
Abstract
To date, the diagnoses of Parkinson syndromes are based on clinical examination. Therefore, these specific diagnoses are made, when the neuropathological process is already advanced. However, disease modification or neuroprotection, is considered to be most effective before marked neurodegeneration has occurred. In recent years, early clinical or prodromal stages of Parkinson syndromes came into focus. Moreover, subtypes of distinct diseases will allow predictions of the individual course of the diseases more precisely. Thereby, patients will be enrolled into clinical trials with more specific disease entities and endpoints. Furthermore, novel fluid and imaging biomarkers that allow biochemical diagnoses are under development. These will lead to earlier diagnoses and earlier therapy in the future as consequence. Furthermore, therapeutic approaches will take the underlying neuropathological process of neurodegenerative Parkinson syndromes more specific into account. Specifically, future therapies will target the aggregation of aggregation-prone proteins such as alpha-synuclein and tau, the degradation of pathological aggregates, and the spreading of pathological protein aggregates throughout the brain. Many of these approaches are already in (pre)clinical development. In addition, anti-inflammatory approaches are in development. Furthermore, drug-repurposing is a feasible approach to shorten the developmental process of new drugs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since James Parkinson described Parkinson’s disease (PD) more than two centuries went by (Parkinson 1817; 2002). Not much changed in the diagnostic process since then. PD is considered a mainly clinical diagnosis (Postuma et al. 2015). Nevertheless, to date, we know much more about Parkinsonism then James Parkinson did in 1817. We know, for example, that Parkinsonism is not only caused by PD but also by other neurodegenerative disorders, considered as atypical Parkinson syndromes (aPS) (Levin et al. 2016a). Furthermore, since the discovery of alpha-synuclein (αSyn) as the main component of Lewy bodies in the late 1990’s (Spillantini et al. 1997), huge progress has been made in the understanding of the pathophysiological processes behind PD and related disorders. Now, we are able to distinguish between synucleinopathies and tauopathies histopathologically, dependent on the type of proteinaceous inclusions found in post-mortem tissue (Dickson 2018; Koga and Dickson 2018; Kovacs et al. 2020). Furthermore, genetic studies revealed that mutations in distinct genes are causative for a small two-digit percentage of cases of diseases previously considered as being sporadic (Bardien et al. 2011; Beavan and Schapira 2013). All these insights in the pathophysiology of Parkinson syndromes (PS) cannot hide that levodopa is still the most effective and best-tolerated drug to treat motor symptoms symptomatically in 2022. Furthermore, there is no drug with a proven disease-modifying effect for any of the distinct Parkinson syndromes. Moreover, non-motor symptoms present in PD and related disorders are more difficult to treat and levodopa is far less effective in many cases of aPS compared to PD.
In the following narrative review, we will discuss the obstacles on the way to disease modification in Parkinsonism. This includes the way how we make the diagnosis and the present absence of objective fluid and imaging biomarkers to confirm the diagnosis and to prove disease modification. Furthermore, we will present ongoing developments for clinical diagnostic criteria for early or even premotor stages of Parkinsonism, novel biomarkers, and ongoing and future therapeutic approaches that will shift the focus away from mainly symptomatic therapy towards the modification of the underlying pathophysiological processes of different diseases with Parkinsonism. Finally, we will discuss some obstacles in clinical care for patients with Parkinsonism.
Diagnostic and pathophysiological aspects of Parkinson’s disease and atypical Parkinson syndromes
Clinical differential diagnosis of Parkinson’s disease and atypical Parkinson syndromes
In clinical routine, the diagnoses of neurodegenerative PS are based on distinct diagnostic criteria. These usually consist of core criteria, supportive criteria, and exclusion criteria (Postuma et al. 2015; Höglinger et al. 2017; Wenning et al. 2022). However, these clinical criteria only allow the prediction of the underlying pathology with a certain probability. To make the definite diagnoses, a post-mortem neuropathological examination of the brain is necessary, which is still the gold standard for the diagnoses of neurodegenerative PS (Kovacs et al. 2020; Dickson 2018; Koga and Dickson 2018).
Histopathologically, PS can be divided in synucleinopathies and tauopathies, according to the presence of aggregates of alpha-synuclein (αSyn) or tau (Levin et al. 2016a). While PD, dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) are all associated with the intracellular inclusions of misfolded αSyn, there are huge differences in the nature and localization of αSyn aggregates present in these disorders (Ayers et al. 2022; Koga et al. 2021). PD and DLB are characterized by the presence of Lewy bodies, which are large cytoplasmic deposits of αSyn in neurons (Goedert et al. 2013). In PD, in early stages, these are located in the brain stem, before they spread throughout the brain in the course of the disease (Braak et al. 2003), while the localization of (cortical) Lewy bodies in DLB is more diffuse (Koga et al. 2021). Moreover, the structure of brainstem and cortical Lewy bodies is different (Koga et al. 2021). MSA on the other hand is characterized by the presence of αSyn deposits in oligodendrocytes, called glial cytoplasmic inclusions (Koga et al. 2021). Progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), are characterized by the accumulation of 4-repeat tau in neurons and astrocytes (Rösler et al. 2019; Kovacs et al. 2020; Zhang et al. 2020). Targeting the different neurodegenerative processes will be essential for future neuroprotective therapies to slow or even halt the neurodegenerative process (Respondek et al. 2019). However, especially in early disease stages, the diagnostic accuracy to make the diagnosis of PD is low even if the diagnosis is made by movement disorder specialists (Adler et al. 2014). Moreover, the diagnostic accuracy further decreases, when the patients show an insufficient response to initial dopaminergic treatment (Adler et al. 2014). Furthermore, the sensitivity of the previous MSA diagnostic criteria was low at first visit (Osaki et al. 2009). Moreover, recent diagnostic criteria for PSP and MSA allow to make diagnoses at earlier staged by the introduction of “suggestive of PSP” (Höglinger et al. 2017) or “possible prodromal multiple system atrophy” (Wenning et al. 2022) as diagnostic certainty levels. However, there is always a trade-off between sensitivity and specificity. Therefore, the prediction of the specific neurodegenerative process by clinical examination alone can be very challenging, emphasizing the need for objective biomarkers to support the diagnostic process.
Furthermore, even typical PD motor symptoms including bradykinesia and rigidity are often misjudged as age-related or orthopedic in the beginning of the disease and it sometimes takes years until the correct diagnosis is made (Gaenslen et al. 2011). Thus, patients that are included in clinical trials that investigate the neuroprotective or disease-modifying potential of new agents might be too far progressed in the course of the disease to allow the observation of relevant changes in the progression rate.
In all neurodegenerative disorders, the pathophysiological process starts years or even decades before the occurrence of first symptoms. In PD, first motor-symptoms occur, when already 50% of the dopaminergic cells in the substantia nigra pars compacta died and 70–80% of striatal dopamine levels are depleted (Fearnley and Lees 1991). Furthermore, neuropathological examinations show that the substantia nigra is not the first structure to be affected by degeneration in PD, emphasizing that the pathophysiological process starts long before first motor symptoms occur (Braak et al. 2003).
Specific subtypes in Parkinson’s disease and atypical Parkinson syndromes
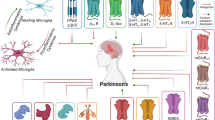
In general, practice patients with Parkinson’s disease were grouped in tremor-dominant (TD), akinetic-rigid (AR) and equivalence type (ET) (Rajput et al. 2009). In PD patients with ET, tremor and akinetic symptoms are present to a similar extent. However, the classification in different subtypes of PD is very imprecise and can change over the course of the disease. Therefore, in the last decade, several groups spent effort in a more precise subtyping system of PD phenotypes (for critical review (Mestre et al. 2021). The more precise definition of subtypes of PD would help with the prediction of clinical disease progression, which would be beneficial for patient counselling. Furthermore, a more precise knowledge of the longitudinal course of the disease would be helpful in the recruitment for clinical trials and would allow recruiting of smaller groups of patients with a more homogenous progression. For the stratification in subtypes motor symptoms (e.g. more severe symptoms, less pronounced levodopa response), non-motor symptoms (e.g. orthostatic hypotension, cognitive decline, REM-sleep behavior disorder (RBD), and others), demographic markers (age at onset, disease duration), and other biomarkers may be used.
Hence, in a longitudinal study of pathological proven PD cases, the initial presentation of TD subtype, ET subtype, and the AR subtype determined disease progression (Rajput et al. 2017). In the study of Rajput et al., the TD subtype was associated with a more benign course of the disease, while the AR subtype showed the most malignant disease trajectory with shortest survival and highest rate of dementia (Rajput et al. 2009). Furthermore, the AR subtype was associated with more pronounced striatal dopamine depletion measured by post-mortem high-performance liquid chromatography compared to TD and ET subtypes (Rajput et al. 2008).
In another cohort of autopsy-confirmed PD patients, Pablo-Fernandez et al. defined subtypes of PD according to specific motor- and non-motor symptoms (Pablo-Fernández et al. 2019). By this approach, three different subtypes of PD with different rates of progression to specific milestones could be defined: the mild-motor, the intermediate, and the diffuse malignant phenotype. All these phenotypes showed a distinct pattern of Lewy body pathology in the brain (Pablo-Fernández et al. 2019). Interestingly, Alzheimer’s disease (AD) co-pathology was not significantly different between the subgroups of PD patients, although the authors observed an age-dependent accumulation of AD pathology in the cohort.
In an early study in this field, two subtypes of PD were determined mainly by motor symptoms in de novo PD patients and early stage PD patients on treatment: the tremor-dominant (TD) subtype and the postural instability and gait difficulties (PIGD) subtype (Jankovic et al. 1990). Already in 2010, a review of several studies identified the existence of several PD subtypes (PIGD, TD, old-onset and rapid progression, and young-onset slow progression) using cluster analysis (van Rooden et al. 2010).
In a recent study of PD subtypes, a biomarker-based approach revealed that plasma neurofilament light chain (NFL) was increased in the diffuse malignant subtype and was also associated with more rapid clinical progression of symptoms (Pilotto et al. 2021). In several cohorts, low amyloid beta 1-42 in the cerebrospinal fluid (CSF) was associated with faster cognitive decline in PD (Stewart et al. 2014). Interestingly, the PIGD subtype of PD was associated with lower amyloid beta 1-42 and higher P181-tau in the CSF compared to the TD subtype (Kang et al. 2013). And also in a more recent study, the diffuse malignant PD subtype was associated with lower amyloid beta 1-42 and amyloid beta/total tau ratio in the CSF compared to other phenotypes (Fereshtehnejad et al. 2017). Moreover, the diffuse malignant phenotype was associated with a higher extent of atrophy in cerebral magnet resonance imaging (Fereshtehnejad et al. 2017). The authors of a recent German study reported that serum NLF levels were increased in the PIGD subgroup and that NFL levels significantly correlated with the Montreal cognitive assessment (MoCA) scale and the MDS-UPDRS part III in PIGD patients (Pötter-Nerger et al. 2022), emphasizing the diagnostic value of this biomarker.
Definition of specific subtypes may also have some implications on treatment regimens in clinical routine. In a recent review, strategies for differential therapy were described in detail (Marras et al. 2020).
Contrary to various subtypes in PD, there are only two major phenotypes in MSA, the cerebellar subtype (MSA-C), and the Parkinsonism subtype (MSA-P) (Fanciulli and Wenning 2015). However, in longitudinal cohorts, there was no evidence of different progression rates between the two subtypes (Foubert-Samier et al. 2020). MSA is a rapidly progressive neurodegenerative disease with relatively constant progression of symptoms in both subtypes (Pérez-Soriano et al. 2021). Clinically, the extent of autonomic symptoms and earlier onset of RBD appear to be predictors for more rapid disease progression (Low et al. 2015; Giannini et al. 2020). Recent data also highlight the value of NFL as biomarker for disease severity and disease progression (Palleis et al. 2020; Zhang et al. 2022b).
In the past, there was mainly one phenotype of PSP (Litvan et al. 1996). However, a retrospective analysis of pathologically confirmed cases demonstrated that only a quarter of patients showed the classical phenotype, also called Richardson-syndrome (Respondek et al. 2014). Therefore, in the new MDS diagnostic criteria for PSP, a classification in different PSP variants was established (Höglinger et al. 2017), taking into account different clinical presentations of pathologically confirmed PSP cases (Respondek et al. 2014; Lopez et al. 2016; Respondek and Höglinger 2016). These new phenotypes help to clinically identify more patients with 4-repeat tau pathology in vivo and allow inclusion of these patients in natural history studies and clinical trials. The new PSP phenotypes can be divided into cortical and subcortical phenotypes. PSP-speech-language, PSP-frontal, PSP-corticobasal syndrome are referred to the cortical phenotypes (Guasp et al. 2021). The subcortical spectrum contains the PSP-parkinsonism, PSP-progressive gait freezing, PSP-oculomotor subtype and PSP-postural instability (Guasp et al. 2021). One obstacle with the diagnostic criteria of PSP is that patients can qualify for multiple phenotypes according to their clinical symptoms. This issue was recently solved by the MAX (multiple allocation extinction)-rules, which help to allocate patients to only one distinct phenotype (Grimm et al. 2019). However, after applying the MAX-rules, in clinically diagnoses PSP patients, the PSP-Richardson syndrome phenotype is relatively predominant compared to other phenotypes (Frank et al. 2020). Nevertheless, the main idea of the MDS-diagnostic criteria is the identification of more patients with cerebral 4-repeat tauopathy and the influence of different phenotypes on the identification threshold appears to be low (Respondek et al. 2017). In an extensive neuropathological investigation, Kovacs et al. were able to show specific patterns of neurodegeneration and tau pathology in a variety of brain regions in PSP patients (Kovacs et al. 2020).
Patients with PSP-Richardson syndrome present a relatively constant worsening of the disease with an annual deterioration of about ten points in the PSP rating scale (Piot et al. 2020; Grötsch et al. 2021). However, there are only very few data available on disease progression in other phenotypes. For example, in PSP-progressive gait freezing, disease progression is slow and cognitive deficits were only mild in most of the clinical cases (Owens et al. 2016). In terms of longitudinal progression, the MoCA test but not the Mini mental state examination (MMSE) was able to detect cognitive impairments in very early PSP patients. However, these tests were both not able to discriminate between specific phenotypes (Pereira et al. 2022).
