
Abstract
We determined the genomic sequence of a Ukrainian strain of fowl adenovirus B (FAdV-B). The isolate (D2453/1) shared 97.2% to 98.4% nucleotide sequence identity with other viruses belonging to the species Fowl aviadenovirus B. Marked genetic divergence was seen in the hexon, fiber, and ORF19 genes, and phylogenetic analysis suggested that recombination events had occurred in these regions. Our analysis revealed mosaicism in the recombination patterns, a finding that has also been described in the genomes of strains of FAdV-D and FAdV-E. The shared recombination breakpoints, affecting the same genomic regions in viruses belonging to different species, suggest that similar selection mechanisms are acting on the key neutralization antigens and epitopes in viruses of different FAdV species.
Similar content being viewed by others


Avian adenoviruses (genus Aviadenovirus, family Adenoviridae) comprise 15 species, five of which are associated primarily with infections of chickens (Fowl aviadenovirus A to E) [1]. Fowl adenoviruses (FAdVs) are characterized by 70- to 90-nm icosahedral, non-enveloped, viral particles and a double-stranded DNA genome of ~42-45 kilobase pairs in length. A variety of diseases have been ascribed to FAdV infections. The best-known FAdV-associated diseases in chickens are inclusion body hepatitis (IBH), hepatitis-hydropericardium syndrome (HHS), and gizzard erosion (GE), which are usually caused by certain serotypes of FAdV-C, FAdV-D or -E, and FAdV-A, respectively [2].
FAdV-B has been isolated from cases associated with lameness, tarsal joint swelling, inclusion body hepatitis, enteric infections, and cardiac and pulmonary diseases. FAdV-B differs from other FAdVs in the following features of its genomic structure: i) it is the longest genome among aviadenoviruses, ii) it lacks the 14B and 14C coding ORFs in the left part of the genome, and iii) it has some unique genes (including ORF57) encoded at the right end of the genome [3, 4]. The two whole genome sequences currently available for FAdV-B are 97.4% identical, with major sequence variations in the ORF19 coding region [4]. In this study, we describe and analyze the genome of a novel FAdV-B strain isolated in Ukraine from an industrial broiler-type chicken in 2013. The affected flock displayed depression from 28 days of age. Necropsy, performed at 28-33 days of age, revealed haemorrhages and atrophy in the bursa. Hemorrhage in the breast muscles and legs was also observed. The excess mortality in the flock was reportedly relatively low (2.5% or less). Upon routine virological examination, infectious bursal disease virus and FAdV were isolated from pools of bursa Fabricii and caecal tonsils in embryonated eggs and on LMH (chicken hepatocellular carcinoma) cells, respectively. The isolated FAdV was identified as FAdV-B using a broad-range FAdV-specific PCR assay that amplifies a fragment of the DNA polymerase gene [5]. Given that genetic information about FAdV-B strains from Ukraine was not available, it seemed to be of interest to further characterize this isolate. The genomic information of the isolate (D2453/1/10,12/13/UA, hereafter D2453/1) can be useful for monitoring virus circulation, as well as for eventual vaccine development efforts.
Supernatants of tissue culture showing cytopathic changes were centrifuged at 10,000 × g for 5 min, and 200 μL of the supernatant was used for nucleic acid extraction using the ZiXpress32 Viral Nucleic Acid Extraction Kit and a ZiXpress32 robot according to the manufacturer’s instructions (Zinexts Life Science). The extracted DNA was measured fluorometrically, and the Illumina® Nextera XT DNA Library Preparation Kit and the Nextera XT Index Kit v2 Set A (Illumina, San Diego, CA, USA) were used to prepare a DNA library. DNA samples were diluted to 0.2 ng/μL in nuclease-free water (Promega, Madison, WI, USA) in a final volume of 2.5 μL. Tagmentation was carried out as recommended by the manufacturer, although at reduced volume. Incubation steps were carried out in a GeneAmp 9700 PCR System (Applied Biosystems, Foster City, CA, USA). The final DNA library was purified using a Gel/PCR DNA Fragments Extraction Kit (Geneaid Biotech Ltd., Taipei, Taiwan). The concentration of the purified DNA samples was determined using a Qubit 2.0 Fluorometer and the Qubit dsDNA HS Assay Kit (Thermo Fischer Scientific, Waltham, MA, USA). The DNA library was pooled with other DNA libraries, and after denaturation and adjustment to the required concentration (1.5 pM), the sample was loaded onto the reagent cartridge of a NextSeq 500/550 Mid Output Flow Cell (Illumina, San Diego, CA, USA) [6]. The 150-cycle sequencing protocol was carried out to obtain 150-nt-long single reads.
