
Abstract
Japanese encephalitis (JE) is a zoonotic epidemic disease caused by Japanese encephalitis virus (JEV), and currently, no medicines are available to treat this disease. Autophagy modulators play an important role in the treatment of tumors, heart disease, and some viral diseases. The aim of this study was to investigate the effects of autophagy modulators on JEV infection and the host response in mice. The experimental mice were grouped as follows: DMEM (control), JEV, JEV+rapamycin (JEV+Rapa), JEV+wortmannin (JEV+Wort), JEV+chloroquine (JEV+CQ), Rapa, Wort, and CQ. The control group was treated with DMEM. The mice in other groups were infected with 105 PFU of JEV, and Rapa, Wort, and CQ were administered 2 h prior to JEV challenge and then administered daily for 10 consecutive days. All mice were monitored for neurological signs and survival. The damage of subcellular structures in the mouse brain was evaluated by transmission electron microscopy. The distribution of virus in the mouse brain was determined by RNAScope staining and immunohistochemical staining. The neuroinflammatory responses in the brain were examined via quantitative real-time PCR, and the signal pathways involved in neuroinflammation were identified by Western blot. The mice in the JEV+Wort and JEV+CQ groups showed milder neurological symptoms, less damage to the mitochondria in the brain tissue, and a higher survival rate than those in the JEV+Rapa and JEV groups. Compared with the JEV+Rapa and JEV groups, the distribution of JEV in the brain of mice in the JEV+Wort and JEV+CQ groups was lower, and the inflammatory response was weaker. No significant difference was observed in the expression of the PI3K/AKT/NF-κB pathway in mouse brain among the different groups. Our study suggests that the autophagy inhibitors Wort and CQ reduce JEV infection and weaken the inflammatory response, which does not depend on the PI3K/AKT/NF-κB pathway in mouse brain.
Similar content being viewed by others

Background
Japanese encephalitis (JE) is a mosquito-borne zoonotic infection caused by Japanese encephalitis virus (JEV). JEV is a neurotropic virus whose clinical manifestations range from hyperthermia syndrome to multifocal central nervous system disease to death [1]. Currently, JEV is mainly prevalent in the Asia-Pacific region, with about 68,000 cases of JEV infection each year and a mortality rate of 25%–30%, and 50% of the survivors are affected by neuropsychiatric sequelae [2,3,4]. In the future, JEV is likely to become an emerging global pathogen [5]. At present, inactivated vaccines are safe and reliable, but the production and purification processes are complicated [6]. Although live attenuated vaccines offer good protection, there is a potential threat of mutant strains reverting to the pathogenic strain. In addition, the source cell line is not recognized by the FDA, and the scope of application is greatly restricted [7]. Therefore, therapeutic drugs for JEV should be investigated. Previous studies have shown that the autophagy pathway positively regulates the replication of JEV in the early stage of viral infection in vitro [8].
Autophagy is a highly conserved homeostatic process through which cytoplasmic macromolecules, excess or damaged organelles, and some pathogens are delivered to lysosomes for degradation [9, 10]. Autophagy usually occurs at a basal level in all cells but is upregulated in response to extracellular or intracellular stress and pathogen infection [11]. Some drugs that regulate autophagy, including rapamycin (Rapa), wortmannin (Wort), and chloroquine (CQ), have been used in basic and clinical research on some diseases. Rapa is a commonly used autophagy inducer that can promote the occurrence of autophagy by inhibiting the MTOR pathway [12, 13]. Wort, a hydrophobic fungal metabolite of the fungus Talaromyces wortmanni, has been widely used as a powerful tool to examine the role of PI3K in cellular signaling [14]. As an early autophagy inhibitor, Wort can prevent autophagosome formation primarily by inhibiting the activity of class III phosphatidylinositide 3 kinases [15, 16]. CQ can inhibit autophagosome and lysosome fusion and/or prevent the degradation of autophagic lysosomal material downstream (in the late stage) of autophagosome formation [17, 18].
In recent years, the role of autophagy in viral infections has also attracted attention. Immune cells can degrade viral proteins in time through enhanced autophagy, thereby inhibiting the replication of related viruses [19]. However, enhanced autophagy can promote the replication of related viruses, including human immunodeficiency virus, herpes simplex virus, enterovirus 71, poliovirus, and coxsackievirus B4 [20]. The administration of the drugs 3-MA or Wort and CQ to inhibit autophagy can reduce the production of influenza virus [21, 22]. Measles virus strains with different virulence properties play obviously different roles in promoting viral replication and antiviral defense in the autophagy pathway [23]. Therefore, the interaction between the virus and the autophagy pathway depends on the cell line used and its intracellular and extracellular environment. Moreover, the effect of the autophagy pathway may be very different in in vivo experiments. In this study, the results suggest that the autophagy inhibitors Wort and CQ reduce JEV infection and weaken inflammatory response in mice.