With regard to biomarkers, it remains unknown if NFL is able to discriminate between specific subtypes of PSP. Nevertheless, NFL can predict disease progression in PSP patients (Jabbari et al. 2017; Rojas et al. 2018). The most established use of biomarkers in this field is the differentiation between PSP-corticobasal syndrome (PSP-CBS) and AD-corticobasal syndrome (AD-CBS). Amyloid beta 1-42 or ratios with total tau in CSF can be used to distinguish between PSP-CBS and AD-CBS (Höglinger et al. 2017). In recent years, also tau-PET imaging showed robust discrimination between PSP-CBS and AD-CBS (Palleis et al. 2021).
Early to prodromal diagnosis of Parkinson’s disease and atypical Parkinson syndromes
As stated above, the clinical diagnosis of PD or aPS is generally based on clinical features. These features develop in the course of the disease. However, the presence of clinical motor-symptoms indicates an already advanced stage of the neuropathology. In PD for example, already 40–70% of substantia nigra dopaminergic neurons have died at time of diagnosis (Fearnley and Lees 1991). Therefore, in recent years, movement disorders experts put their focus more into prodromal or even preclinical phases of PD (Berg et al. 2015; Postuma and Berg 2016). Non-motor symptoms that precede the typical motor features of PD became more important, including hyposmia, depression, REM-sleep behavior disorder, and others (Postuma and Berg 2016). The first notion of RBD as a predictor of a future synucleinopathy was made in a cohort of RBD patients, in which the vast majority of patients developed PD or MSA over the time of a decade (Boeve et al. 2001; Iranzo et al. 2021).
Further work established constellations of specific symptoms e.g. mainly non-motor-symptoms of PD, which pre-manifest to PD motor symptoms (Heinzel et al. 2019). These symptoms were classified with their predictive value in the MDS PD risk calculator, which is available as an online tool on the MDS website (Heinzel et al. 2019). The likelihood of developing PD is mainly driven by a polysomnographic diagnosed RBD. However, there were several other factors driving the individual risk for the development of PD. A recent international multicenter study assessed the predictive value of additional factors for the pheno-conversion of RBD to a neurodegenerative synucleinopathy (PD, DLB, MSA). The factors with the greatest hazard ratio were quantitative motor testing or objective motor examination using the Unified Parkinson’s disease rating scale, followed by olfactory deficits, cognitive impairment, erectile dysfunction, motor symptoms, and an abnormal dopamine transporter SPECT (Postuma et al. 2019). Also others found that slight motor signs and abnormal dopamine transporter SPECT have a high predictive value for the development of PD (Simonet et al. 2021; Chahine et al. 2021a). Orthostatic hypotension had an intermediary predictive value, urge symptoms, constipation, and hyposmia had a very low predictive value (Shi et al. 2021). Using this risk assessment, premotor cohorts of PD can be built and early disease biomarkers may be further established (Iranzo et al. 2021). Still, one of the most relevant questions that remains unanswered so far is the prediction of the time point of pheno-conversion to motor PD (Miglis et al. 2021; Zhang et al. 2022a). Arnaldi and colleagues recently developed an additional tool for patient stratification to predict pheno-conversion to motor stages of PD (Arnaldi et al. 2022).
In stark contrast to the widely established and validated concept of recognizing PD in premotor stages, this concept is far less established in aPS. In the MDS diagnostic criteria for PSP, the new level of certainty “suggestive of” may help to make an earlier diagnosis of PSP (Höglinger et al. 2017). In a recent retrospective study using autopsy-confirmed cases of PSP, the time to the correct diagnosis could be reduced by the application of these criteria (Grimm et al. 2020). However, these data have to be confirmed in a prospective manner, which will be done in national PSP cohorts in Germany (Respondek and Höglinger 2021). Unfortunately, also, the criteria suggestive of the diagnosis of PSP rest up on motor features. A recent study tried to establish earlier symptoms of PSP patients by looking at general practitioner’s data (Viscidi et al. 2021). These data indicated that patients that later develop a PSP have more consultations because of movement disorders, gait disorders, falls, cognitive symptoms and depression compared to a matched control group. Furthermore, these patients more often complained about speech and visual problems (Viscidi et al. 2021). This study provides useful data for further studies on earlier identification of PSP patients.
In the new diagnostic criteria for multiple system atrophy, a novel “possible prodromal MSA” stage was established (Wenning et al. 2022). This stage mainly relies on the presence of RBD and has to be prospective evaluated for sensitivity and specificity of detection of patients eventually developing possible or probable MSA. The assumption of including only RBD patients in this stage seems to be unspecific at first. However, in the future, biomarker enhanced methods may improve specificity.
In these early neuropathological disease stages of PD and aPS, neuroprotective or disease-modifying drugs would have the highest impact on reducing disease progression resulting in sustained quality of life of the patients.
The biggest challenge in the next decade will be the standardized detection of people at risk for the development of a neurodegenerative Parkinsonian syndrome. The incidence of patients with prodromal PD or aPS in the population is low and prodromal symptoms are mainly unspecific (Heinzel et al. 2019). Usually, prodromal PD or aPS patients do not consider consulting a neurologist at first. Therefore, biomarker driven approaches have to be developed. Biomarker approaches may include digital biomarkers, fluid biomarkers, imaging biomarkers, or a combination of these. In addition, training of other clinical disciplines will be important to raise attention to early premotor signs of Parkinsonian syndromes.
Genetic aspects
In general, neurodegenerative synucleinopathies and tauopathies are considered to be sporadic diseases. Monogenetic forms caused by mutations in SNCA, encoding αSyn or MAPT, encoding tau are very rare (Magistrelli et al. 2021) and are, therefore, of low relevance for most of the patients. However, genome-wide association studies (GWAS) showed that distinct single-nucleotide-polymorphisms (SNP) increase the risk to develop neurodegenerative Parkinsonism. In PD, over 90 SNPs have been identified in the most recent meta-analysis of GWAS (Nalls et al. 2019). In a PSP GWAS, SNPs in four genes including MAPT were identified to be associated with a higher risk to develop PSP. One of the genes, in which distinct SNPs were associated with a higher risk to develop PSP was EIF2AK3, encoding for the protein PERK that is part of the unfolded protein response (Höglinger et al. 2011). In MSA so far, no GWAS unequivocally identified a risk SNP with a statistically significant p value. However, a recent MSA GWAS of only histopathologically confirmed MSA cases found SNPs that were borderline significant in close neighborhood to ZIC1 and ZIC4 (Hopfner et al. 2022). Since GWAS in general identify common variants with a low risk, the presence of individual SNPs in a patient has no huge influence on the risk to develop PD or an aPS. However, the compilation of variants into a polygenic risk score can be used to determine the risk of distinct individuals (Dehestani et al. 2021). In the future, these data might be used to specifically enroll patients at risk into clinical trials. Furthermore, data about the accumulation of risk SNPs in the same biological pathway could help to implement therapies that are tailored to the specific biological situation of the individual patient. Furthermore, in PD, mutations in genes other than SNCA play an important role. 5–10% of PD patients have a heterozygous mutation in the gene GBA, encoding the lysosomal protein glucocerebrosidase (Beavan and Schapira 2013), and up to 6% of sporadic PD patients have a mutation in LRRK2, encoding leucine-rich repeat kinase 2 (Bardien et al. 2011). To date, these specific genetic constellations have not been taken into consideration in most clinical trials and they are not at all considered in routine clinical care.
Pathophysiological aspects
The pathological aggregation of αSyn and tau are the main pathophysiological events that lead to cell death in neurodegenerative Parkinson syndromes. In synucleinopathies, small oligomers of αSyn that form on the way from monomeric αSyn to the large aggregates found in Lewy bodies, are considered to be toxic for the cells (Bengoa-Vergniory et al. 2017; Alam et al. 2019). Also, in tauopathies, oligomers of the tau protein are considered to be toxic (Niewiadomska et al. 2021). Protein degradation mechanisms that are involved in the degradation of pathological protein aggregates are the ubiquitin–proteasome-system (Türker et al. 2021), the autophagy-lysosome-system consistent of chaperone-mediated autophagy and macroautophagy (Fleming et al. 2022), and the unfolded protein response (Johnston and Samant 2021). For example, in GBA mutation associated PD, one pathophysiological concept is that the lack of glucocebrosidase function leads to the formation of complex sphingolipids, which impair lysosomal function and thereby reduce degradation of pathological αSyn aggregates (Taguchi et al. 2017). Furthermore, cell culture experiments showed that activation of macroautophagy rescued human dopaminergic neurons from αSyn induced cell death (Höllerhage et al. 2014, 2019). Moreover, it was shown that activation of PERK, a protein involved in unfolded protein response, the gene of which (EIF2AK3) was associated with PSP in a GWAS (Höglinger et al. 2011), rescued cultured human neurons in vitro and spinal cord neurons in mice in vivo from tau-induced toxicity in tauopathy models (Bruch et al. 2017).
Furthermore, cell-to-cell transmission of distinct pathological species of αSyn (Jan et al. 2021; Brás and Outeiro 2021) and tau (Brunello et al. 2020) in a so-called “prion like manner” is considered to be the mechanism behind the progression of synucleinopathies and tauopathies throughout the brain (Uemura et al. 2020). Histopathological studies showed that PD pathology can be divided in six stages. While in stage one and two, pathology is limited to deeper brainstem structures and the olfactory bulb, in stage three and four the pathological process reaches the midbrain including the substantia nigra leading to the typical motor symptoms (Braak et al. 2003). A similar staging system was established for tau pathology in PSP (Kovacs et al. 2020).
Biomarkers
In the past years, biomarkers for PD including real-time quaking induced conversion (RT-QuIC) from the CSF and other biosamples (Poggiolini et al. 2021), presence of (posttranslationally modified) αSyn in the CSF (Kwon et al. 2022), auto-antibodies against αSyn (Folke et al. 2021), αSyn content in exosomes (Jiang et al. 2020, 2021), and αSyn deposits in the skin (Doppler 2021), were extensively investigated. However, none of these markers is already established in clinical routine care and methods to evaluate pathological signals vary between different laboratories. Nevertheless, many of these biomarkers are very promising. The RT-QuIC was first used in the prion field (Candelise et al. 2017). Is this assay, pathological protein seeds from biomaterial of patients are used to induce aggregation of recombinant protein. By shaking, these aggregates are disrupted again and then act as newly formed seeds for further aggregation. The continuous aggregation can be visualized by fluorescence labelling of beta-sheets within the aggregates (Atarashi 2022). In Creutzfeld-Jakob disease, the RT-QuIC was included into the most recent diagnostic criteria (Hermann et al. 2021). In addition, in the field of synucleinopathies, the usage of the RT-QuIC assay is advancing in recent years. A recent study provides evidence for distinct conformations of αSyn in PD and MSA by RT-QuIC analysis (Shahnawaz et al. 2020), and also others showed that αSyn RT-QuIC is suitable to distinguish between PD and aPS (Bargar et al. 2021; Russo et al. 2021; Singer et al. 2021). Thus, in the near future, RT-QuIC will help to distinguish between PD and aPS earlier in the course of the diseases, which can be difficult by clinical assessment alone (see above). Furthermore, it has also been shown that αSyn RT-QuIC reveals a positive signal in the CSF of most patients with idiopathic RBD (iRBD), considered as prodromal stage of PD. In a small series 18 out of 18 (100%) patients with idiopathic iRBD had a positive αSyn RT-QuIC signal in the CSF (Rossi et al. 2020). In a larger series, 47 out of 52 (90%) patients with iRBD had a positive αSyn RT-QuIC signal in the CSF (Iranzo et al. 2021). These data suggest that eventually, RT-QuIC will help to make the diagnosis of PD in a prodromal stage, before the occurrence of typical motor symptoms. Another αSyn-based biomarker is the content of αSyn in brain-derived exsomes in the plasma. Recent studies showed that exosomal αSyn is elevated in PD but not in healthy controls, patients with MSA, or other neurodegenerative PS that are not associated with αSyn pathology. However, clusterin was elevated in patients with a primary tauopathy (Jiang et al. 2020, 2021). Interestingly, exosomal αSyn was also elevated in patients with iRBD (Jiang et al. 2020). These data suggest that the investigation of proteins in the exosomes will be useful to distinguish between synucleinopathies and tauopathies in the future. Furthermore, existing data suggest that exosomal αSyn is a suitable marker to detect prodromal stages of PD.
Moreover, it was shown that the detection of phosphorylated αSyn in the skin can support the diagnosis of PD (Doppler 2021) and that skin αSyn can be used to distinguish between PD and tauopathies (Giannoccaro et al. 2022). In addition, more recently, RT-QuIC was used to investigate pathological αSyn aggregates in the skin, suggesting that this method was able to discriminate between synucleinopathies and tauopathies (Martinez-Valbuena et al. 2022). Interestingly, pathological αSyn deposits in the skin were also found in 10 out of 18 patients with RBD and in 20 out of 25 patients with early PS, but none of the investigated healthy control persons (Doppler et al. 2017). These data suggest that skin biopsy can be useful to prove αSyn pathology in a part of the patients and potentially can help to select patients for disease-modifying therapies in the future.
Another unspecific biomarker for neurodegeneration that was established in the past years is neurofilament light chain (NLF) (Gaetani et al. 2019). This marker can be used to distinguish PD from aPS because of the larger amount of neurodegeneration in aPS compared to PD (Rojas et al. 2018; Zhang et al. 2022b). The most promising fluid biomarkers and their added value are displayed in Table 1.