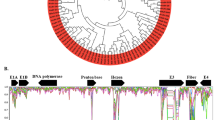
FastQ files collected from the four flow cell lanes were used to assemble the adenoviral genome sequence, using Genious v 9.1.8 (https://www.geneious.com). A combination of de novo sequencing and reference mapping was used to obtain the consensus genome sequence (accession number: MT500572). Genome annotation was carried out as implemented in Geneious v 9.1.8 using the genome sequence of the FAdV-B strain 40440-M/2015 (accession no. MG953201). The genome of the Ukrainian FAdV-B isolate D2453/1 was sequenced at an average depth of ~1175X (358,847 of 1,550,667 sequence reads mapped on the final consensus genome sequence). The length of the nearly complete genome sequence was 45,762 bp, corresponding to 99.98% coverage, with only five nucleotides missing in the inverted terminal repeat sequences. The GC content of the newly sequenced genomic DNA of the Ukrainian FAdV isolate was 56.3%. It shared 98.4% genome-wide sequence identity with a Hungarian FAdV-B strain (40440-M/2015) whose genome sequence was published recently [4], and 97.2% identity with a prototype strain (340) of FAdV-B [7]. The genome of D2453/1 was found to contain 37 ORFs (Fig. 1A). A range of 0 to 5.6% and 0 to 20% pairwise nucleotide sequence divergence was seen between D2453/1 and 40440-M/2015 and between D2453/1 and 340, respectively (data not shown).

Map of the genome of strain D2453/1 (top). Similarity plot of D2453/1 and two FAdV-B reference genomes (parameters: window size, 200 bp; step, 20 bp) (middle). Phylogenetic trees based on the sequences of selected genes of FAdV-B strains (neighbor-joining method; p-distance); the outgroup sequences were derived from a serotype 8b FAdV strain (strain name, 764; accession number, KT862811) (bottom).
To further analyze the genome of D2453/1, we performed Simplot analysis [8] using a genome sequence alignment of the three FAdV-B isolates (Fig. 1B). We observed genomic regions where mutations tended to be more frequent. These regions included, but were not limited to, the hexon gene, the fiber gene, and the putative ORF19 coding region. Next, we performed phylogenetic analysis based on these individual genes as well as another region with lower sequence divergence (ORF14A). Neighbor-joining (NJ) trees were generated using the p-distance method to calculate gene-specific distance matrices [7]. The phylogenetic relationships in the highlighted genomic regions gave intriguing results that were consistent with Simplot analysis and permitted us to conclude that the three FAdV-B strains had a peculiar pattern of recombination events. The Ukrainian FAdV-B strain shared a closer relationship with strain 340 in the fiber gene and with 40440-M/2015 in ORF19. In the hexon gene, it differed significantly for both of the other strains (94.2% and 94.4% nt sequence identity to 340 and 40440-M/2015, respectively) (Fig. 1C).