Materials and methods
JEV strain
JEV (P3 strain) was stored at the State Key Laboratory of Agricultural Microbiology, Huazhong Agricultural University. The JEV P3 strain was amplified in the brains of neonatal mice, and virulence was determined using a plaque test in BHK-21 cells [24].
Establishment of the JE mouse model and treatment
Six-week-old female BALB/c mice were used in this study. The experiment was conducted in strict accordance with the Regulations on the Administration of Laboratory Animals in Hubei Province. The use of laboratory animals was approved by the Scientific Ethic Committee of Huazhong Agricultural University (number HZAUMO-2018-059). A total of 305 mice were divided into eight groups: 272 mice in the infection groups, including JEV (105 PFU, 0.1 mL), JEV (105 PFU, 0.1 mL) + Rapa (5 mg/kg [0.5 mg in 1 mL DMEM per mouse], 0.2 mL), JEV (105 PFU, 0.1 mL) + Wort (1 mg/kg [0.2 mg in 1 mL DMEM per mouse], 0.2 mL), and JEV (105 PFU, 0.1 mL) + CQ (50 mg/kg [5 mg in 1 mL DMEM per mouse], 0.2 mL). The remaining mice were divided among the control groups, including DMEM (0.1 mL), Rapa (5 mg/kg, 0.2 mL), Wort (1 mg/kg, 0.2 mL), and CQ (50 mg/kg, 0.2 mL). In particular, Rapa, Wort, and CQ were administered 2 h prior to JEV challenge and then administered daily for 10 consecutive days. The mice were given continuous access to food for 20 days, with daily observation and recording of their clinical signs. Clinical signs were scored based on a previously reported scale for JEV-infected mice [24]. All experiments were conducted using the protocol recommended by the Research Ethics Committee of the College of Veterinary Medicine, Huazhong Agricultural University, Hubei Province, China.
Collection of mouse brain samples
Five mice in each group were sacrificed at 5, 7, 10, and 15 days after JEV infection. The remaining mice were sacrificed at 20 days after JEV infection. The mice were sacrificed by cervical dislocation prior to collection of brain tissue samples. The left brain was frozen. Portions of the right cerebral cortex were taken (about the size of a sesame seed) and fixed in 2.5% glutaraldehyde, with the remaining portions fixed in 4% formaldehyde.
Transmission electron microscopy (TEM)
After the small pieces of tissue were completely fixed in 2.5% glutaraldehyde, the tissues were embedded using a pure embedding medium (anhydrous acetone mixed with an embedding agent at a volume ratio of 1:1). After the tissue boundaries were trimmed, the tissues were sliced into ultra-thin sections (80–100 nm), which were stained sequentially with 4% uranyl acetate and lead citrate. The samples were observed and photographed using a TECNA110 transmission electron microscope (Philips, Netherlands).
Paraffin sectioning
After the brain tissues were fixed with 4% formaldehyde for 48 h, they were dehydrated using an ethanol gradient, embedded in paraffin with the cut surface down, and serially sectioned at 5 μm. Subsequently, the sections were subjected to different staining methods.
Hematoxylin–eosin (HE) staining
The standard HE staining method was adopted to stain selected tissues: the nuclei were stained with hematoxylin, whereas the cytosol and extracellular matrix were stained with eosin, followed by mounting with neutral gum.
Immunohistochemical (IHC) staining
Paraffin sections were dewaxed and placed in 3% H2O2 to quench endogenous peroxidase and were heated in citrate buffer at 96 °C to complete antigen retrieval. After washing, the sections were blocked in 5% BSA for 1 h. Then, they were incubated with a mouse anti-JEV primary antibody (1:100, provided by the State Key Laboratory of Agricultural Microbiology, Huazhong Agricultural University) and a secondary antibody (HRP-labelled goat anti-mouse/rabbit IgG, Gene Tech Co., Ltd., Shanghai, China). Finally, color development was performed with DAB, and hematoxylin was counterstained. All IHC-stained sections were scanned using a Leica Aperio CS2 section scanning system.
RNAScope staining
RNAScope staining was performed according to the instructions of the RNAScope staining kit as follows. The sections were first dewaxed and dried completely, and RNAscope hydrogen peroxide was then added dropwise. Afterward, the slides were placed in boiling RNAscope target retrieval reagent for antigen retrieval. Next, the slides were incubated with the appropriate probe at 40 °C. After washing, the samples were detected with a Fast Red substrate, counterstained with hematoxylin, and mounted in glycerogelatin. All IHC-stained sections were scanned using a Leica Aperio CS2 scanner.