Imaging
Today, neuroimaging is one of the most essential diagnostic instruments to distinguish different etiologies of Parkinsonism. In a recent instructive review of the literature, Peralta et al. discuss imaging modalities and their contribution to making the differential diagnoses of PD and aPS (Peralta et al. 2022). The most relevant obstacle in neuroimaging is the detection of the underlying pathology in patients with Parkinsonism. Of course, magnet resonance imaging can rule out symptomatic lesions and structural changes in the brain, but structural changes as consequence of the neurodegenerative process appear quite late in the progression of PD and aPS and also do not occur in all patients (Mahlknecht et al. 2010). Imaging modalities, therefore, have to focus more on functional aspects. One promising method may be MR spectroscopy allowing the detection of specific regional metabolic changes (Ciurleo et al. 2014). The major limitation of MR spectroscopy was the regional restriction to one or several regions of interest. With whole-brain MR spectroscopy, this limitation has been overcome and this technique may offer interesting insights in early stages of PD and aPS (Ding and Lanfermann 2015). In a pilot study, metabolic alterations in early PD patients were detected compared to healthy controls (Klietz et al. 2019a). In addition, quantitative cerebral microstructural MR imaging might offer promising insight in the pathophysiology of PD and aPS. In a recent study in early PD patients, a distinct profile of quantitative microstructural changes in PD patients compared to healthy controls was described (Klietz et al. 2021c).
However, the in vivo detection of a synucleinopathy or tauopathy by imaging is still challenging. In the past years, tau PET tracers became available for research purposes, allowing the in vivo detection of hyperphosphorylated and misfolded intracellular tau protein. While the first generation of tau tracers showed unspecific binding patterns (Villemagne et al. 2018), the second generation was promising in recent studies as cross-sectional imaging biomarker (Brendel et al. 2020). For patients with CBS the combination of tau and amyloid PET imaging proved effective in determination of the underlying 4-repeat tau or AD pathology (Palleis et al. 2021; Song et al. 2021). Nevertheless, these tracers are not available in the clinical routine diagnostic yet. Despite these advances in the imaging of tau pathology, imaging of αSyn in vivo is still not possible (Korat et al. 2021).
In the future, a combination of radiological and fluid biomarkers will allow making early and correct diagnoses of PD and specific aPS. This will improve patient counseling, selection of patients for specific therapies, and more efficient inclusion of suitable patients in clinical trials.
Studies with the aim to investigate neuroprotection
Since a neuroprotective therapy would be most efficacious before the neurodegenerative process is too advanced, an inclusion of patients in clinical trials as early as possible in the disease process is desirable. However, due to the lack of established clinical features, biomarkers, or imaging parameters that predict the occurrence of a neurodegenerative PS with a high likelihood, all studies so far enrolled patient that already fulfilled the diagnostic criteria of the distinct disorders. Furthermore, subtypes of PD were not considered in most PD trials (Pagano et al. 2021; Rascol et al. 2011). Recent trials in PSP enrolled patients according to the previous NINDS-SPSP diagnosis criteria (Höglinger et al. 2021) or by the presence of postural instability and vertical gaze palsy (Dam et al. 2021) and thereby de facto only included patients with a Richardson-syndrome without taking into account that these only comprise a quarter of all PSP patients (Respondek and Höglinger 2016). In MSA, some trial only included patients with predominant Parkinsonism (Dodel et al. 2010), whereas other trials included patients with MSA-C and MSA-P (Bensimon et al. 2009; Levin et al. 2016b, 2019).
Since at time of diagnoses the pathophysiological process is already advanced, the observation of strong clinical effects by potentially disease-modifying drugs might be compromised. Given that in PD 50% of dopaminergic neurons and 70–80% of striatal dopamine are already demised before typical motor symptoms occur (Jankovic et al. 1990), only 50% of the originally present dopaminergic neurons are left to be rescued in clinically established PD. A hypothetical disease-modifying therapy that would be able to rescue 50% of the dopaminergic neurons that are left, would lead to a further decline to 25% of the originally present cells, if therapy started at the time of diagnosis of clinically established PD. This would be accompanied by further increase in disability. On the other hand, if a therapy started at a stage, where 90% of the cells would be still present, the rescue of 50% of dopaminergic neurons would lead to a decline to only 45% of the originally present dopaminergic neurons, meaning that motor symptoms would occur much later or only very mildly. However, the rescue of 50% of the neurons would be an extremely huge achievement and is probably beyond any realistic expectations in the near future. Nevertheless, with a rescue of only 25% of the neurons left, one probably cannot expect much of a clinical benefit in patient that already lost 50% of the cells at the beginning of the treatment, since a rescue of 25% of the cells left would mean that only 12.5% of the originally present cells would survive in the long run. PD and aPS have a disease course of many years before motor symptoms occur (Gaenslen et al. 2011; Postuma and Berg 2016). If one assumed a demise of 10% of the dopaminergic cells in PD every year, within 1 year, the part of dopaminergic cells would decline from 50 to 45% in clinically established PD, even though natural history data suggest that annual clinical decline is measurable. However, given that there are inter-individual differences, it is conceivable that even complete or almost complete rescue of all cells that would have died in 1 year by treatment with a neuroprotective agent, would not lead to a relevant clinical difference compared to untreated patients. Nevertheless, in most recent trials, the duration of the treatment was limited to 1 year (Pagano et al. 2021) (NCT01560754). Furthermore, it is possible that agents that interfere with the pathological process would not be effective immediately, but only after a delay. Assuming that compounds that reduce aggregation of αSyn or tau, would immediately prevent further aggregation of any protein, it is still possible that already existent toxic aggregates would still do further harm to the cells. In light of the abovementioned assumptions, a trial duration of one year in already clinically established patients is probably too short to be able to observe relevant clinical effects and longer treatment and observation phases would be desirable. In line with that, in a previous trial with MSA patients, there was a tendency to improvement in the treatment group towards the end of the treatment period (Levin et al. 2019), which theoretically could have been more pronounced, if the trial duration had been longer. Furthermore, in many trials, patients with symptomatic co-medication with rasagiline were enrolled. Therefore, mild symptomatic effects which could be stronger with less endogenous dopamine left might have diluted small neuroprotective effects On the other hand, it is difficult to enroll patients with clinically established Parkinsonism without therapy in a trial with a duration of longer than a year, because many of the participants would eventually need effective symptomatic treatment. Another obstacle might be the reliance on clinical outcomes. Symptomatic effects of the treatment may confound clinical readouts. Furthermore, clinical scores have at least some interrater variability (Richards et al. 1994). Therefore, novel, objective outcome measures would be helpful. In a previous PD trial, a digital, completely objective outcome measure was used, showing that digital outcome measures are feasible (Pagano et al. 2021). In future clinical trials, it will be important to include patients in prodromal stages of PS, when neurodegeneration is not too far advanced. In addition to clinical readouts, also novel digital, objective readouts should be considered. Furthermore, since neuroprotective therapies might take longer until they have a relevant effect in comparison to symptomatic therapies, longer treatment and observation periods are desirable.
Novel neuroprotective approaches
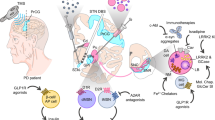
In future approaches for neuroprotective therapies, the molecular mechanisms behind neurodegeneration will move more into focus. These will comprise the reduced formation of pathological aggregates of tau and αSyn, the increased degradation of pathological aggregates, and the inhibition of cell-to-cell spreading of pathological protein aggregates.
Reduction of the formation of pathological protein aggregates
Since pathological aggregates of αSyn and tau are involved in the pathophysiology of neurodegenerative Parkinson’s syndromes, one approach for a disease-modifying therapy would be the reduction of the formation of these pathological aggregates. One approach to achieve this is the reduction of the translation of the underlying proteins αSyn and tau. The idea behind this concept is that the presence of too much aggregation-prone protein (i.e. αSyn or tau) increases the risk of the formation of toxic oligomers that are harmful for the cells (Alam et al. 2019; Niewiadomska et al. 2021). Therefore, recently, antisense-nucleotides (ASO) were investigated. These are small nucleic acid sequences with a homology to a target messenger RNA (mRNA). The formation of double-strand RNA, that is not physiological in eukaryotic cells, leads to the degradation of the targeted mRNA and thus reduced protein translation. Phase I clinical trials investigating ASOs against αSyn in MSA (NCT04165486), and tau in PSP (NCT04539041) are ongoing. Furthermore, also a clinical trial targeting LRRK2 with ASOs is ongoing (NCT03976349).
Since, tau aggregates in PSP are mainly consistent of 4-repeat tau, another therapeutical approach would be to modify the splicing process of MAPT, to shift the presence of tau-protein from 4-repeat- to 3-repeat isoforms. Recently, a reporter system was developed that allows screening for specific splicing modifiers of MAPT (Truong et al. 2021). Thus, in the future, pharmacological interventions that modify MAPT-splicing might be discovered and eventually used in clinical care.
In addition to approaches that target the translation of aggregation-prone proteins αSyn and tau, other approaches aim for the inhibition of the aggregation process itself. The green tea extract epigallocatechin gallate (EGCG) is one compound that inhibits the aggregation of αSyn and other aggregation-prone proteins (Fernandes et al. 2021). Recently, in an investigator initiated double-blind placebo-controlled trial, EGCG was investigated in patients with MSA over one year. Interestingly, EGCG treatment led to a significant reduction in atrophy in the striatum and the precentral gyrus in a subgroup of patients that were investigated with MRI scans. Furthermore, EGCG treatment led to a tendency in the reduction of clinical progression compared to placebo (Levin et al. 2019). Even though the clinical benefit was not statistically significant, data from the MRI subgroup and the tendency to clinical benefit are promising. For once, these data suggest that a treatment of one year might have been too short to observe a statistical significant clinical benefit. Furthermore, these data suggest that inhibition of aggregation is a feasible approach and therefore might be more effective, if used in earlier or prodromal disease stages in the future.
Another interesting anti-aggregatory compound is anle138b. The lead structure of this compound was identified in a cell-free screening assay, in which 20.000 compounds were investigated for their potential to reduce αSyn aggregation in vitro. After the primary screening, the lead structure was chemically modified in order to improve its anti-aggregatory properties (Wagner et al. 2013). Anle138b proved to be efficient in mouse models of PD (Levin et al. 2014), MSA (Lemos et al. 2020; Heras-Garvin et al. 2019; Fellner et al. 2016), tauopathies (Wagner et al. 2015), and Alzheimer’s disease (Brendel et al. 2019). Recently, a phase I-trial with healthy person investigating safety and tolerability of anle138b was successfully finished (NCT04208152) and further trials in patients are planned.
Increased degradation of pathological protein aggregates
Another approach to reduce the burden of pathological protein aggregates is increased degradation. Mechanisms that are involved in the degradation of pathological αSyn and tau-aggregates are the ubiquitin–proteasome-system (Türker et al. 2021), autophagy, which comprises macroautophagy, chaperone-mediated autophagy, and microautophagy (Fleming et al. 2022), and the unfolded protein response (Johnston and Samant 2021). In macroautophagy, large protein aggregates and defective organelles are engulfed by a double-membrane structure called the autophagosome. In the process of macroautophagy the autophagosome is fused with the lysosome, in which degradation occurs (Feng et al. 2014). Activators of macroautophagy proved to be protective against αSyn induced toxicity in a PD cell model (Höllerhage et al. 2014, 2019). However, also reduction of the formation of autophagosomes as one of the first steps of macroautophagy protected dopaminergic neurons in vitro from αSyn-induced toxicity and bypassing of macroautophagy led to increased exosomal release of αSyn (Fussi et al. 2018). Thus, the role of macroautophagy as a therapeutic target is not yet fully understood. Previously, it was suggested that a pulsatile activation of macroautophagy might be a feasible approach as therapy for PD (Fowler and Moussa 2018). While autophagy is an essential intracellular process, an overstimulation might be harmful for the cells, which is supported by observations that mutated αSyn stimulated macroautophagy and thereby led to cell death (Choubey et al. 2011). However, others showed that wild-type αSyn led to impaired macroautophagy which was also harmful (Winslow and Rubinsztein 2011). In a pulsatile therapeutic regimen, by activation of autophagy one could intermittently remove pathological protein aggregates, while autophagy function would be left to its physiological states in the intervals.
In PD caused by heterozygous GBA mutations, one assumption is that impaired function of the lysosomal protein glucocerebrosidase leads to the formation of complex sphingolipids from glycosylceramide and these then lead to impaired lysosomal function. Consequently, toxic αSyn species accumulate and lead to cell death. Physiologically glycosylceramide is metabolized by glucocerebrosidase into ceramide and glucose. In a previous trial (MOVES-PD, NCT02906020), the glucosylceramide-synthase inhibitor venglustat was investigated in PD patients with a heterozygous GBA-mutation with the aim to lower the substrate for the formation of complex sphingolipids and thereby prevent impairment of lysosomal function and accumulation of toxic αSyn species. Interestingly, patients treated with venglustat showed a linear decline in motor function as measured by an increase of the score of part II and III of the MDS-UPDRS, while placebo-treated patients stayed stable first, before also a linear decline started. This result was interpreted as a mild negative symptomatic effect of venglustat. Another therapeutical approach in GBA-mutation associated PD would be the activation of glucocerebrosidase. Currently, a phase II trial with a glucocerebrosidase activator (BIA 28-6156 / LTI-291) is in preparation.
The unfolded protein response (UPR) is a cellular mechanism that is involved in protein quality control and degradation of misfolded proteins. Interestingly, EIF2AK3, the gene encoding for PERK, a protein involved in UPR, was identified as a risk gene for PSP in a previous GWAS (Höglinger et al. 2011). Furthermore, mutations in EIF2AK3 lead Wolcott-Rallison-syndrome (WRS), an extremely rare condition with complete absence of PERK which is associated with neurodegeneration. Moreover, histopathological examination of a post-mortem WRS brain showed changes in autophagic flux in absence of PERK, suggesting an interplay between the UPR and autophagy (Bruch et al. 2015). Interestingly, experimental data showed that pharmacological activation of PERK rescued neuronal cells in in vitro and in vivo models of tauopathies (Bruch et al. 2017), suggesting that PERK activation might be a rational approach for a neuroprotective therapy of PSP and other tauopathies.