A closer look at the hexon gene revealed intriguing genetic relationships among FAdV-B strains. A distance matrix (and an NJ phylogenetic tree) prepared from a 405-nt-long fragment within the loop-1 region of the hexon gene (corresponding to nt 417 to 821 of the hexon gene of D2453/1) showed that the Ukrainian FAdV-B isolate was more similar to the homologous sequence of a West African chicken-origin FAdV (99.5% nt sequence identity) detected in 2012 [9] than to the prototype FAdV-B strain 340 (79.8% nt sequence identity) or the variant FAdV-B strain 40440-M/2015 (80.7% nt sequence identity) (data not shown) [3, 4]. Interestingly, recombination analysis suggested that the breakpoints within the hexon gene involved the loop region where the major antigenic epitopes are located; therefore, changes affecting this region may result in altered antigenicity.
Altogether, the pattern of recombination distribution shows a mosaicism affecting a small fraction of the genome. Although the recombination landscape may become more complex once additional full-length FAdV-B genome sequences become available, the findings of the present study are consistent with a recently published work on the analysis of FAdV-D and FAdV-E genome sequences [10]. Schachner and co-workers analyzed numerous FAdV-A, -C, -D and -E genomes and identified mosaicism in the patterns of recombination in FAdV-D and -E genomes, mainly affecting the hexon gene, the fiber gene, and ORF19. As we identified major recombination breakpoints in the same regions of the few FAdV-B genomes currently available, our study supplements these recent observations, demonstrating that similar selection processes may act on the key neutralization antigens and epitopes within FAdV-B. Because the present study implies greater genetic variation among FAdV-B isolates than previously thought, a more complete picture of the genetic diversity of FAdV-B requires additional sequence data to be analyzed.
References
Harrach B, Kajan GL (2011) Aviadenovirus. In: Tidona CA, Darai G (eds) The springer index of viruses, 2nd edn. Springer, New York, pp 13–28
Schachner A, Matos M, Grafl B, Hess M (2018) Fowl adenovirus-induced diseases and strategies for their control—a review on the current global situation. Avian Pathol 47:111–126
Marek A, Kosiol C, Harrach B, Kaján GL, Schlötterer C, Hess M (2013) The first whole genome sequence of a Fowl adenovirus B strain enables interspecies comparisons within the genus Aviadenovirus. Vet Microbiol 166:250–256
Kaján GL, Affranio I, Tóthné Bistyák A, Kecskeméti S, Benkő M (2019) An emerging new fowl adenovirus genotype. Heliyon 5:e01732
Kaján GL, Sameti S, Benko M (2011) Partial sequence of the DNA-dependent DNA polymerase gene of fowl adenoviruses: a reference panel for a general diagnostic PCR in poultry. Acta Vet Hung 59:279–285
Olasz F, Mészáros I, Marton S, Kaján GL, Tamás V, Locsmándi G, Magyar T, Bálint Á, Bányai K, Zádori Z (2019) A simple method for sample preparation to facilitate efficient whole-genome sequencing of African swine fever virus. Viruses 11:1129
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC (1999) Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160
Pauly M, Akoua-Koffi C, Buchwald N, Schubert G, Weiss S, Couacy-Hymann E, Anoh AE, Mossoun A, Calvignac-Spencer S, Leendertz SA, Leendertz FH, Ehlers B (2015) Adenovirus in rural Côte D’Ivoire: high diversity and cross-species detection. EcoHealth 12:441–452
Schachner A, Gonzalez G, Endler L, Ito K, Hess M (2019) Fowl adenovirus (FAdV) recombination with intertypic crossovers in genomes of FAdV-D and FAdV-E, displaying hybrid serological phenotypes. Viruses 11:1094
Acknowledgements
This study was funded by the National Research, Development and Innovation Office (Grant no. K120201).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Graciela Andrei.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Homonnay, Z., Jakab, S., Bali, K. et al. Genome sequencing of a novel variant of fowl adenovirus B reveals mosaicism in the pattern of homologous recombination events. Arch Virol 166, 1477–1480 (2021). https://doi.org/10.1007/s00705-021-04972-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-04972-9