Tissue immunofluorescence
Paraffin sections were dewaxed in water and placed in 3% H2O2 to quench endogenous peroxidase. Antigen retrieval was performed by heating the sections in citrate buffer at 96 °C. After washing, the sections were blocked with 5% BSA for 1 h. Then, they were incubated with primary antibodies (mouse anti-JEV-E antibody, 1:100, State Key Laboratory of Agricultural Microbiology, Huazhong Agricultural University; rabbit anti-LC3A/B antibody, 1:100, Seville Biotech Co., Ltd.) and secondary antibodies (FITC goat anti-mouse IgG; Cy3 goat anti-rabbit IgG). After washing, DAPI staining, and mounting with an anti-fluorescence quencher, the fluorescence signals were detected using a fluorescence confocal microscope.
Quantitative real-time PCR (qPCR)
The total RNA of brain tissue was extracted with TRIzol Reagent, following the manufacturer’s instructions, and then reverse transcribed into cDNA using a TAKARA reverse transcription kit. Afterward, the qPCR reaction was carried out using a TAKARA qPCR kit. The primer sequences for the qPCR reaction are shown in Table 1. The reaction conditions for qPCR were as follows: pre-denaturation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s, and melting curve analysis at 95 °C for 15 s, 60 °C for 30 s, and 95 °C for 15 s. The relative expression levels of target genes in each sample were calculated using the 2-ΔΔCt analysis method.
Western blot
Total tissue protein was extracted using RIPA lysate, and protein quantitation was performed using the BCA method, with each sample adjusted to have the same protein content. After a prepared gel of a suitable concentration was fixed in an electrophoresis tank, protein samples and a marker were added to the sample wells using a micropipette for electrophoresis. Each excised protein band (gel slice) was transferred to a PVDF membrane, blocked with 5% skim milk at 37 °C, and incubated with TBST-diluted primary antibodies overnight at 4 °C. The primary antibodies were as follows: PI3 Kinase P85 alpha (ABclonal), Phospho-AKT (BOSTER), Phospho-JNK1/2 (ABclonal), Phospho-ERK1 (ABclonal), NF-κB (ABclonal), and GAPDH (Servicebio). Next, TBST-diluted secondary antibodies were added for incubation at 37 °C, and a color-developing solution was used to produce colored bands, whose grey values were analyzed using ImageJ software. The relative expression levels of PI3K, P-AKT, P-ERK, P-JNK, and P65 proteins in brain tissues were measured using GAPDH as an internal reference. Quantitative statistics of the grey values of related proteins were performed using ImageJ software.
Statistical analysis
Data are expressed as the mean ± SD, and intergroup differences were analyzed using one-way ANOVA, with P < 0.05* and P < 0.01** indicating significance and extreme significance, respectively.
Results
Neurological symptoms in mice infected with JEV
Neurological symptoms in mice infected with JEV were observed and scored [24]. The JEV+Rapa group had a long duration of clinical signs (5–20 days after JEV infection), and noticeable phenotypic signs of piloerection, arched back, and motor dysfunction were present. The JEV group had a short duration of signs (5–12 days after JEV infection) and showed significant neurological signs. The JEV+Wort group showed only mild mental depression and piloerection at 7–10 days post JEV infection and then returned to normal. No significant neurological signs were observed in the JEV+CQ, DMEM control, and drug control groups (Figs. 1A and B). The JEV+Rapa group had a 65.5% morbidity rate and the lowest survival rate among all groups. The JE incidence in the JEV group was 32.7%. The JEV+Wort group had a lower morbidity rate and a survival rate of 90%. The survival rate in the JEV+CQ group was nearly 100%. No clinical signs were observed in the DMEM and drug control groups (Fig. 1C and D).

Autophagy inhibitors alleviate neurological signs in Japanese encephalitis virus (JEV)-infected mice. (A) The JEV and JEV+ Rapa groups developed neurological signs at 5-12 days after JEV infection. (B) Behavioural scores showed that the neurological signs disappeared in the JEV group at 12 days after JEV infection. The mice in the JEV+Rapa group had obvious neurological signs 5-20 days after virus infection, and those in the JEV+ Wort group exhibited piloerection. The other groups did not show evident neurological signs. Behaviour score: 0 = asymptomatic; 1 = mental depression and piloerection; 2 = mental depression, piloerection, and arched back; 3 = mental depression, piloerection, arched back, and motor disorder; 4 = mental depression, piloerection, arched back, motor disorder, and eyelid swelling. (C) Statistical analysis of JEV prevalence in mice (the ratio of the number of symptomatic mice to the total number of mice). (D) Statistical analysis of survival rate of mice infected with virus after 20 days (the ratio of the number of surviving mice to the total number of mice).