Inhibition of spreading of aggregation-prone proteins
Cell-to-cell spreading in a so-called “prion-like” manner plays an important role in the progression of neurodegenerative disorders, including synucleinopathies and tauopathies, throughout the brain (Jucker and Walker 2018). Therefore, prevention of cell-to-cell spreading is a rational approach for disease-modifying therapies. Cell-to-cell spreading includes the release of toxic protein species from diseased cells, the presence of toxic protein species in the extracellular space, and the uptake of toxic protein species in previously healthy cells. All three parts are potential targets for neuroprotective therapies. In the past years, the understanding of the specific uptake mechanisms of pathological protein aggregates increased a lot. Previously it was shown that reduced levels of BIN1-amphysin2, a negative regulator of endocytosis led to increased tau propagation in cell culture (Calafate et al. 2016). Others found that heparin sulfate proteoglycans are involved in the uptake of tau-fibrils (Holmes et al. 2013). Recently, it was published that receptor-related protein 1 (LRP1), a member of the LDL receptors family is involved in tau uptake (Rauch et al. 2020). Furthermore, some studies suggest that receptors on the cell surface such as lymphocyte activating gene 3 (LAG3) and amyloid precursor-like protein 1 (APLD1) are involved in intracellular internalization of distinct αSyn species (Mao et al. 2016; Zhang et al. 2021). However, more recently, it was questioned, if LAG3 was expressed in human cells at all (Emmenegger et al. 2021). While approaches that target specific uptake mechanisms are still in early stages of preclinical development, approaches that aim on the scavenging of pathological protein species from the extracellular space using specific antibodies are more advanced. Previously, two phase II trials were conducted in PSP investigating the disease-modifying potential of tau-antibodies with an N-terminal epitope. However, both trials failed to meet the primary efficacy endpoint which was clinical improvement compared to placebo measured with the PSP rating scale (Höglinger et al. 2021; Dam et al. 2021). This led to the discussion, if either the epitope of the investigated antibodies was wrong, or the therapies came too late, or if tau was the correct target at all (Jabbari and Duff 2021). However, new tau antibodies with other tau epitopes are in the development pipeline, one of these (UCB0107, bepranemab), is already under investigation in early phase clinical trials (NCT04658199).
Furthermore, also in PD, trials with antibodies against αSyn were conducted. While, a trial with the N-terminal anti-αSyn antibody cinpanemab (NCT03318523) failed to meet primary and secondary endpoints, another trial with the C-terminal anti-αSyn antibody prasinezumab (NCT03100149) (Pagano et al. 2021) showed promising results with a trend towards improvement in the treatment group compared to the placebo group in the MDS-UPDRS part III. Furthermore, this trial is in particular interesting, because in addition to the MDS-UPDRS, a digital motor score was used as outcome parameter. Interestingly, in this score there was a significant improvement in the treatment group compared to placebo with a slightly higher effect in the group with the lower dose of prasinezumab. Nevertheless, these data are very interesting, because they show the feasibility of a completely objective bias-free readout in a clinical trial.
Anti-inflammatory approaches
In the past years, the contribution of inflammation in neurodegenerative disorders came more into focus (Pajares et al. 2020; Nichols et al. 2019). Inflammatory processes in the central nervous system involve the activation of microglia. Amongst other causes, reactive oxygen species (ROS) generated by an enzyme called myeloperoxidase lead to activation of microglia. Therefore, previously a myeloperoxidase inhibitor (verdiperstat) was investigated in a clinical trial in MSA (NCT03952806). Even though the trial failed to meet its primary and key secondary outcome measures, anti-inflammatory approaches might play a role in future therapies of neurodegenerative PS.
Drug repurposing
Another approach towards novel therapies for PS would be the repurposing of already approved drugs. Drug repurposing has the advantage of saving costly and time-consuming steps in the development of completely new drugs, since data about compatibility, dosage, and pharmacokinetics of approved drugs are readily available. For instance, we previously performed a drug-screening of 1,600 FDA-approved drugs in an αSyn cell model and identified that the phosphodiesterase I inhibitor, vinpocetine, protected human neurons from αSyn-induced toxicity in vitro and murine neurons in vivo (Höllerhage et al. 2017). Since vinpocetine is approved in many European countries for indications other than Parkinsonism or synucleinopathies, and the drug is generally well-tolerated, it would be reasonable to investigate a potential neuroprotective effect in patients suffering from a neurodegenerative synucleinopathy (e.g. iRBD, PD, or MSA).
Clinical care for patients with Parkinson’s disease and atypical Parkinson syndromes
There are several obstacles in care for people with PD or aPS. In this part of the review, we will mainly focus on later disease stages, although we know that there are also several issues at the beginning of the disease as well. These are the psychological impact of communicating the diagnosis of prodromal PD or aPS to previously healthy individuals and management of their coping with this information.
Patient management: drug safety
In advanced stages of PD and aPS, most of the patients are in a geriatric age and display several characteristics of geriatric patients like multimorbidity and polypharmacy (Greten et al. 2021; Klietz et al. 2019b). These patients are extremely vulnerable for side effects of drug treatment (Lingor et al. 2016). In PD, patients display a distinct profile of comorbidities including hypertension, heart failure, polyneuropathy, diabetes mellitus, sarcopenia, frailty, and cerebral small vessel disease (Greten et al. 2021; Klietz et al. 2019b). Treatment of these patients needs a careful monitoring of drug effects and side effects. In a recent review, a strategy for monitoring of these patients has been proposed (Klietz et al. 2019b). Furthermore, geriatric visits should include specific assessments and strategies including discontinuation of medication (Scott et al. 2015). The usage of drug prescribing lists to detect potentially inadequate medication can be helpful in these patient groups. In a recent study, the FORTA classification system has been described in geriatric PD in-patients as a promising tool to increase drug-safety (Greten et al. 2021). In two studies, typical comorbidities with high prevalence in PSP patients were arterial hypertension and diabetes (Papapetropoulos et al. 2005; Rabadia et al. 2019). This has also been reported in a recent study that used PSP patients’ data from general practitioners in Germany (Kwasny et al. 2021). Furthermore, German health care data report common cardiovascular, muscular, neurological, and urogenital comorbidities in PSP (Zella et al. 2020). However, typical drug constellations in the treatment of PSP and MSA patients are not clear and should be determined in larger cohorts. In summary, patients with advanced PD or aPS are more likely to present with geriatric strains like multimorbidity and polypharmacy. Furthermore, these patients often present with cognitive impairments (Klietz et al. 2019b). Therefore, these patients have a higher likelihood of medical emergencies and need drug-safety concepts (Moßhammer et al. 2016; Okunoye et al. 2020). In the future, drug safety concepts should be digital. However, in the meantime measures like the emergency box might help to cope with drug-safety issues in acute emergency referral (Krey et al. 2021).
Palliative care
Patients with PD have an only mildly reduced life expectancy (Macleod et al. 2014). However, an increasing number of symptoms lead to reduced quality of life over a long disease duration. In patients with aPS life expectancy is dramatically reduced and also symptom burden severely reduces quality of life. The international multicenter prospective CLASP study examined late-stage PD and aPS patients and described an extensive reduction in the activities of daily living and quality of life (Balzer-Geldsetzer et al. 2018). Advanced PD and aPS patients suffer from severe motor and non-motor symptoms (Schrag et al. 2020). In particular, neuropsychiatric symptoms contribute to reduced quality of life (Hommel et al. 2020; Eichel et al. 2022). As also discussed earlier, there is still no cure for neurodegenerative PS. Therefore, palliative care approaches are extremely important in the holistic treatment of these patients (Miyasaki 2013; Miyasaki and Kluger 2015; Oliver et al. 2016). Unfortunately, only a very small proportion of patients with PD received palliative care interventions (Klietz et al. 2018; Safarpour et al. 2015). In a case series, Bükki et al. reported palliative care interventions in patients with aPS in a hospital setting. Most patients could be discharged after acute interventions with improved symptom control and increased quality of life (Bükki et al. 2016). Interestingly, about 72% of these advanced PD patients have an advance directive (Klietz et al. 2018). Investigation of the specific content of these advance directives revealed that the vast majority did not address disease specific complications (Klietz et al. 2019c). In a consensus-based Delphi panel of German palliative care and PD experts, disease specific recommendations for specific advance directives for people with PD have been developed (Klietz et al. 2020a). One explanation for the low-grade implementation of palliative care interventions in PD patients may be the low level of palliative care training in neurologists compared to general practitioners (Jensen et al. 2022). Cooperation between neurologists and palliative care specialists mainly focuses on patients with brain tumors and amyotrophic lateral sclerosis. Unfortunately, concepts for other progressive neurological diseases are rare (Oliver et al. 2020). In a recent international multicenter study, concepts for palliative care interventions were studied and palliative care training programs were developed (Gatsios et al. 2021; Meinders et al. 2021). A survey on aPS patients revealed that this patient group prefers an early conversation about diagnosis with information about the disease, natural history, prognosis, and treatment options (Saranza et al. 2021). These aPS patients also want to have a conversation about end of life and advance care planning in later stages of the disease (Saranza et al. 2021). Advance care planning concepts are necessary to improve clinical care (Lum et al. 2019; Seeber et al. 2019). Moreover, in aPS, early psychological interventions should be employed, because of the large proportion of patients undergoing assisted suicide (Nuebling et al. 2021; Lum et al. 2019; Churm et al. 2022). In the future, palliative care interventions must become an essential part of PD and, even more importantly, aPS therapy (Miyasaki et al. 2022). In addition, more neurologists need specific training in palliative care, not only for PD and aPS patients.
Caregiver burden
Finally, to optimize patient care, the caregiver must also be considered. Caregiver burden is an extremely important issue in PD and also aPS (Mosley et al. 2017). In a recent study, caregiver burden and caregiver burnout was identified as a relevant factor contributing to transition to institutional care (Jensen et al. 2021). Furthermore, caregivers provide a huge quantity of informal care which is a huge economic factor in PD (Martinez-Martin et al. 2019). Measurement of caregiver burden can use generic scales such as the Zarit burden scale or disease specific scales such as the very well-validated Parkinson’s disease caregiver burden questionnaire (Zarit et al. 1986; Zhong et al. 2013; Klietz et al. 2019d, 2021a). The usage of disease specific questionnaires can assess the relationship of specific symptoms of a specific disease to the perceived caregiver burden. However, in the clinical routine, a fast assessment can also be performed by using visual analogue scales or other simple tools with good correlations to more detailed questionnaires (Klietz et al. 2019d; Jensen et al. 2021). A very interesting tool is the recently published caregiver task questionnaire, which allows to assess the perceived burden of specific caregiving tasks in a specific caregiver setting (Klietz et al. 2021b). Caregiver burden is mainly determined by the patient’s loss of autonomy and increasing care-dependency. General factors contributing to caregiver burden are exacerbated by the quantity of caregiving hours per day (Grün et al. 2016; Tessitore et al. 2018; Genç et al. 2019). More specific aspects influencing caregiver burden in PD were motor symptoms, cognitive decline, and other neuropsychiatric symptoms (Klietz et al. 2020b, c; Eichel et al. 2022; Bruno et al. 2016; Chahine et al. 2021b; Martinez-Martin et al. 2015). In the future, physicians must include caregiver burden and plans for interventions aiming to reduce this burden and allow sustained care at home in the evaluation of PD and aPS patients with complex symptoms. In patients with huge symptom load and caregiver burnout, a transition to institutional care might be a good option to reduce caregiver burden, improve overall quality of care for these patients, and even improve relationship satisfaction (Heine et al. 2021; Jensen et al. 2021; Trapp et al. 2019). Management programs for patients and their caregivers might improve coping with complex symptoms for both the patients and their caregivers (Hellqvist et al. 2018, 2020; Tennigkeit et al. 2020). Treatment of complex patients in specific PD networks may improve overall care and efficacy of PD treatment by including all relevant stakeholders (Prell et al. 2020a, b).
Conclusion
Disease modification in Parkinsonism and other neurodegenerative conditions is still a holy grail, since there is no therapy yet with a proven disease-modifying effect. However, many very promising approaches are in development. Theses comprise the inhibition of the formation of pathological protein aggregates by lowering total protein levels or by pharmacological inhibition of the aggregation process. Furthermore, other approaches aim to reduce the burden of pathological protein aggregates by inducing their degradation. Other approaches target the cell-to-cell spreading of the neurodegenerative process by inhibiting the uptake of pathological protein aggregates on the one hand or by scavenging pathological protein species from the extracellular space using antibodies on the other hand. A different, less mechanistic approach is drug-repurposing with the advantage that time-consuming and costly steps in the development of new drugs can be skipped, because data regarding dosage, pharmacokinetics, and tolerability of already approved drugs are readily available. Furthermore, in the future, it will be of utmost importance to identify persons at risk to develop PD or a related disorder as early as possible to start the investigation of potentially neuroprotective treatments in a stage before neurodegeneration is too marked to achieve a relevant clinical effect. Tools like the MDS risk score as well as novel biomarkers can help with that. Furthermore, novel readouts including biomarkers representing the ongoing pathological process and bias-free digital clinical readouts will support the ongoing quest for disease modification in Parkinsonism. We are very optimistic that disease modification is on the verge to become reality and that we will experience huge changes in the way how we treat Parkinsonism within the 20’s of the twenty-first century. Nevertheless, despite all treatment options, there will still be large groups of patients reaching advanced disease stages in need for excellent clinical care. These patients will profit from drug-safety interventions, palliative care and interventions to reduce caregiver burden.
