by ellipticine [32], BaP [14], and Sudan I [20, 29] activated with microsomes isolated from rat liver consisted of 50 mmol dm−3 potassium phosphate buffer (pH 7.4), 1 mmol dm−3 NADPH, 0.5 mg of microsomal proteins, 0.1 mmol dm−3 ellipticine, BaP, or Sudan I (all dissolved in 0.0075 cm3 dimethyl sulfoxide), and 0.5 mg of calf thymus DNA in a final volume of 0.75 cm3. The reaction was initiated by adding 0.1 mmol dm−3 ellipticine, BaP, or Sudan I. Incubations at 37 °C containing ellipticine, BaP, or Sudan I were carried out for 60, 90, and 60 min, respectively. Ellipticine-, BaP-, and Sudan I-DNA adduct formation has been shown to be linear up to 90 min [14, 16, 20, 29]. Control incubations were carried out either without microsomes, without NADPH, without DNA, or without ellipticine, BaP, or Sudan I. After the incubation, DNA was isolated from the residual water phase by the phenol/chloroform extraction method. DNA adducts were analyzed with the nuclease P1 version of the 32P-postlabeling technique [32]. Resolution of the adducts by thin-layer chromatography using polyethylenimine-cellulose plates (Macherey and Nagel, Düren, Germany) was carried out as follows: (1) Ellipticine-DNA adducts as reported [13, 32]; (2) BaP-DNA adducts as described [14, 26]; and (3) Sudan I-DNA adducts as reported [27, 29]. DNA adduct levels (RAL, relative adduct labeling) were calculated as described [46].
Statistical analyses
For statistical data analysis we used Student’s t test. All P values are two-tailed and considered significant at the 0.05 level.
Change history
26 July 2017
An erratum to this article has been published.
References
Velick SF, Strittmatter P (1956) J Biol Chem 221:265
Vergeres G, Ramsden J, Waskell L (1995) J Biol Chem 270:3414
Vergeres G, Waskell L (1995) Biochimie 77:604
Porter TD (2002) J Biochem Mol Toxicol 16:311
Durr UH, Waskell L, Ramamoorthy A (2007) Biochim Biophys Acta 1768:3235
Durr UH, Yamamoto K, Im SC, Waskell L, Ramamoorthy A (2007) J Am Chem Soc 129:6670
Schenkman JB, Jansson I (2003) Pharmacol Ther 97:139
Sobrado P, Goren MA, James D, Amundson CK, Fox BG (2008) Protein Expr Purif 58:229
Clarke TA, Im SC, Bidwai A, Waskell L (2004) J Biol Chem 279:36809
Guengerich F (2005) Arch Biochem Biophys 440:204
Schenkman JB, Jansson I (1999) Drug Metab Rev 31:351
Kotrbova V, Aimova D, Ingr M, Borek-Dohalska L, Martinek V, Stiborova M (2009) Protein Expr Purif 66:203
Aimová D, Svobodová L, Kotrbová V, Mrázová B, Hodek P, Hudeček J, Václavíková R, Frei E, Stiborová M (2007) Drug Metab Dispos 35:1926
Arlt VM, Stiborova M, Henderson CJ, Thiemann M, Frei E, Aimova D, Singhs R, da Costa GG, Schmitz OJ, Farmer PB, Wolf CR, Philips DH (2008) Carcinogenesis 29:656
Kotrbova V, Mrazova B, Moserova M, Martinek V, Hodek P, Hudecek J, Frei E, Stiborova M (2011) Biochem Pharmacol 82:669
Stiborova M, Martinek V, Schmeiser HH, Frei E (2006) Neuro Endocrinol Lett 27(Suppl 2):35
Stiborova M, Indra R, Moserova M, Cerna V, Rupertova M, Martinek V, Eckschlager T, Kizek R, Frei E (2012) Chem Res Toxicol 25:1075
Stiborova M, Moserova M, Cerna V, Indra R, Dracinsky M, Šulc M, Henderson CJ, Wolf CR, Schmeiser HH, Phillips DH, Frei E, Arlt VM (2014) Toxicology 318:1
Vranová I, Moserová M, Hodek P, Kizek R, Frei E, Stiborová M (2013) Int J Electrochem Sci 8:1586
Stiborova M, Martinek V, Rydlova H, Hodek P, Frei E (2002) Cancer Res 62:5678
Stiborová M, Stiborová-Rupertová M, Bořek-Dohalská L, Wiessler M, Frei E (2003) Chem Res Toxicol 16:38
Stiborová M, Breuer A, Aimová D, Stiborová-Rupertová M, Wiessler M, Frei E (2003) Int J Cancer 107:885
Stiborova M, Sejbal J, Bořek-Dohalská L, Aimová D, Poljaková J, Forsterová K, Rupertová M, Wiesner J, Hudeček J, Wiessler M, Frei E (2004) Cancer Res 64:8374
Stiborova M, Frei E (2014) Current Med Chem 21:575
Indra R, Moserova M, Sulc M, Frei E, Stiborova M (2013) Neuro Endocrinol Lett 34(Suppl 2):55
Hodek P, Koblihova J, Kizek R, Frei E, Arlt VM, Stiborova M (2013) Environ Toxicol Pharmacol 36:989
Stiborová M, Dračínská H, Martínek V, Svášková D, Hodek P, Milichovský J, Hejduková Ž, Brotánek J, Schmeiser HH, Frei E (2013) Chem Res Toxicol 26:290
Stiborová M, Bořek-Dohalská L, Aimová D, Kotrbová V, Kukačková K, Janouchová K, Rupertová M, Ryšlavá H, Hudeček J, Frei E (2006) Gen Physiol Biophys 25:245
Stiborová M, Asfaw B, Frei E, Schmeiser HH, Wiessler M (1995) Chem Res Toxicol 8:489
Stiborova M, Rupertová M, Aimová D, Ryšlavá H, Frei E (2007) Toxicology 236:50
Stiborova M, Poljaková J, Ryšlavá H, Dračínský M, Eckschlager T, Frei E (2007) Int J Cancer 120:243
Stiborová M, Rupertová M, Frei E (2011) Biochim Biophys Acta 1814:175
Stiborová M, Bieler CA, Wiessler M, Frei E (2001) Biochem Pharmacol 62:1675
Krais AM, Speksnijder EN, Melis JP, Indra R, Moserova M, Godschalk RW, van Schooten FJ, Seidel A, Kopka K, Schmeiser HH, Stiborova M, Phillips DH, Luijten M, Arlt VM (2015) Arch Toxicol. doi:10.1007/s00204-015-1531-8
Refat NA, Ibrahim ZS, Moustafa GG, Sakamoto KQ, Ishizuka M, Fujita S (2008) J Biochem Mol Toxicol 22:77
Stiborová M, Asfaw B, Anzenbacher P, Hodek P (1988) Cancer Lett 40:327
Indra R, Moserova M, Kroftova N, Sulc M, Martinkova M, Adam V, Eckschlager T, Kizek R, Arlt VM, Stiborova M (2014) Neuro Endocrinol Lett 35(Suppl 2):105
Lubet RA, Connolly G, Kouri RE, Nebert DW, Bigelow SW (1983) Biochem Pharmacol 32:3053
Gasiewicz TA, Kende AS, Rucci G, Whitney B, Willey JJ (1996) Biochem Pharmacol 52:1787
Chang CY, Puga A (1998) Mol Cell Biol 18:525
Nebert DW, Dalton TP, Okey AB, Gonzalez FJ (2004) J Biol Chem 279:23847
Endo K, Uno S, Seki T, Ariga T, Kusumi Y, Mitsumata M, Yamada S, Makishima M (2008) Toxicol Appl Pharmacol 230:135
Wiechelman KJ, Braun RD, Fitzpatrick JD (1988) Anal Biochem 75:231
Arlt VM, Poirier MC, Sykes SE, Kaarthik J, Moserova M, Stiborova M, Wolf R, Henderson CJ, Phillips DH (2012) Toxicol Lett 213:160
Moserova M, Kotrbova V, Aimova D, Sulc M, Frei E, Stiborova M (2009) Interdisc Toxicol 2:239
Schmeiser HH, Stiborova M, Arlt VM (2013) Methods Mol Biol 1044:389
Acknowledgments
Supported by GACR (Grant 14-8344S) and Charles University (Grant UNCE 204025/2012). Work at King’s College London is also supported by Cancer Research UK (Grant Number C313/A14329).
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at https://doi.org/10.1007/s00706-017-2011-z.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stiborová, M., Moserová, M., Mrízová, I. et al. Induced expression of microsomal cytochrome b 5 determined at mRNA and protein levels in rats exposed to ellipticine, benzo[a]pyrene, and 1-phenylazo-2-naphthol (Sudan I). Monatsh Chem 147, 897–904 (2016). https://doi.org/10.1007/s00706-015-1636-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-015-1636-z