Relationship between autophagy and viral infection
The brain tissues of three mice in different treatment groups were subjected to TEM to observe the damage of the subcellular structure, and co-localization of the autophagy factor LC3 with the JEV E protein was observed using tissue immunofluorescence. These results showed that severe mitochondrial damage had occurred in the brain tissue of mice in the JEV and JEV+Rapa groups, while the damage in the brain tissue of mice in the JEV+Wort and JEV+CQ groups was mild (Fig. 2). Meanwhile, co-localization of LC3 and the E protein occurred in more neurons in the brain tissue of the JEV and JEV+Rapa groups. However, a small number of neurons in the brain tissue of JEV+Wort and JEV+CQ groups exhibited co-localization signals (Fig. 3).

Autophagy inhibitors have a protective effect on the subcellular structure of brain tissue. Transmission electron microscopy (TEM) was used to observe the pathological changes of subcellular structure in mouse brain tissue. Severe mitochondrial damage to brain tissue was observed in the JEV+Rapa and JEV groups, and slight damage to mitochondria of brain tissue was observed in the JEV+Wort and JEV+CQ groups (red arrow, mitochondria; scale bar, 2 µm).

Autophagy inhibitors reduce the colocalization of the autophagy factor LC3 and Japanese encephalitis virus (JEV) E protein in brain tissues of mice infected with JEV. (A) Confocal immunofluorescence images of the LC3 protein (red) and the JEV E protein (green) with a scale bar of 50 μm. Co-localization was observed in a large number of neurons in the brain of mice in the JEV+Rapa and JEV groups in fewer neurons in the brain of the mice in the JEV+Wort and JEV+CQ groups. (B) Average number of cells with co-localized LC3 protein and JEV E protein in multiple fields of view. Each error bar represents the standard deviation (SD) of the number of cells in three independent experiments, with each experiment involving 10 fields of view and each field containing about 150 cells.
Distribution of JEV in the brain tissues of JEV-infected mice
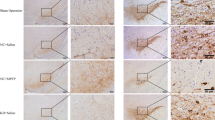
Brain tissues were collected at 10 days postinfection. Then, RNAScope staining and IHC staining were used to observe the distribution of the JEV-nucleic-acid-positive signal and to detect the distribution of the JEV-antigen-positive signal, respectively. RNAScope staining revealed that the viral nucleic acids of the JEV group were mainly distributed in the cerebral cortex and thalamus in brain tissues. In the JEV+Rapa group, JEV nucleic acids were mainly distributed in the cerebral cortex, olfactory tubercle, thalamus, mesencephalon, pons, and medulla oblongata. No obvious JEV-nucleic-acid-positive signal was seen in the brain tissues of the JEV+Wort and JEV+CQ groups. IHC staining showed that the JEV antigen in brain tissues of the JEV, JEV+Wort, and JEV+CQ groups was mainly distributed in the cerebral cortex, whereas that in the JEV+Rapa group was mainly concentrated in the cerebral cortex, thalamus, hypothalamus, mesencephalon, and pons (Fig. 4A).

Autophagy inhibitors reduce the distribution of Japanese encephalitis virus (JEV) in brain tissues of mice infected with JEV. (A) 10 days after JEV infection, RNAScope staining was used to observe the distribution of JEV nucleic acid positive signal (red), and immunohistochemical (IHC) staining was used to observe the distribution of JEV antigen (brown) (400 X). (B) The JEV load in brain tissues was measured using qPCR. Left, the viral load 10 days after JEV infection; right, the viral load 20 days after JEV infection. Total RNA was extracted from JEV-infected cells using TRIzol Reagent and reverse transcribed into cDNA using a TAKARA PrimeScriptTM RT reagent Kit with gDNA Eraser, followed by CT value determination based on the fluorescent dye in the TAKARA TB GreenTM Premix Ex TaqTM II kit and appropriate primers. Each error bar represents the standard deviation of three independent measurements for three mice in a group. One-way ANOVA was performed using Graph Pad Prism 6 software. **, p < 0.01; *, p < 0.05, compared with each group.
To confirm that autophagy inhibitor treatment might alleviate JEV infection in JEV-infected mice, qPCR was performed to determine the JEV load in mouse brain tissues at 10 and 20 days after JEV infection. The results showed that the JEV and JEV+Rapa groups had a significantly higher JEV load than the JEV+Wort and JEV+CQ groups at 10 days postinfection (P < 0.01). The JEV load in the brain of different groups of mice decreased 20 days after infection, but the JEV+Rapa group had a significantly higher JEV load than the JEV, JEV+Wort, and JEV+CQ groups (P < 0.01) (Fig. 4B).
Inflammatory responses
To evaluate the effectiveness of autophagy inhibitors in JEV-induced encephalitis, mouse brain tissues were observed at 10 and 20 days after JEV infection. The brain tissues of the JEV and JEV+Rapa groups showed obvious vascular inflammatory responses and microglial proliferation. Mild vascular inflammatory responses were found in brain tissues of the JEV+Wort and JEV+CQ groups, but obvious microglial proliferation was observed. Pathological changes in brain tissues of the control and single-administration groups were not evident (Fig. 5).

Autophagy inhibitors alleviate histopathological changes in the brain tissues of Japanese encephalitis virus (JEV)-infected mice. Significant perivascular cuffing was observed in the brain tissues of the JEV and JEV+Rapa groups at 10 days after JEV infection (blue arrows). Glial cell proliferation occurred in the brain tissues of the JEV and JEV+Rapa groups at 20 days after JEV infection (red arrows). The JEV+Wort and JEV+CQ groups showed mild vasculitis and obvious glial cell proliferation after JEV infection. The control and drug control groups showed normal brain tissue morphology (400X).
To confirm that autophagy inhibitors might alleviate inflammatory responses in mouse brain tissues, qPCR was performed to determine the expression levels of the pro-inflammatory cytokines IL-6, IL-1β, and TNF-α in the mouse brain tissues of different treatment groups. The results showed that the secretion of pro-inflammatory cytokines in brain tissues of the JEV and JEV+Rapa groups was significantly higher than that in the JEV+Wort and JEV+CQ groups at 10 days after JEV infection (P < 0.01). The secretion of pro-inflammatory cytokines in the brain of different groups was obviously decreased 20 days after infection, but the expression levels of IL-1β and TNF-α in the brain was significantly higher in the JEV+Rapa group than that in other groups (P < 0.01) (Fig. 6).

Autophagy inhibitors reduce the secretion of pro-inflammatory cytokines in the brain tissues of mice infected with JEV. Brain tissues were sampled at 10 and 20 days after JEV infection, and total RNA was extracted. The expression levels of IL-6, IL-1β, and TNF-α in the brain tissues of mice infected with JEV were determined using Q-PCR. Each error bar represents the standard deviation of three independent measurements for three mice in a group. One-way ANOVA was performed using Graph Pad Prism 6 software. **, p < 0.01; *, p < 0.05, as compared with each group.
Role of the PI3K/AKT pathway in brain tissues of JEV-infected mice
Wort is an inhibitor of PI3K, so the effect of autophagy inhibitors on the PI3K/AKT pathway was investigated. Mouse brain tissues were collected at 10 days after JEV infection and subjected to protein extraction, followed by Western blot analysis to evaluate the regulation of downstream signals by the PI3K/AKT pathway and the effects of this pathway on the nuclear translocation of NF-κB. The levels of PI3K, P-AKT, P-JNK, and P65 proteins in the JEV and JEV+Rapa groups were not significantly different from those of the other groups (P < 0.05) (Fig. 7).

Autophagy inhibitors may reduce the secretion of inflammatory factors by reducing the activation of the PI3K/AKT/NF-кB pathway. Brain tissues were sampled at 10 days after JEV infection, followed by separation of cytoplasmic proteins and nucleoproteins. The expression levels of PI3K, P-AKT, P-JNK, P-ERK, and P65 proteins were measured using western blot and scanning densitometry and normalized to GAPDH levels. (A) A representative image. (B) Quantitative data. Each error bar represents the standard deviation of three independent measurements for three mice in a group. One-way ANOVA was performed using Graph Pad Prism 6 software. **, p < 0.01; *, p < 0.05, compared with each group.
Discussion
JEV, a neurotropic virus, causes severe inflammatory reactions in the central nervous system [25, 26]. Previous studies have shown that the autophagy pathway positively regulates the replication of JEV in the early stage of viral infection in vitro [8]. Due to the complexity of the body’s response, the effect of autophagy-regulating drugs on JEV infection and host response in mice infected with JEV should be studied further. In this study, a JEV mouse model was established, and the mice were injected intraperitoneally with an autophagy inducer and inhibitors to investigate the effect of autophagy modulators on the severity of JEV infection in mice and their mechanisms of action.
When infected with JEV, mice develop notable neurological symptoms such as ataxia and dyskinesia [24]. After the mice began to show symptoms, we observed their symptoms every day and scored the symptoms [24]. The mice in the Rapa+JEV group showed clinical manifestations of varying severity at 5–20 days after JEV infection, including piloerection, arched back, eye congestion, and hind limb paralysis. The mice in the JEV group without any other treatment developed clinical signs at 5–10 days after JEV infection but recovered to normal in the late stage of infection. Some of the JEV-infected mice treated with Wort and CQ developed early mild signs. The mice in the Wort+JEV group had a survival rate of nearly 90%, and those in the CQ+JEV group had a survival rate of nearly 100%. In addition, we found that the brain tissues of mice in the JEV+Wort and JEV+CQ groups had less mitochondrial damage. This is consistent with the analysis of clinical signs in mice.
Among the autophagy-related ATG proteins, microtubule-associated proteins (LC3I, LC3II), homologs of mammalian ATG8, were identified as a marker of autophagosomes [27]. Early studies have shown that non-lipid LC3 in the autophagy pathway is a key host factor that affects the replication life cycle of JEV, and it mainly plays a role in the early stages of JEV infection [11]. Compared with mice in the JEV+Wort and JEV+CQ groups, the co-localization of LC3 and JEV-E appeared in more neurons in the brain tissue of mice in the JEV and JEV+Rapa groups. Autophagy is functional early during infection, but as the infection progresses, autophagosome maturation is inhibited, which causes dysfunctional autophagy [11]. Combined with our experiments, the results suggested that Wort and CQ might reduce the prevalence of mice being infected with JEV by weakening the interaction between autophagy and JEV infection. To determine whether the early stage or late stage of autophagy has a stronger influence on viral infection, better research methods and larger clinical trials are needed due to the complexity of the body’s response and the complex mechanism of autophagy.
JEV mainly acts on the central nervous system and causes neurological symptoms [28]. JEV is mainly distributed in the cerebral cortex, basal ganglia, thalamus, mesencephalon, pons, and medulla oblongata [28]. To determine whether autophagy inhibitors weakened the neurological signs in mice by reducing the degree of JEV infection, we assessed the degree of JEV infection in mouse brain. The results showed that JEV was distributed in the cerebral cortex, thalamus, or even the whole brain of mice in the JEV+Rapa group, while JEV was mainly distributed in the cerebral cortex and thalamus in brain tissues of mice in the JEV group. Moreover, JEV was mainly distributed in the cerebral cortex in brain tissues of mice in the JEV+Wort and JEV+CQ groups. In addition, we found that the viral load in the brain tissue of mice in the JEV+Wort and JEV+CQ groups was significantly lower than that in the JEV and JEV+Rapa groups at 10 days after virus infection, which was consistent with the above test results. This result further confirmed that Wort and CQ might reduce the degree of virus infection in brain tissue of mice infected with JEV.
As a neurotropic virus, JEV infection mainly affects brain tissue, inducing pathological changes in brain tissue to varying degrees. These are mainly manifested as degeneration and necrosis of neurons, glial cell proliferation, and perivascular cuffing [29]. The histopathological results showed that the brain tissue of mice in the JEV and JEV+Rapa groups had obvious vascular inflammation and late glial cell proliferation, while the mice in the JEV+Wort and JEV+CQ groups had slight vascular inflammatory response and obvious glial cell proliferation. The number of reactive astrocytes and activated microglia in mice increases after infection. In rodents, astrocytes produce cytokines such as IL-6, IL-1, and IL-18, and they release only CCL5 chemokine [26, 30]. To confirm that autophagy inhibitors might alleviate the vascular inflammatory responses in brain tissues in JEV-infected mice, we measured the expression levels of IL-1, IL-6, and TNF in brain tissues of mice in the JEV+Wort and JEV+CQ groups. The results showed that the expression levels of these cytokines were significantly lower than those in the JEV and JEV+Rapa groups. Therefore, combined with our experiments, the results suggested that autophagy inhibitors might reduce the degree of inflammation in brain tissues of mice infected with JEV, which may be related to the suppression of JEV infection in mice by autophagy inhibitors. On the other hand, the ability of pathogens to invade cells depends on their ability to bind to the corresponding cellular receptor [31]. Autophagy can be activated as an innate immune mechanism to control infection by intracellular pathogens [32,33,34]. Previous research has demonstrated that autophagy-deficient A549 cells show increased levels of IFNB, IL6, and IP10 mRNA in response to JEV infection [8]. Wort and CQ might inhibit JEV infection by enhancing the immune response in the peripheral or central nervous system, which inhibits the immune response caused by JEV infection and exerts a certain protective effect on brain tissue in JEV-infected mice.
The PI3K/AKT pathway plays an important role in regulating various inflammatory responses [35]. As an early autophagy inhibitor, Wort can prevent autophagosome formation, primarily by inhibiting the activity of PI3K [13, 14]. In addition, the endoplasmic reticulum is an organelle for viral replication and maturation, and a growing body of evidence indicates that endoplasmic reticulum stress induces autophagy [36]. Early studies have found that hepatitis C virus and hepatitis B virus promote autophagosome formation by inducing ER stress, and the UPR signaling pathway is involved in activating autophagy pathways [37]. In this study, the results showed that the PI3K/AKT, PERK, and NF-κB signaling pathways did not exhibit significant activation, which may be related to the complexity of the body’s response and the antiviral mechanism of CQ drugs on enveloped RNA viruses such as SARS-CoV [38]. We found that the antiviral effect of CQ drugs on JEV may be related to the drug’s interference with the terminal glycation of cell receptors to weaken the ability of the virus to bind to the receptor. However, large-scale experiments are still needed to determine the protective mechanism of Wort and CQ and their ability to weaken inflammatory response in the brain of mice infected with JEV.
Conclusions
In summary, this study initially showed that the autophagy inhibitors wortmannin and chloroquine slowed the occurrence of neurological symptoms and reduced the degree of destruction of the subcellular structure of brain tissues in JEV-infected mice. In addition, the autophagy inhibitors wortmannin and chloroquine reduced the distribution of JEV in the brain of mice and weakened the inflammatory response in the brain of mice infected with JEV. Further study is needed to determine the protective mechanism.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- CNS:
-
Central nervous system
- CQ:
-
Chloroquine
- HCQ:
-
Hydroxychloroquine
- JEV:
-
Japanese encephalitis virus
- JE:
-
Japanese encephalitis
- LC3:
-
Microtubule-associated proteins
- MHC:
-
Major histocompatibility complex
- MTORC1:
-
Rapamycin complex 1
- NF-кB:
-
Nuclear factor-κB
- PI3K III:
-
Class III phosphatidylinositol 3-kinase
- Rapa:
-
Rapamycin
- ULK1/2 complex:
-
UNC-51-like kinase 1/2 complex
- UPR:
-
Unfolded protein response
- Wort:
-
Wortmannin
References
Sips GJ, Wilschut J, Smit JM (2012) Neuroinvasive flavivirus infections. Rev Med Virol 22:69–87
Lord JS, Gurley ES, Pulliam JRC (2015) Rethinking Japanese encephalitis virus transmission: a framework for implicating host and vector species. Plos Neglect Trop D. 9:e0004074
Xu QQ, Zhu NW, Chen SL, Zhao P, Ren H, Zhu SY, Tang H, Zhu YZ, Qi ZT (2017) E3 Ubiquitin Ligase Nedd4 Promotes Japanese encephalitis virus replication by suppressing autophagy in human neuroblastoma cells. Sci Rep 7:45375
Park SL, Huang YS, Lyons AC, Ayers VB, Hettenbach SM, McVey DS, Burton KR, Higgs S, Vanlandingham DL (2018) North American domestic pigs are susceptible to experimental infection with Japanese encephalitis virus. Sci Rep 8:7951
Weaver SC, Reisen WK (2010) Present and future arboviral threats. Antiviral Res 85:328–345
Srivastava AK, Putnak JR, Lee SH, Hong SP, Moon SB, Barvir DA, Zhao B, Olson RA, Kim S-O, Yoo W-D, Towle AC, Vaughn DW, Innis BL, Eckels KH (2001) A purified inactivated Japanese encephalitis virus vaccine made in vero cells. Vaccine 19:4557–4565
Lin CW, Wu S-C (2003) A functional epitope determinant on domain III of the Japanese encephalitis virus envelope protein interacted with neutralizing-antibody combining sites. J Virol 77(4):2600–2606
Li J, Liang J, Liao C, Lin Y (2012) Autophagy is involved in the early step of Japanese encephalitis. Microbes Infect 14:159–168
Wang DY, Zhang JB, Jiang WK, Cao ZP, Zhao F, Cai TG, Aschner M, Luo WJ (2017) The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy 13:914
Deretic V, Levine B (2018) Autophagy balances inflammation in innate immunity. Autophagy 14:243–251
Sharma M, Bhattacharyya S, Nain M, Kaur M, Sood V, Gupta V, Khasa R, Abdin MZ, Vrati S, Kalia M (2014) Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy 10:1637–1651
Dunlop EA, Tee AR (2014) mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol 36:121–129
Hwang JY, Gertner M, Pontarelli F, Court-Vazquez B, Bennett MV, Ofengeim D, Zukin RS (2017) Global ischemia induces lysosomal-mediated degradation of mTOR and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ 24:317–329
Budziszewska B, Zajc A, Basta-Kaim A, Leœkiewicz M, Steczkowska M, Lasoñ W, Kaciñski M (2010) Effects of neurosteroids on the human corticotropin-releasing hormone gene. Pharmacol Rep 62:1030–1040
Agarwal S, Mirzoeva S, Readhead B, Dudley JT, Budunoval I (2019) PI3K inhibitors protect against glucocorticoid-induced skin atrophy. EBio Med. 41:526–537
Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM (2010) Dual role of 3methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 285:10850–10861
Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ (2007) Potential therapeutic applications of autophagy. Nat Rev Drug Disc 6:304–312
Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema K, Coppes RP, Engedal N, Mari M, Reggiori F (2018) Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14:1435–1455
Karla K (2009) Subversion of the cellular autophagy pathway by viruses. Curr Top Microbiol Immunol 335(1):323–333
Lee YR, Wang PS, Wang JR, Liu HS (2014) Enterovirus 71-induced autophagy increases viral replication and pathogenesis in a suckling mouse model. J Biomed Sci 21(1):1–11
Killian MS (2012) Dual role of autophagy in HIV-1 replication and pathogenesis. AIDS Res Ther 9(1):16
Zhang R, Chi X, Wang S, Qi B, Chen JL (2014) The regulation of autophagy by influenza A virus. BioMed Res Int 2014(7):498083
Rozières A, Viret C, Faure M (2017) Autophagy in measles virus infection. Viruses 9:359
Ye J, Jiang R, Cui M, Zhu BB, Sun LQ, Wang YY, Zohaib A, Dong Q, Ruan XD, Song YF et al (2014) Etanercept reduces neuroinflammation and lethality in mouse model of Japanese encephalitis. J Infect Dis 210:875–889
Yang CM, Lin CC, Lee IT, Lin YH, Yang CM, Chen WJ, Jou MJ, Hsiao LD (2012) Japanese encephalitis virus induces matrix metalloproteinase-9 expression via a ROS/c-Src/PDGFR/PI3K/Akt/MAPKs-dependent AP-1 pathway in rat brain astrocytes. J Neuroinflammation 9(1):12–12
Potokar M, Jorgačevski J, Zorec R (2019) Astrocytes in flavivirus infections. Int J Mol Sci 20:691
Kuma A, Komatsu M, Mizushima N (2017) Autophagy-monitoring and autophagy-deficient mice. Autophagy 13:1619–1628
Myint KS, Raengsakulrach B, Young GD, Gettayacamin M, Ferguson LM, Innis BL, Hoke ChJ, Vaughn DW (1999) Production of lethal infection that resembles fatal human disease by intranasal inoculation of macaques with Japanese encephalitis virus. Am J Trop Med Hyg 60:338–342
Lien JC, Wang CY, Lai HC, Lu CY, Lin YF, Gao GY, Chen KC, Huang AC, Huang SH, Lin CW (2018) Structure analysis and antiviral activity of CW-33 analogues against Japanese encephalitis virus. Sci Rep 8:16595
Liu ZY, Yao XQ, Jiang WS, Li W, Zhu SY, Liao CR, Zou L, Ding RT, Chen JT (2020) Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. J Neuroinflammation 17:90
Joubert PE, Meiffren G, Grégoire IP, Pontini G, Richetta C, Flacher M, Azocar O, Vidalain PO, Vidal M, Lotteau V et al (2009) Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 6:354–366
Hu BL, Zhang YN, Jia L, Wu HS, Fan CF, Sun YT, Ye CJ, Liao M, Zhou JY (2015) Binding of the pathogen receptor HSP90AA1 to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR pathway. Autophagy 11:503–515
Chaumorcel M, Souquère S, Pierron G, Codogno P, Esclatine A (2008) Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 4:46–53
Delpeut S, Rudd PA, Labonté P, Messling V (2012) Membrane fusion-mediated autophagy induction enhances morbillivirus cell-to-cell spread. J Virol 86:8527–8535
Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B (2010) The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 15:329–341
Wang K, Li SF, Worku T, Hao XJ, Yang LH, Zhang SJ (2017) Rab11a is required for porcine reproductive and respiratory syndrome virus induced autophagy to promote viral replication. Biochem Biophys Res Commun 492:236–242
Lee YR, Kuo SH, Lin CY, Fu PJ, Lin YS, Yeh TM, Liu HS (2018) Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci Rep 8:489
Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, Seidah NG, Nichol ST (2005) Chloroquine is a potent inhibitor of SARS coronavirus. Infection and spread. Virol J 2(1):69
Funding
This work was supported by National Key Research and Development Program of China (2016YFD0500407).
Author information
Authors and Affiliations
Contributions
ZJ and GC designed the research and wrote the manuscript, and ZJ performed the majority of the study, analyzed the data, and generated the figures. CG, LX, ZW and HX provided advice in the design of the study. CS provided experimental materials. XC provided technical advice on the experiments. HW, GM, and WX conducted parts of the animal surgery and performed experiments. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
All experiments were conducted using the protocol recommended by the Research Ethics Committee of the College of Veterinary Medicine, Huazhong Agricultural University, Hubei Province, China. The use of laboratory animals was approved by the Scientific Ethics Committee of Huazhong Agricultural University (no. HZAUMO-2018-059).
Additional information
Handling Editor: Zhenhai Chen.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, J., Han, W., Xie, C. et al. Autophagy inhibitors alleviate Japanese encephalitis virus-induced cerebral inflammation in mice. Arch Virol 167, 849–859 (2022). https://doi.org/10.1007/s00705-021-05283-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-05283-9