References
Adler CH, Beach TG, Hentz JG, Shill HA, Caviness JN, Driver-Dunckley E, Sabbagh MN, Sue LI, Jacobson SA, Belden CM, Dugger BN (2014) Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology 83(5):406–412
Alam P, Bousset L, Melki R, Otzen DE (2019) α-synuclein oligomers and fibrils: a spectrum of species, a spectrum of toxicities. J Neurochem 150(5):522–534
Arnaldi D, Mattioli P, Famà F, Girtler N, Brugnolo A, Pardini M, Donniaquio A, Massa F, Orso B, Raffa S, Bauckneht M, Morbelli S, Nobili F (2022) Stratification tools for disease-modifying trials in prodromal synucleinopathy. Movement Disord 37(1):52–61
Atarashi R (2022) RT-QuIC as ultrasensitive method for prion detection. Cell Tissue Res. https://doi.org/10.1007/s00441-021-03568-8
Ayers JI, Lee J, Monteiro O, Woerman AL, Lazar AA, Condello C, Paras NA, Prusiner SB (2022) Different α-synuclein prion strains cause dementia with Lewy bodies and multiple system atrophy. Proc Nat Acad Sci USA 119(6):e2113489119
Balzer-Geldsetzer M, Ferreira J, Odin P, Bloem BR, Meissner WG, Lorenzl S, Wittenberg M, Dodel R, Schrag A (2018) Study protocol: care of late-stage Parkinsonism (CLaSP): a longitudinal cohort study. BMC Neurol 18(1):185
Bardien S, Lesage S, Brice A, Carr J (2011) Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Parkinsonism Relat Disord 17(7):501–508
Bargar C, Wang W, Gunzler SA, LeFevre A, Wang Z, Lerner AJ, Singh N, Tatsuoka C, Appleby B, Zhu X, Xu R, Haroutunian V, Zou W-Q, Ma J, Chen SG (2021) Streamlined alpha-synuclein RT-QuIC assay for various biospecimens in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun 9(1):62
Beavan MS, Schapira AHV (2013) Glucocerebrosidase mutations and the pathogenesis of Parkinson disease. Ann Med 45(8):511–521
Bengoa-Vergniory N, Roberts RF, Wade-Martins R, Alegre-Abarrategui J (2017) Alpha-synuclein oligomers: a new hope. Acta Neuropathol 134(6):819–838
Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN (2009) Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain 132(Pt 1):156–171
Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, Gasser T, Goetz CG, Halliday G, Joseph L, Lang AE, Liepelt-Scarfone I, Litvan I, Marek K, Obeso J, Oertel W, Olanow CW, Poewe W, Stern M, Deuschl G (2015) MDS research criteria for prodromal Parkinson’s disease. Movement Disord 30(12):1600–1611
Boeve BF, Silber MH, Ferman TJ, Lucas JA, Parisi JE (2001) Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Movement Disord 16(4):622–630
Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211
Brás IC, Outeiro TF (2021) Alpha-Synuclein: mechanisms of release and pathology progression in synucleinopathies. Cells 10(2):375
Brendel M, Barthel H, van Eimeren T, Marek K, Beyer L, Song M, Palleis C, Gehmeyr M, Fietzek U, Respondek G, Sauerbeck J, Nitschmann A, Zach C, Hammes J, Barbe MT, Onur O, Jessen F, Saur D, Schroeter ML, Rumpf J-J, Rullmann M, Schildan A, Patt M, Neumaier B, Barret O, Madonia J, Russell DS, Stephens A, Roeber S, Herms J, Bötzel K, Classen J, Bartenstein P, Villemagne V, Levin J, Höglinger GU, Drzezga A, Seibyl J, Sabri O (2020) Assessment of 18F-PI-2620 as a biomarker in progressive supranuclear palsy. JAMA Neurol 77(11):1408–1419
Brendel M, Deussing M, Blume T, Kaiser L, Probst F, Overhoff F, Peters F, von Ungern-Sternberg B, Ryazanov S, Leonov A, Griesinger C, Zwergal A, Levin J, Bartenstein P, Yakushev I, Cumming P, Boening G, Ziegler S, Herms J, Giese A, Rominger A (2019) Late-stage Anle138b treatment ameliorates tau pathology and metabolic decline in a mouse model of human Alzheimer’s disease tau. Alzheimer’s Res Ther 11(1):67
Bruch J, Kurz C, Vasiljevic A, Nicolino M, Arzberger T, Höglinger GU (2015) Early neurodegeneration in the brain of a child without functional PKR-like endoplasmic reticulum kinase. J Neuropathol Exp Neurol 74(8):850–857
Bruch J, Xu H, Rösler TW, de Andrade A, Kuhn P-H, Lichtenthaler SF, Arzberger T, Winklhofer KF, Müller U, Höglinger GU (2017) PERK activation mitigates tau pathology in vitro and in vivo. EMBO Mol Med 9(3):371–384
Brunello CA, Merezhko M, Uronen R-L, Huttunen HJ (2020) Mechanisms of secretion and spreading of pathological tau protein. Cell Mol Life Sci 77(9):1721–1744
Bruno V, Mancini D, Ghoche R, Arshinoff R, Miyasaki JM (2016) High prevalence of physical and sexual aggression to caregivers in advanced Parkinson’s disease. Experience in the Palliative Care Program. Parkinsonism Relat Disord 24:141–142
Bükki J, Nübling G, Lorenzl S (2016) Managing advanced progressive supranuclear palsy and corticobasal degeneration in a palliative care unit: admission triggers and outcomes. Am J Hosp Palliat Care 33(5):477–482
Calafate S, Flavin W, Verstreken P, Moechars D (2016) Loss of Bin1 promotes the propagation of tau pathology. Cell Rep 17(4):931–940
Candelise N, Schmitz M, Da Silva Correia SM, Arora AS, Villar-Piqué A, Zafar S, Llorens F, Cramm M, Zerr I (2017) Applications of the real-time quaking-induced conversion assay in diagnosis, prion strain-typing, drug pre-screening and other amyloidopathies. Expert Rev Mol Diagn 17(10):897–904
Chahine LM, Brumm MC, Caspell-Garcia C, Oertel W, Mollenhauer B, Amara A, Fernandez-Arcos A, Tolosa E, Simonet C, Hogl B, Videnovic A, Hutten SJ, Tanner C, Weintraub D, Burghardt E, Coffey C, Cho HR, Kieburtz K, Poston KL, Merchant K, Galasko D, Foroud T, Siderowf A, Marek K, Simuni T, Iranzo A (2021a) Dopamine transporter imaging predicts clinically-defined α-synucleinopathy in REM sleep behavior disorder. Anna Clin Transl Neurol 8(1):201–212
Chahine LM, Feldman R, Althouse A, Torsney B, Alzyoud L, Mantri S, Edison B, Albert S, Daeschler M, Kopil C, Marras C (2021b) Contribution of neuropsychiatric symptoms in Parkinson’s disease to different domains of caregiver burden. J Neurol 268(8):2961–2972
Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, Zharkovsky A, Kaasik A (2011) Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 286(12):10814–10824
Churm D, Dickinson C, Robinson L, Paes P, Cronin T, Walker R (2022) Understanding how people with Parkinson’s disease and their relatives approach advance care planning. Eur Geriatr Med 13(1):109–117
Ciurleo R, Di Lorenzo G, Bramanti P, Marino S (2014) Magnetic resonance spectroscopy: an in vivo molecular imaging biomarker for Parkinson’s disease? Biomed Res Int 2014:519816
Dam T, Boxer AL, Golbe LI, Höglinger GU, Morris HR, Litvan I, Lang AE, Corvol J-C, Aiba I, Grundman M, Yang L, Tidemann-Miller B, Kupferman J, Harper K, Kamisoglu K, Wald MJ, Graham DL, Gedney L, O’Gorman J, Haeberlein SB (2021) Safety and efficacy of anti-tau monoclonal antibody gosuranemab in progressive supranuclear palsy: a phase 2, randomized, placebo-controlled trial. Nat Med 27(8):1451–1457
Dehestani M, Liu H, Gasser T (2021) Polygenic risk scores contribute to personalized medicine of Parkinson’s Disease. J Personalized Med 11(10):1030
Dickson DW (2018) Neuropathology of Parkinson disease. Parkinsonism Relat Disord 46(Suppl 1):S30–S33
Ding X-Q, Lanfermann H (2015) Whole brain 1H-Spectroscopy: a developing technique for advanced analysis of cerebral metabolism. Clin Neuroradiol 25(Suppl 2):245–250
Dodel R, Spottke A, Gerhard A, Reuss A, Reinecker S, Schimke N, Trenkwalder C, Sixel-Döring F, Herting B, Kamm C, Gasser T, Sawires M, Geser F, Köllensperger M, Seppi K, Kloss M, Krause M, Daniels C, Deuschl G, Böttger S, Naumann M, Lipp A, Gruber D, Kupsch A, Du Y, Turkheimer F, Brooks DJ, Klockgether T, Poewe W, Wenning G, Schade-Brittinger C, Oertel WH, Eggert K (2010) Minocycline 1-year therapy in multiple-system-atrophy: effect on clinical symptoms and (11)C (R)-PK11195 PET (MEMSA-trial). Movement Disord 25(1):97–107
Doppler K (2021) Detection of dermal alpha-synuclein deposits as a biomarker for Parkinson’s Disease. J Parkinsons Dis 11(3):937–947
Doppler K, Jentschke H-M, Schulmeyer L, Vadasz D, Janzen A, Luster M, Höffken H, Mayer G, Brumberg J, Booij J, Musacchio T, Klebe S, Sittig-Wiegand E, Volkmann J, Sommer C, Oertel WH (2017) Dermal phospho-alpha-synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson’s disease. Acta Neuropathol 133(4):535–545
von Eichel H, Heine J, Wegner F, Rogozinski S, Stiel S, Groh A, Krey L, Höglinger GU, Klietz M (2022) Neuropsychiatric symptoms in Parkinson’s disease patients are associated with reduced health-related quality of life and increased caregiver burden. Brain Sci 12(1):89
Emmenegger M, de Cecco E, Hruska-Plochan M, Eninger T, Schneider MM, Barth M, Tantardini E, de Rossi P, Bacioglu M, Langston RG, Kaganovich A, Bengoa-Vergniory N, Gonzalez-Guerra A, Avar M, Heinzer D, Reimann R, Häsler LM, Herling TW, Matharu NS, Landeck N, Luk K, Melki R, Kahle PJ, Hornemann S, Knowles TPJ, Cookson MR, Polymenidou M, Jucker M, Aguzzi A (2021) LAG3 is not expressed in human and murine neurons and does not modulate α-synucleinopathies. EMBO Mol Med 13(9):e14745
Fanciulli A, Wenning GK (2015) Multiple-system atrophy. N Engl J Med 372(3):249–263
Fearnley JM, Lees AJ (1991) Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114(Pt 5):2283–2301
Fellner L, Kuzdas-Wood D, Levin J, Ryazanov S, Leonov A, Griesinger C, Giese A, Wenning GK, Stefanova N (2016) Anle138b partly ameliorates motor deficits despite failure of neuroprotection in a model of advanced multiple system atrophy. Front Neurosci 10:99
Feng Y, He D, Yao Z, Klionsky DJ (2014) The machinery of macroautophagy. Cell Res 24(1):24–41
Fereshtehnejad S-M, Zeighami Y, Dagher A, Postuma RB (2017) Clinical criteria for subtyping Parkinson’s disease: biomarkers and longitudinal progression. Brain 140(7):1959–1976
Fernandes L, Cardim-Pires TR, Foguel D, Palhano FL (2021) Green tea polyphenol epigallocatechin-gallate in amyloid aggregation and neurodegenerative diseases. Front Neurosci 15:718188
Fleming A, Bourdenx M, Fujimaki M, Karabiyik C, Krause GJ, Lopez A, Martín-Segura A, Puri C, Scrivo A, Skidmore J, Son SM, Stamatakou E, Wrobel L, Zhu Y, Cuervo AM, Rubinsztein DC (2022) The different autophagy degradation pathways and neurodegeneration. Neuron 110(6):935–966
Folke J, Rydbirk R, Løkkegaard A, Hejl A-M, Winge K, Starhof C, Salvesen L, Pedersen LØ, Aznar S, Pakkenberg B, Brudek T (2021) Cerebrospinal fluid and plasma distribution of anti-α-synuclein IgMs and IgGs in multiple system atrophy and Parkinson’s disease. Parkinsonism Relat Disord 87:98–104
Foubert-Samier A, Pavy-Le Traon A, Guillet F, Le-Goff M, Helmer C, Tison F, Rascol O, Proust-Lima C, Meissner WG (2020) Disease progression and prognostic factors in multiple system atrophy: a prospective cohort study. Neurobiol Dis 139:104813
Fowler AJ, Moussa CE-H (2018) Activating autophagy as a therapeutic strategy for Parkinson’s disease. CNS Drugs 32(1):1–11
Frank A, Peikert K, Linn J, Brandt MD, Hermann A (2020) MDS criteria for the diagnosis of progressive supranuclear palsy overemphasize Richardson syndrome. Ann Clin Transl Neurol 7(9):1702–1707
Fussi N, Höllerhage M, Chakroun T, Nykänen N-P, Rösler TW, Koeglsperger T, Wurst W, Behrends C, Höglinger GU (2018) Exosomal secretion of α-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis 9(7):757
Gaenslen A, Swid I, Liepelt-Scarfone I, Godau J, Berg D (2011) The patients’ perception of prodromal symptoms before the initial diagnosis of Parkinson’s disease. Movement Disord 26(4):653–658
Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H (2019) Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry 90(8):870–881
Gatsios D, Antonini A, Gentile G, Konitsiotis S, Fotiadis D, Nixina I, Taba P, Weck C, Lorenzl S, Lex KM, Paal P (2021) Education on palliative care for Parkinson patients: development of the “Best care for people with late-stage Parkinson’s disease” curriculum toolkit. BMC Med Educ 21(1):538
Genç F, Yuksel B, Tokuc FEU (2019) Caregiver burden and quality of life in early and late stages of idiopathic Parkinson’s disease. Psychiatry Investig 16(4):285–291
Giannini G, Mastrangelo V, Provini F, Droghini A, Cecere A, Barletta G, Mignani F, Guaraldi P, Cortelli P, Calandra-Buonaura G (2020) Progression and prognosis in multiple system atrophy presenting with REM behavior disorder. Neurology 94(17):e1828–e1834
Giannoccaro MP, Avoni P, Rizzo G, Incensi A, Infante R, Donadio V, Liguori R (2022) Presence of skin α-Synuclein deposits discriminates Parkinson’s disease from progressive supranuclear palsy and corticobasal syndrome. J Parkinsons Dis 12(2):585–591
Goedert M, Spillantini MG, Del Tredici K, Braak H (2013) 100 years of Lewy pathology. Nat Rev Neurol 9(1):13–24
Greten S, Müller-Funogea JI, Wegner F, Höglinger GU, Simon N, Junius-Walker U, Gerbel S, Krause O, Klietz M (2021) Drug safety profiles in geriatric patients with Parkinson’s disease using the FORTA (Fit fOR The Aged) classification: results from a mono-centric retrospective analysis. J Neural Trans (vienna, Austria: 1996) 128(1):49–60
Grimm M-J, Respondek G, Stamelou M, Arzberger T, Ferguson L, Gelpi E, Giese A, Grossman M, Irwin DJ, Pantelyat A, Rajput A, Roeber S, van Swieten JC, Troakes C, Antonini A, Bhatia KP, Colosimo C, van Eimeren T, Kassubek J, Levin J, Meissner WG, Nilsson C, Oertel WH, Piot I, Poewe W, Wenning GK, Boxer A, Golbe LI, Josephs KA, Litvan I, Morris HR, Whitwell JL, Compta Y, Corvol J-C, Lang AE, Rowe JB, Höglinger GU (2019) How to apply the movement disorder society criteria for diagnosis of progressive supranuclear palsy. Movement Disord 34(8):1228–1232
Grimm M-J, Respondek G, Stamelou M, Arzberger T, Ferguson L, Gelpi E, Giese A, Grossman M, Irwin DJ, Pantelyat A, Rajput A, Roeber S, van Swieten JC, Troakes C, Meissner WG, Nilsson C, Piot I, Compta Y, Rowe JB, Höglinger GU (2020) Clinical conditions “suggestive of progressive supranuclear palsy”-diagnostic performance. Movement Disord 35(12):2301–2313
Grötsch M-T, Respondek G, Colosimo C, Compta Y, Corvol JC, Ferreira J, Huber MK, Klietz M, Krey LFM, Levin J, Jecmenica-Lukic M, Macías-García D, Meissner WG, Mir P, Morris H, Nilsson C, Rowe JB, Seppi K, Stamelou M, van Swieten JC, Wenning G, Del Ser T, Golbe LI, Höglinger GU (2021) A modified progressive supranuclear palsy rating scale. Movement Disord 36(5):1203–1215
Grün D, Pieri V, Vaillant M, Diederich NJ (2016) Contributory factors to caregiver burden in Parkinson disease. J Am Med Dir Assoc 17(7):626–632
Guasp M, Molina-Porcel L, Painous C, Caballol N, Camara A, Perez-Soriano A, Sánchez-Gómez A, Garrido A, Muñoz E, Marti MJ, Valldeoriola F, Grau O, Gelpí E, Respondek G, Höglinger GH, Compta Y (2021) Association of PSP phenotypes with survival: a brain-bank study. Parkinsonism Relat Disord 84:77–81
Heine J, von Eichel H, Staege S, Höglinger GU, Wegner F, Klietz M (2021) Relationship satisfaction in people with Parkinson’s disease and their caregivers: a cross-sectional observational study. Brain Sci 11(6):822
Heinzel S, Berg D, Gasser T, Chen H, Yao C, Postuma RB (2019) Update of the MDS research criteria for prodromal Parkinson’s disease. Movement Disord 34(10):1464–1470
Hellqvist C, Berterö C, Dizdar N, Sund-Levander M, Hagell P (2020) Self-management education for persons with parkinson’s disease and their care partners: a quasi-experimental case-control study in clinical practice. Parkinson’s Dis 2020:6920943
Hellqvist C, Dizdar N, Hagell P, Berterö C, Sund-Levander M (2018) Improving self-management for persons with Parkinson’s disease through education focusing on management of daily life: patients’ and relatives’ experience of the Swedish National Parkinson School. J Clin Nurs 27(19–20):3719–3728
Heras-Garvin A, Weckbecker D, Ryazanov S, Leonov A, Griesinger C, Giese A, Wenning GK, Stefanova N (2019) Anle138b modulates α-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Movement Disord 34(2):255–263
Hermann P, Appleby B, Brandel J-P, Caughey B, Collins S, Geschwind MD, Green A, Haïk S, Kovacs GG, Ladogana A, Llorens F, Mead S, Nishida N, Pal S, Parchi P, Pocchiari M, Satoh K, Zanusso G, Zerr I (2021) Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol 20(3):235–246
Höglinger GU, Litvan I, Mendonca N, Wang D, Zheng H, Rendenbach-Mueller B, Lon H-K, Jin Z, Fisseha N, Budur K, Gold M, Ryman D, Florian H, Ahmed A, Aiba I, Albanese A, Bertram K, Bordelon Y, Bower J, Brosch J, Claassen D, Colosimo C, Corvol J-C, Cudia P, Daniele A, Defebvre L, Driver-Dunckley E, Duquette A, Eleopra R, Eusebio A, Fung V, Geldmacher D, Golbe L, Grandas F, Hall D, Hatano T, Honig L, Hui J, Kerwin D, Kikuchi A, Kimber T, Kimura T, Kumar R, Ljubenkov P, Lorenzl S, Ludolph A, Mari Z, McFarland N, Meissner W, Mir Rivera P, Mochizuki H, Morgan J, Munhoz R, Nishikawa N, O’Sullivan J, Oeda T, Oizumi H, Onodera O, Ory-Magne F, Peckham E, Postuma R, Quattrone A, Quinn J, Ruggieri S, Sarna J, Schulz PE, Slevin J, Tagliati M, Wile D, Wszolek Z, Xie T, Zesiewicz T (2021) Safety and efficacy of tilavonemab in progressive supranuclear palsy: a phase 2, randomised, placebo-controlled trial. Lancet Neurol 20(3):182–192
Höglinger GU, Melhem NM, Dickson DW, Sleiman PMA, Wang L-S, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, Singleton AB, Farrer MJ, Yu C-E, Golbe LI, Revesz T, Hardy J, Lees AJ, Devlin B, Hakonarson H, Müller U, Schellenberg GD (2011) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43(7):699–705
Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, Mollenhauer B, Müller U, Nilsson C, Whitwell JL, Arzberger T, Englund E, Gelpi E, Giese A, Irwin DJ, Meissner WG, Pantelyat A, Rajput A, van Swieten JC, Troakes C, Antonini A, Bhatia KP, Bordelon Y, Compta Y, Corvol J-C, Colosimo C, Dickson DW, Dodel R, Ferguson L, Grossman M, Kassubek J, Krismer F, Levin J, Lorenzl S, Morris HR, Nestor P, Oertel WH, Poewe W, Rabinovici G, Rowe JB, Schellenberg GD, Seppi K, van Eimeren T, Wenning GK, Boxer AL, Golbe LI, Litvan I (2017) Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Movement Disord 32(6):853–864
Höllerhage M, Fussi N, Rösler TW, Wurst W, Behrends C, Höglinger GU (2019) Multiple molecular pathways stimulating macroautophagy protect from alpha-synuclein-induced toxicity in human neurons. Neuropharmacology 149:13–26
Höllerhage M, Goebel JN, de Andrade A, Hildebrandt T, Dolga A, Culmsee C, Oertel WH, Hengerer B, Höglinger GU (2014) Trifluoperazine rescues human dopaminergic cells from wild-type α-synuclein-induced toxicity. Neurobiol Aging 35(7):1700–1711
Höllerhage M, Moebius C, Melms J, Chiu W-H, Goebel JN, Chakroun T, Koeglsperger T, Oertel WH, Rösler TW, Bickle M, Höglinger GU (2017) Protective efficacy of phosphodiesterase-1 inhibition against alpha-synuclein toxicity revealed by compound screening in LUHMES cells. Sci Rep 7(1):11469
Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci USA 110(33):E3138–E3147
Hommel ALAJ, Meinders MJ, Lorenzl S, Dodel R, Coelho M, Ferreira JJ, Laurens B, Spampinato U, Meissner W, Rosqvist K, Timpka J, Odin P, Wittenberg M, Bloem PhD BR, Koopmans RT, Schrag A (2020) The prevalence and determinants of neuropsychiatric symptoms in late-stage Parkinsonism. Movement Disord Clin Pract 7(5):531–542
Hopfner F, Tietz AK, Ruf V, Ross OA, Koga S, Dickson D, Aguzzi A, Attems J, Beach T, Beller A, van Deerlin V, Desplats P, Deuschl G, Duyckaerts C, Ellinghaus D, Evsyukov V, Flanagan ME, Franke A, Frosch MP, Gearing M, Gelpi E, Ghetti B, Glass JD, Grinberg LT, Halliday G, Helbig I, Höllerhage M, Huitinga I, Irwin DJ, Keene DC, Kovacs GG, Lee EB, Levin J, Martí MJ, Mackenzie I, McKeith I, McLean C, Mollenhauer B, Neumann M, Newell KL, Pantelyat A, Pendziwiat M, Peters A, Porcel LM, Rabano A, Matěj R, Rajput A, Rajput A, Reimann R, Scott WK, Seeley W, Selvackadunco S, Simuni T, Stadelmann C, Svenningsson P, Thomas A, Trenkwalder C, Troakes C, Trojanowski JQ, White CL, Xie T, Ximelis T, Yebenes J, Müller U, Schellenberg GD, Herms J, Kuhlenbäumer G, Höglinger G (2022) Common variants near ZIC1 and ZIC4 in autopsy-confirmed Multiple System Atrophy. Movement Disord (in press)
Iranzo A, Fairfoul G, Ayudhaya ACN, Serradell M, Gelpi E, Vilaseca I, Sanchez-Valle R, Gaig C, Santamaria J, Tolosa E, Riha RL, Green AJE (2021) Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol 20(3):203–212
Jabbari E, Duff KE (2021) Tau-targeting antibody therapies: too late, wrong epitope or wrong target? Nat Med 27(8):1341–1342
Jabbari E, Zetterberg H, Morris HR (2017) Tracking and predicting disease progression in progressive supranuclear palsy: CSF and blood biomarkers. J Neurol Neurosurg Psychiatry 88(10):883–888
Jan A, Gonçalves NP, Vaegter CB, Jensen PH, Ferreira N (2021) The prion-like spreading of alpha-synuclein in Parkinson’s disease: update on models and hypotheses. Int J Mol Sci 22(15):8338
Jankovic J, McDermott M, Carter J, Gauthier S, Goetz C, Golbe L, Huber S, Koller W, Olanow C, Shoulson I (1990) Variable expression of Parkinson’s disease: a base-line analysis of the DATATOP cohort. The Parkinson Study Group. Neurology 40(10):1529–1534
Jensen I, Bretschneider A, Stiel S, Wegner F, Höglinger GU, Klietz M (2022) Analysis of Parkinson’s disease outpatient counselling for advance directive creation: a cross-sectional questionnaire-based survey of german general practitioners and neurologists. Brain Sci 12:749
Jensen I, Lescher E, Stiel S, Wegner F, Höglinger G, Klietz M (2021) Analysis of transition of patients with Parkinson’s disease into institutional care: a retrospective pilot study. Brain Sci 11(11):1470
Jiang C, Hopfner F, Berg D, Hu MT, Pilotto A, Borroni B, Davis JJ, Tofaris GK (2021) Validation of α-Synuclein in L1CAM-immunocaptured exosomes as a biomarker for the stratification of Parkinsonian syndromes. Movement Disord 36(11):2663–2669
Jiang C, Hopfner F, Katsikoudi A, Hein R, Catli C, Evetts S, Huang Y, Wang H, Ryder JW, Kuhlenbaeumer G, Deuschl G, Padovani A, Berg D, Borroni B, Hu MT, Davis JJ, Tofaris GK (2020) Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J Neurol Neurosurg Psychiatry 91(7):720–729
Johnston HE, Samant RS (2021) Alternative systems for misfolded protein clearance: life beyond the proteasome. FEBS J 288(15):4464–4487
Jucker M, Walker LC (2018) Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21(10):1341–1349
Kang J-H, Irwin DJ, Chen-Plotkin AS, Siderowf A, Caspell C, Coffey CS, Waligórska T, Taylor P, Pan S, Frasier M, Marek K, Kieburtz K, Jennings D, Simuni T, Tanner CM, Singleton A, Toga AW, Chowdhury S, Mollenhauer B, Trojanowski JQ, Shaw LM (2013) Association of cerebrospinal fluid β-amyloid 1–42, T-tau, P-tau181, and α-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA Neurol 70(10):1277–1287
Klietz M, Berndt JM, Wegner F, Schneider N, Höglinger GU, Eggers C, Stiel S (2020a) Consensus-based recommendations for advance directives of people with Parkinson’s Disease in regard to typical complications by German movement disorder specialists. J Clin Med 9(2):449
Klietz M, Bronzlik P, Nösel P, Wegner F, Dressler DW, Dadak M, Maudsley AA, Sheriff S, Lanfermann H, Ding X-Q (2019a) Altered neurometabolic profile in early Parkinson’s disease: a study with short echo-time whole brain MR spectroscopic imaging. Front Neurol 10:777
Klietz M, Eichel HV, Staege S, Kutschenko A, Respondek G, Huber MK, Greten S, Höglinger GU, Wegner F (2021a) Validation of the Parkinson’s disease caregiver burden questionnaire in progressive supranuclear palsy. Parkinson’s Dis 2021:9990679
Klietz M, von Eichel H, Schnur T, Staege S, Höglinger GU, Wegner F, Stiel S (2021b) One year trajectory of caregiver burden in Parkinson’s disease and analysis of gender-specific aspects. Brain Sci 11(3):295
Klietz M, Elaman MH, Mahmoudi N, Nösel P, Ahlswede M, Wegner F, Höglinger GU, Lanfermann H, Ding X-Q (2021c) Cerebral microstructural alterations in patients with early Parkinson’s disease detected with quantitative magnetic resonance measurements. Front Aging Neurosci 13:763331
Klietz M, Greten S, Wegner F, Höglinger GU (2019b) Safety and tolerability of pharmacotherapies for Parkinson’s disease in geriatric patients. Drugs Aging 36(6):511–530
Klietz M, Öcalan Ö, Schneider N, Dressler D, Stiel S, Wegner F (2019c) Advance directives of german people with Parkinson’s disease are unspecific in regard to typical complications. Parkinson’s Dis 2019:2107821
Klietz M, Rippena L, Lange F, Tulke A, Paracka L, Dressler D, Wegner F (2019d) Validating the Parkinson’s disease caregiver burden questionnaire (PDCB) in German caregivers of advanced Parkinson’s disease patients. Int Psychogeriatr 31(12):1791–1800
Klietz M, Schnur T, Drexel S, Lange F, Tulke A, Rippena L, Paracka L, Dressler D, Höglinger GU, Wegner F (2020b) Association of motor and cognitive symptoms with health-related quality of life and caregiver burden in a German cohort of advanced Parkinson’s disease patients. Parkinson’s Dis 2020:5184084
Klietz M, Schnur T, Drexel SC, Lange F, Paracka L, Huber MK, Dressler D, Höglinger GU, Wegner F (2020c) Alexithymia is associated with reduced quality of life and increased caregiver burden in Parkinson’s disease. Brain Sci 10(6):401
Klietz M, Tulke A, Müschen LH, Paracka L, Schrader C, Dressler DW, Wegner F (2018) Impaired quality of life and need for palliative care in a German cohort of advanced Parkinson’s disease patients. Front Neurol 9:120
Koga S, Dickson DW (2018) Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy. J Neurol Neurosurg Psychiatry 89(2):175–184
Koga S, Sekiya H, Kondru N, Ross OA, Dickson DW (2021) Neuropathology and molecular diagnosis of Synucleinopathies. Mol Neurodegener 16(1):83
Korat Š, Bidesi NSR, Bonanno F, Di Nanni A, Hoàng ANN, Herfert K, Maurer A, Battisti UM, Bowden GD, Thonon D, Vugts D, Windhorst AD, Herth MM (2021) Alpha-synuclein PET tracer development-an overview about current efforts. Pharmaceuticals (basel, Switzerland) 14(9):847
Kovacs GG, Lukic MJ, Irwin DJ, Arzberger T, Respondek G, Lee EB, Coughlin D, Giese A, Grossman M, Kurz C, McMillan CT, Gelpi E, Compta Y, van Swieten JC, Laat LD, Troakes C, Al-Sarraj S, Robinson JL, Roeber S, Xie SX, Lee VM-Y, Trojanowski JQ, Höglinger GU (2020) Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol 140(2):99–119
Krey L, Lange P, Tran AT, Greten S, Höglinger GU, Wegner F, Krause O, Klietz M (2021) Patient safety in a box: implementation and evaluation of the emergency box in geriatric and Parkinson patients. J Clin Med 10(23):5618
Kwasny MJ, Oleske DM, Zamudio J, Diegidio R, Höglinger GU (2021) Clinical features observed in general practice associated with the subsequent diagnosis of progressive supranuclear palsy. Front Neurol 12:637176
Kwon EH, Tennagels S, Gold R, Gerwert K, Beyer L, Tönges L (2022) Update on CSF biomarkers in Parkinson’s disease. Biomolecules 12(2):329
Lemos M, Venezia S, Refolo V, Heras-Garvin A, Schmidhuber S, Giese A, Leonov A, Ryazanov S, Griesinger C, Galabova G, Staffler G, Wenning GK, Stefanova N (2020) Targeting α-synuclein by PD03 AFFITOPE® and Anle138b rescues neurodegenerative pathology in a model of multiple system atrophy: clinical relevance. Transl Neurodegeneration 9(1):38
Levin J, Kurz A, Arzberger T, Giese A, Höglinger GU (2016a) The Differential diagnosis and treatment of atypical Parkinsonism. Deutsches Arzteblatt Int 113(5):61–69
Levin J, Maaß S, Schuberth M, Giese A, Oertel WH, Poewe W, Trenkwalder C, Wenning GK, Mansmann U, Südmeyer M, Eggert K, Mollenhauer B, Lipp A, Löhle M, Classen J, Münchau A, Kassubek J, Gandor F, Berg D, Egert-Schwender S, Eberhardt C, Paul F, Bötzel K, Ertl-Wagner B, Huppertz H-J, Ricard I, Höglinger GU, André E, Blankenstein C, Canelo M, Düring M, Ebentheuer J, Fricke C, Gerbes A, Groiss S, Gruber D, Hartmann C, Kirchner T, Kroneberg D, Kunz M, Lorenzl S, Moldovan A, Noda A, Pape H, Respondek G, Schäffer E, Schneider M, Schnitzler A, Schulz-Schaeffer W, Schwarz J, Skowronek C, Storch A, Tadic V, Vadász D, Zimmermann B (2019) Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): a randomised, double-blind, placebo-controlled trial. Lancet Neurol 18(8):724–735
Levin J, Maaß S, Schuberth M, Respondek G, Paul F, Mansmann U, Oertel WH, Lorenzl S, Krismer F, Seppi K, Poewe W, Wenning G, Giese A, Bötzel K, Höglinger G (2016b) The PROMESA-protocol: progression rate of multiple system atrophy under EGCG supplementation as anti-aggregation-approach. J Neural Transmission (vienna, Austria: 1996) 123(4):439–445
Levin J, Schmidt F, Boehm C, Prix C, Bötzel K, Ryazanov S, Leonov A, Griesinger C, Giese A (2014) The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol 127(5):779–780
Lingor P, Csoti I, Koschel J, Schrader C, Winkler C, Wolz M, Reichmann H (2016) Der geriatrische Parkinson-Patient - eine neurologische Herausforderung (The Geriatric Patient with Parkinson’s Disease - a Neurological Challenge). Fortschr Neurol Psychiatr 84(Suppl 1):S41–S47
Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, Goetz CG, Golbe LI, Grafman J, Growdon JH, Hallett M, Jankovic J, Quinn NP, Tolosa E, Zee DS (1996) Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 47(1):1–9
Lopez G, Bayulkem K, Hallett M (2016) Progressive supranuclear palsy (PSP): Richardson syndrome and other PSP variants. Acta Neurol Scand 134(4):242–249
Low PA, Reich SG, Jankovic J, Shults CW, Stern MB, Novak P, Tanner CM, Gilman S, Marshall FJ, Wooten F, Racette B, Chelimsky T, Singer W, Sletten DM, Sandroni P, Mandrekar J (2015) Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol 14(7):710–719
Lum HD, Jordan SR, Brungardt A, Ayele R, Katz M, Miyasaki JM, Hall A, Jones J, Kluger B (2019) Framing advance care planning in Parkinson disease: patient and care partner perspectives. Neurology 92(22):e2571–e2579
Macleod AD, Taylor KSM, Counsell CE (2014) Mortality in Parkinson’s disease: a systematic review and meta-analysis. Movement Disord 29(13):1615–1622
Magistrelli L, Contaldi E, Comi C (2021) The Impact of SNCA variations and its product alpha-synuclein on non-motor features of Parkinson’s disease. Life (Basel, Switzerland) 11(8):804
Mahlknecht P, Hotter A, Hussl A, Esterhammer R, Schocke M, Seppi K (2010) Significance of MRI in diagnosis and differential diagnosis of Parkinson’s disease. Neurodegener Dis 7(5):300–318
Mao X, Ou MT, Karuppagounder SS, Kam T-I, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin J-H, Kang HC, Zhang J, Xu J, Chen R, Park H, Andrabi SA, Kang SU, Gonçalves RA, Liang Y, Zhang S, Qi C, Lam S, Keiler JA, Tyson J, Kim D, Panicker N, Yun SP, Workman CJ, Vignali DAA, Dawson VL, Ko HS, Dawson TM (2016) Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science (new York, NY) 353(6307):3374
Marras C, Chaudhuri KR, Titova N, Mestre TA (2020) Therapy of Parkinson’s disease subtypes. Neurotherapeutics 17(4):1366–1377
Martinez-Martin P, Macaulay D, Jalundhwala YJ, Mu F, Ohashi E, Marshall T, Sail K (2019) The long-term direct and indirect economic burden among Parkinson’s disease caregivers in the United States. Movement Disord 34(2):236–245
Martinez-Martin P, Rodriguez-Blazquez C, Forjaz MJ, Frades-Payo B, Agüera-Ortiz L, Weintraub D, Riesco A, Kurtis MM, Chaudhuri KR (2015) Neuropsychiatric symptoms and caregiver’s burden in Parkinson’s disease. Parkinsonism Relat Disord 21(6):629–634
Martinez-Valbuena I, Visanji NP, Olszewska DA, Sousa M, Bhakta P, Vasilevskaya A, Anastassiadis C, Tartaglia MC, Kovacs GG, Lang AE (2022) Combining Skin α-synuclein real-time quaking-induced conversion and circulating neurofilament light chain to distinguish multiple system atrophy and Parkinson’s disease. Mov Disord 37(3):648–650
Meinders MJ, Gentile G, Schrag AE, Konitsiotis S, Eggers C, Taba P, Lorenzl S, Odin P, Rosqvist K, Chaudhuri KR, Antonini A, Bloem BR, Groot MM (2021) Advance care planning and care coordination for people with Parkinson’s disease and their family caregivers-study protocol for a multicentre, randomized controlled trial. Front Neurol 12:673893
Mestre TA, Fereshtehnejad S-M, Berg D, Bohnen NI, Dujardin K, Erro R, Espay AJ, Halliday G, van Hilten JJ, Hu MT, Jeon B, Klein C, Leentjens AFG, Marinus J, Mollenhauer B, Postuma R, Rajalingam R, Rodríguez-Violante M, Simuni T, Surmeier DJ, Weintraub D, McDermott MP, Lawton M, Marras C (2021) Parkinson’s disease subtypes: critical appraisal and recommendations. J Parkinsons Dis 11(2):395–404
Miglis MG, Adler CH, Antelmi E, Arnaldi D, Baldelli L, Boeve BF, Cesari M, Dall’Antonia I, Diederich NJ, Doppler K, Dušek P, Ferri R, Gagnon J-F, Gan-Or Z, Hermann W, Högl B, Hu MT, Iranzo A, Janzen A, Kuzkina A, Lee J-Y, Leenders KL, Lewis SJG, Liguori C, Liu J, Lo C, Ehgoetz Martens KA, Nepozitek J, Plazzi G, Provini F, Puligheddu M, Rolinski M, Rusz J, Stefani A, Summers RLS, Yoo D, Zitser J, Oertel WH (2021) Biomarkers of conversion to α-synucleinopathy in isolated rapid-eye-movement sleep behaviour disorder. Lancet Neurol 20(8):671–684
Miyasaki JM (2013) Palliative care in Parkinson’s disease. Curr Neurol Neurosci Rep 13(8):367
Miyasaki JM, Kluger B (2015) Palliative care for Parkinson’s disease: has the time come? Curr Neurol Neurosci Rep 15(5):26
Miyasaki JM, Lim S-Y, Chaudhuri KR, Antonini A, Piemonte M, Richfield E, Alburquerque Gonzalez D, Lorenzl S, Walker R, Bhidayasiri R, Bouca R, McConvey V (2022) Access and attitudes toward palliative care among movement disorders clinicians. Movement Disord 37(1):182–189
Mosley PE, Moodie R, Dissanayaka N (2017) Caregiver burden in Parkinson disease: a critical review of recent literature. J Geriatr Psychiatry Neurol 30(5):235–252
Moßhammer D, Haumann H, Mörike K, Joos S (2016) Polypharmacy-an upward trend with unpredictable effects. Deutsches Arzteblatt Int 113(38):627–633
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, Bras J, Young E, von Coelln R, Simón-Sánchez J, Schulte C, Sharma M, Krohn L, Pihlstrøm L, Siitonen A, Iwaki H, Leonard H, Faghri F, Gibbs JR, Hernandez DG, Scholz SW, Botia JA, Martinez M, Corvol J-C, Lesage S, Jankovic J, Shulman LM, Sutherland M, Tienari P, Majamaa K, Toft M, Andreassen OA, Bangale T, Brice A, Yang J, Gan-Or Z, Gasser T, Heutink P, Shulman JM, Wood NW, Hinds DA, Hardy JA, Morris HR, Gratten J, Visscher PM, Graham RR, Singleton AB, Adarmes-Gómez AD, Aguilar M, Aitkulova A, Akhmetzhanov V, Alcalay RN, Alvarez I, Alvarez V, Barrero FJ, Bergareche Yarza JA, Bernal-Bernal I, Billingsley K, Blazquez M, Bonilla-Toribio M, Botía JA, Boungiorno MT, Brockmann K, Bubb V, Buiza-Rueda D, Cámara A, Carrillo F, Carrión-Claro M, Cerdan D, Chelban V, Clarimón J, Clarke C, Compta Y, Cookson MR, Craig DW, Danjou F, Diez-Fairen M, Dols-Icardo O, Duarte J, Duran R, Escamilla-Sevilla F, Escott-Price V, Ezquerra M, Feliz C, Fernández M, Fernández-Santiago R, Finkbeiner S, Foltynie T, Garcia C, García-Ruiz P, Gomez Heredia MJ, Gómez-Garre P, González MM, Gonzalez-Aramburu I, Guelfi S, Guerreiro R, Hardy J, Hassin-Baer S, Hoenicka J, Holmans P, Houlden H, Infante J, Jesús S, Jimenez-Escrig A, Kaishybayeva G, Kaiyrzhanov R, Karimova A, Kinghorn KJ, Koks S, Kulisevsky J, Labrador-Espinosa MA, Leonard HL, Lewis P, Lopez-Sendon JL, Lovering R, Lubbe S, Lungu C, Macias D, Manzoni C, Marín J, Marinus J, Marti MJ, Martínez Torres I, Martínez-Castrillo JC, Mata M, Mencacci NE, Méndez-del-Barrio C, Middlehurst B, Mínguez A, Mir P, Mok KY, Muñoz E, Narendra D, Ojo OO, Okubadejo NU, Pagola AG, Pastor P, Perez Errazquin F, Periñán-Tocino T, Pihlstrom L, Plun-Favreau H, Quinn J, R’Bibo L, Reed X, Rezola EM, Rizig M, Rizzu P, Robak L, Rodriguez AS, Rouleau GA, Ruiz-Martínez J, Ruz C, Ryten M, Sadykova D, Schreglmann S, Shashkin C, Sierra M, Suarez-Sanmartin E, Taba P, Tabernero C, Tan MX, Tartari JP, Tejera-Parrado C, Tolosa E, Trabzuni D, Valldeoriola F, van Hilten JJ, van Keuren-Jensen K, Vargas-González L, Vela L, Vives F, Williams N, Zharkinbekova N, Zharmukhanov Z, Zholdybayeva E, Zimprich A, Ylikotila P, Reich S, Savitt J, Agee M, Alipanahi B, Auton A, Bell RK, Bryc K, Elson SL, Fontanillas P, Furlotte NA, Huber KE, Hicks B, Jewett EM, Jiang Y, Kleinman A, Lin K-H, Litterman NK, McCreight JC, McIntyre MH, McManus KF, Mountain JL, Noblin ES, Northover CA, Pitts SJ, Poznik GD, Sathirapongsasuti JF, Shelton JF, Shringarpure S, Tian C, Tung J, Vacic V, Wang X, Wilson CH, Anderson T, Bentley S, Dalrymple-Alford J, Fowdar J, Halliday G, Henders AK, Hickie I, Kassam I, Kennedy M, Kwok J, Lewis S, Mellick G, Montgomery G, Pearson J, Pitcher T, Sidorenko J, Silburn PA, Wallace L, Wray NR, Zhang F (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18(12):1091–1102
Nichols MR, St-Pierre M-K, Wendeln A-C, Makoni NJ, Gouwens LK, Garrad EC, Sohrabi M, Neher JJ, Tremblay M-E, Combs CK (2019) Inflammatory mechanisms in neurodegeneration. J Neurochem 149(5):562–581
Niewiadomska G, Niewiadomski W, Steczkowska M, Gasiorowska A (2021) Tau oligomers neurotoxicity. Life (basel, Switzerland) 11(1):28
Nuebling GS, Butzhammer E, Lorenzl S (2021) Assisted suicide in Parkinsonian disorders. Front Neurol 12:656599
Okunoye O, Kojima G, Marston L, Walters K, Schrag A (2020) Factors associated with hospitalisation among people with Parkinson’s disease - a systematic review and meta-analysis. Parkinsonism Relat Disord 71:66–72
Oliver D, Borasio GD, Veronese S, Voltz R, Lorenzl S, Hepgul N (2020) Current collaboration between palliative care and neurology: a survey of clinicians in Europe. BMJ Support Palliative Care. https://doi.org/10.1136/bmjspcare-2020-002322
Oliver DJ, Borasio GD, Caraceni A, de Visser M, Grisold W, Lorenzl S, Veronese S, Voltz R (2016) A consensus review on the development of palliative care for patients with chronic and progressive neurological disease. Eur J Neurol 23(1):30–38
Osaki Y, Ben-Shlomo Y, Lees AJ, Wenning GK, Quinn NP (2009) A validation exercise on the new consensus criteria for multiple system atrophy. Movement Disord 24(15):2272–2276
Owens E, Josephs KA, Savica R, Hassan A, Klassen B, Bower J, Maraganore D, Matsumoto J, Ahlskog JE (2016) The clinical spectrum and natural history of pure akinesia with gait freezing. J Neurol 263(12):2419–2423
de Pablo-Fernández E, Lees AJ, Holton JL, Warner TT (2019) Prognosis and neuropathologic correlation of clinical subtypes of Parkinson disease. JAMA Neurol 76(4):470–479
Pagano G, Boess FG, Taylor KI, Ricci B, Mollenhauer B, Poewe W, Boulay A, Anzures-Cabrera J, Vogt A, Marchesi M, Post A, Nikolcheva T, Kinney GG, Zago WM, Ness DK, Svoboda H, Britschgi M, Ostrowitzki S, Simuni T, Marek K, Koller M, Sevigny J, Doody R, Fontoura P, Umbricht D, Bonni A (2021) A phase II study to evaluate the safety and efficacy of prasinezumab in early Parkinson’s Disease (PASADENA): rationale, design, and baseline data. Front Neurol 12:705407
Pajares M, Rojo I, A, Manda G, Boscá L, Cuadrado A, (2020) Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells 9(7):1687
Palleis C, Brendel M, Finze A, Weidinger E, Bötzel K, Danek A, Beyer L, Nitschmann A, Kern M, Biechele G, Rauchmann B-S, Häckert J, Höllerhage M, Stephens AW, Drzezga A, van Eimeren T, Villemagne VL, Schildan A, Barthel H, Patt M, Sabri O, Bartenstein P, Perneczky R, Haass C, Levin J, Höglinger GU (2021) Cortical 18 FPI-2620 binding differentiates corticobasal syndrome subtypes. Movement Disord 36(9):2104–2115
Palleis C, Morenas-Rodriguez E, Murcia FJM, Giese A, Nuscher B, Haass C, Höglinger G, Bötzel K, Levin J (2020) Longitudinal correlation between neurofilament light chain and UMSARS in multiple system atrophy. Clin Neurol Neurosurg 195:105924
Papapetropoulos S, Singer C, McCorquodale D, Gonzalez J, Mash DC (2005) Cause, seasonality of death and co-morbidities in progressive supranuclear palsy (PSP). Parkinsonism Relat Disord 11(7):459–463
Parkinson J (1817; 2002) An essay on the shaking palsy. 1817. J Neuropsychiatr Clin Neurosci 14(2):223–36; discussion 222
Peralta C, Strafella AP, van Eimeren T, Ceravolo R, Seppi K, Kaasinen V, Arena JE, Lehericy S (2022) Pragmatic approach on neuroimaging techniques for the differential diagnosis of Parkinsonisms. Movement Disord Clin Pract 9(1):6–19
Pereira MF, Buchanan T, Höglinger GU, Bogdanovic M, Tofaris G, Prangnell S, Sarangmat N, FitzGerald JJ, Antoniades CA (2022) Longitudinal changes of early motor and cognitive symptoms in progressive supranuclear palsy: the OxQUIP study. BMJ Neurol Open 4(1):e000214
Pérez-Soriano A, Giraldo DM, Ríos J, Muñoz E, Compta Y, Martí MJ (2021) Progression of motor and non-motor symptoms in multiple system atrophy: a prospective study from the Catalan-MSA registry. J Parkinsons Dis 11(2):685–694
Pilotto A, Imarisio A, Conforti F, Scalvini A, Masciocchi S, Nocivelli S, Turrone R, Gipponi S, Cottini E, Borroni B, Rizzetti MC, Pizzi M, Bonanni L, Sturchio A, Espay AJ, Zetterberg H, Ashton NJ, Hye A, Padovani A (2021) Plasma NfL, clinical subtypes and motor progression in Parkinson’s disease. Parkinsonism Relat Disord 87:41–47
Piot I, Schweyer K, Respondek G, Stamelou M, Sckopke P, Schenk T, Goetz CG, Stebbins GT, Höglinger GU (2020) The progressive supranuclear palsy clinical deficits scale. Movement Disord 35(4):650–661
Poggiolini I, Gupta V, Lawton M, Lee S, El-Turabi A, Querejeta-Coma A, Trenkwalder C, Sixel-Döring F, Foubert-Samier A, Le Traon AP, Plazzi G, Biscarini F, Montplaisir J, Gagnon J-F, Postuma RB, Antelmi E, Meissner WG, Mollenhauer B, Ben-Shlomo Y, Hu MT, Parkkinen L (2021) Diagnostic value of cerebrospinal fluid alpha-synuclein seed quantification in synucleinopathies. Brain 145(2):584–595
Postuma RB, Berg D (2016) Advances in markers of prodromal Parkinson disease. Nat Rev Neurol 12(11):622–634
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, Obeso J, Marek K, Litvan I, Lang AE, Halliday G, Goetz CG, Gasser T, Dubois B, Chan P, Bloem BR, Adler CH, Deuschl G (2015) MDS clinical diagnostic criteria for Parkinson’s disease. Movement Disord 30(12):1591–1601
Postuma RB, Iranzo A, Hu M, Högl B, Boeve BF, Manni R, Oertel WH, Arnulf I, Ferini-Strambi L, Puligheddu M, Antelmi E, Cochen De Cock V, Arnaldi D, Mollenhauer B, Videnovic A, Sonka K, Jung K-Y, Kunz D, Dauvilliers Y, Provini F, Lewis SJ, Buskova J, Pavlova M, Heidbreder A, Montplaisir JY, Santamaria J, Barber TR, Stefani A, St Louis EK, Terzaghi M, Janzen A, Leu-Semenescu S, Plazzi G, Nobili F, Sixel-Doering F, Dusek P, Bes F, Cortelli P, Ehgoetz Martens K, Gagnon J-F, Gaig C, Zucconi M, Trenkwalder C, Gan-Or Z, Lo C, Rolinski M, Mahlknecht P, Holzknecht E, Boeve AR, Teigen LN, Toscano G, Mayer G, Morbelli S, Dawson B, Pelletier A (2019) Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain 142(3):744–759
Pötter-Nerger M, Dutke J, Lezius S, Buhmann C, Schulz R, Gerloff C, Kuhle J, Choe C-U (2022) Serum neurofilament light chain and postural instability/gait difficulty (PIGD) subtypes of Parkinson’s disease in the MARK-PD study. J Neural Transmission (vienna, Austria: 1996) 129(3):295–300
Prell T, Siebecker F, Lorrain M, Eggers C, Lorenzl S, Klucken J, Warnecke T, Buhmann C, Tönges L, Ehret R, Wellach I, Wolz M (2020a) Recommendations for standards of network care for patients with Parkinson’s disease in Germany. J Clin Med 9(5):1455
Prell T, Siebecker F, Lorrain M, Tönges L, Warnecke T, Klucken J, Wellach I, Buhmann C, Wolz M, Lorenzl S, Herbst H, Eggers C, Mai T (2020b) Specialized staff for the care of people with Parkinson’s disease in Germany: an overview. J Clin Med 9(8):2581
Rabadia SV, Litvan I, Juncos J, Bordelon Y, Riley DE, Standaert D, Reich SG, Hall DA, Kluger B, Shprecher D, Marras C, Jankovic J (2019) Hypertension and progressive supranuclear palsy. Parkinsonism Relat Disord 66:166–170
Rajput AH, Rajput ML, Ferguson LW, Rajput A (2017) Baseline motor findings and Parkinson disease prognostic subtypes. Neurology 89(2):138–143
Rajput AH, Sitte HH, Rajput A, Fenton ME, Pifl C, Hornykiewicz O (2008) Globus pallidus dopamine and Parkinson motor subtypes: clinical and brain biochemical correlation. Neurology 70(16 Pt 2):1403–1410
Rajput AH, Voll A, Rajput ML, Robinson CA, Rajput A (2009) Course in Parkinson disease subtypes: a 39-year clinicopathologic study. Neurology 73(3):206–212
Rascol O, Fitzer-Attas CJ, Hauser R, Jankovic J, Lang A, Langston JW, Melamed E, Poewe W, Stocchi F, Tolosa E, Eyal E, Weiss YM, Olanow CW (2011) A double-blind, delayed-start trial of rasagiline in Parkinson’s disease (the ADAGIO study): prespecified and post-hoc analyses of the need for additional therapies, changes in UPDRS scores, and non-motor outcomes. Lancet Neurol 10(5):415–423
Rauch JN, Luna G, Guzman E, Audouard M, Challis C, Sibih YE, Leshuk C, Hernandez I, Wegmann S, Hyman BT, Gradinaru V, Kampmann M, Kosik KS (2020) LRP1 is a master regulator of tau uptake and spread. Nature 580(7803):381–385
Respondek G, Höglinger GU (2016) The phenotypic spectrum of progressive supranuclear palsy. Parkinsonism Relat Disord 22(Suppl 1):S34–S36
Respondek G, Höglinger GU (2021) DescribePSP and ProPSP: German multicenter networks for standardized prospective collection of clinical data, imaging data, and biomaterials of patients with progressive supranuclear palsy. Front Neurol 12:644064
Respondek G, Kurz C, Arzberger T, Compta Y, Englund E, Ferguson LW, Gelpi E, Giese A, Irwin DJ, Meissner WG, Nilsson C, Pantelyat A, Rajput A, van Swieten JC, Troakes C, Josephs KA, Lang AE, Mollenhauer B, Müller U, Whitwell JL, Antonini A, Bhatia KP, Bordelon Y, Corvol J-C, Colosimo C, Dodel R, Grossman M, Kassubek J, Krismer F, Levin J, Lorenzl S, Morris H, Nestor P, Oertel WH, Rabinovici GD, Rowe JB, van Eimeren T, Wenning GK, Boxer A, Golbe LI, Litvan I, Stamelou M, Höglinger GU (2017) Which ante mortem clinical features predict progressive supranuclear palsy pathology? Movement Disord 32(7):995–1005
Respondek G, Stamelou M, Höglinger GU (2019) Classification of atypical Parkinsonism per pathology versus phenotype. Int Rev Neurobiol 149:37–47
Respondek G, Stamelou M, Kurz C, Ferguson LW, Rajput A, Chiu WZ, van Swieten JC, Troakes C, Al Sarraj S, Gelpi E, Gaig C, Tolosa E, Oertel WH, Giese A, Roeber S, Arzberger T, Wagenpfeil S, Höglinger GU (2014) The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Movement Disord 29(14):1758–1766
Richards M, Marder K, Cote L, Mayeux R (1994) Interrater reliability of the Unified Parkinson’s disease rating scale motor examination. Movement Disord 9(1):89–91
Rojas JC, Bang J, Lobach IV, Tsai RM, Rabinovici GD, