
Abstract
A recent proposal that the genus Rymovirus be assimilated into the genus Potyvirus is examined, discussed, and rejected. It illustrates the danger of using ‘sequence identity’ as a proxy for phylogenetic relatedness to distinguish closely related but distinct groups of viruses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In his paper entitled “Is it time to retire the genus Rymovirus from the family Potyviridae?” Colin Ward [12] questions whether rymoviruses should be a separate potyvirid genus. The most important of the facts he uses to support his suggestion is that the pairwise identities (PIs) of rymovirus and potyvirus gene sequences overlap, and hence do not distinguish unequivocally between them. He also notes that these two virus groups have different vectors; rymoviruses are mite-borne and potyviruses aphid-borne, and he argues that this is taxonomically irrelevant. His suggestion, however, raises some important and interesting questions.
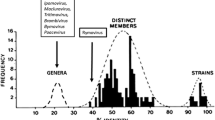
Ward bases his case on data from Shukla and Ward [11], which was published a quarter century ago, and so the first question is whether his claim about pairwise identities is still correct when the calculations are based on the more abundant and longer gene sequences now available in Genbank. We have therefore calculated the pairwise identities of the main ORFs of 102 potyvirus and six rymovirus genome sequences using the SDT program [9]. The sequences were downloaded from Genbank, edited and assembled using BioEdit [6], aligned using the TranslatorX server [1] with its MAFFT option; they became 16206 nts long including alignment indels, and contained no recombinant sequences as tested by RDP4 [8]. Fig. 1 is a ‘histograph’ of the PIs; the mean PI of the ORFs of the most different rymoviruses (i.e. all pairwise comparisons of three ryegrass mosaic virus sequences with three hordeum mosaic virus sequences) is 56.36% +/- 0.25%, which is little different from the mean PI of 55.09% +/- 1.04% for the ORFs of the most different clade of potyviruses (Narcissus degeneration, Onion yellow dwarf, Shallot yellow stripe and Vallota speciosa viruses) versus all the others. Thus the rymoviruses cannot be distinguished from the potyviruses by their PIs, and the basis of Ward’s claim is still absolutely correct.

Pairwise identity ‘histograph’ of 102 potyvirus and six rymovirus main ORF sequences showing only the comparisons connected through the basal phylogenetic nodes. All pairwise comparisons are in green, potyviruses versus potyvirus comparisons in blue, rymovirus versus rymovirus in mauve and rymovirus versus potyvirus in red. The Y-axis is of 100 comparisons/division for all except rymovirus versus rymovirus comparisons, which are of four comparisons/division. The Accession Codes of the sequences used are given in Supplementary File 1
The use of PI estimates as a surrogate for distinguishing the natural groups, or more specifically phylogenetic groups, of potyviruses by Shulka and Ward [11] was expanded by Adams et al. [2], but subsequently questioned by Duffy and Seah [3], who highlighted the problem that “When researchers use the standard technique of per cent nucleotide identity to determine that the new sequence is closely related to another sequence, potentially erroneous conclusions can be drawn from the results.”
PI estimates are only tangentially linked to phylogenetic relatedness, and to determine whether rymoviruses and potyviruses are distinct groups worth designating as genera requires phylogenetic analyses. Phylogenetic algorithms assume that related organisms have descended from shared ancestors by a process of independent genetic divergence and selection, whereas PI measures do not, and are analogous to the measures used by cosmologists when defining the motions of stars, which although derived from a single ‘big bang’, continue to influence one another at a distance by gravitation and ‘dark matter’ (https://en.wikipedia.org/wiki/Dark_matter). Therefore we checked whether two standard, but different, phylogenetic methods distinguish between rymoviruses and potyviruses using the same 108 ORF sequences checked in the SDT analysis above, together with 16 tritimovirus sequences as an outgroup. One phylogeny was calculated by PhyML 3.0 [5] using the GTR+I+Ƭ4 substitution model, and statistical support for individual nodes was assessed using the SH option [10]. Fig. 2 shows the branching pattern of the resulting tree; it was drawn using Figtree 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/) and a commercial computer illustration package. It can be seen that the sequences fall into three monophyletic clusters, the largest contains only potyvirus sequences, the smallest only rymovirus sequences and the third only tritimovirus sequences, with the basal nodes of all three clusters having 1.0 SH statistical support. A tree calculated from the same sequences by the neighbor-joining method in ClustalX [7] was topologically the same as the ML tree with the sequences again forming monophyletic clusters with 100% bootstrap support (1000 replicates). Thus phylogenetic methods distinguish the three potyvirid genera, whereas PI analyses do not as they have problems with closely related clusters, just as they have with outliers [3].

Branching pattern of a ML phylogeny of 102 potyvirus, six rymovirus and 16 tritimovirus ORFs. Arrows indicate the basal nodes of the three lineages. The truncated branch to the tritimoviruses had a length of c. 5.2 substitutions/site. The Accession Codes of the sequences used are given in Supplementary File 1
Recently the Code of the International Committee on Taxonomy of Viruses (ICTV; https://talk.ictvonline.org/information/w/ictv-information/383/ictv-code) updated its Rule 3.20, to define species as “a monophyletic group of viruses whose properties can be distinguished from those of other species by multiple criteria.”, but did not similarly update Rule 3.23 for genera, which merely states that “A genus is a group of species sharing certain common characters.”. Thus the ICTV has now adopted phylogenetics as the basis for defining species, but not yet genera despite the widespread reported use of phylogenetic methods at both taxonomic levels. The ICTV also leaves the sorts of “multiple criteria” or “certain common characters” to be used for distinguishing species and genera to the imagination! The ICTV should clarify the situation by declaring that both species and genera to be monophyletic groupings. It could also aid the choice of characters for defining such groupings by explicitly stating that “virus species include strains/isolates, that are so similar there is no value in giving them separate names”, whereas virus genera are groups of viruses that “it is especially useful to define by their shared properties because the groupings thus formed help with such problems as identifying newly found viruses and predicting their properties” [4].
In summary, the members of the Potyvirus and Rymovirus genera are phylogenetically distinct, they also have different vectors, and they should continue to be recognised as members of distinct genera within the Potyviridae. Furthermore we suggest that, for publication, the monophyly of species and also genera should be established using phylogenetic methods, not sequence identities, and we note that several such methods (e.g. [5] and [7]) require no more computational skill than calculating sequence identities [9].
References
Abascal F, Zardoya R, Telford MJ (2010) TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucl Acids Res 38:W7–W13
Adams MJ, Antoniw JF, Fauquet CM (2005) Molecular criteria for genus and species discrimination within the family Potyviridae. Arch Virol 150:459–479
Duffy S, Seah YM (2010) 98% identical, 100% wrong: per cent nucleotide identity can lead plant virus epidemiology astray. Phil Trans R Soc B 365:1891–1897
Gibbs A, Harrison B (1976) “Plant virology: the principles”. Edward Arnold, London, p 292
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ (1998) Multiple sequence alignment with Clustal X. Trends Biochem Sci 23:403–405
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 1(1):vev003
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9(9):e108277
Shimodaira H, Hasegawa M (1999) Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol 16:1114–1116
Shukla DD, Ward CW (1988) Amino acid sequence homology of coat proteins as a basis for identification and classification of the potyvirus group. J Gen Virol 69:2703–2710
Ward CW (2017) Is it time to retire the genus Rymovirus from the family Potyviridae? Arch Virol 162:2175–2179
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study received no funding.
Conflicts of interests
Neither author has a conflict of interest.
Ethical approval
This article does not report studies that involved the participation of human beings or other animals.
Additional information
Handling Editor: Sead Sabanadzovic.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gibbs, A.J., Gibbs, M.J. Rymovirus: a cautionary tale. Arch Virol 163, 815–817 (2018). https://doi.org/10.1007/s00705-017-3676-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3676-7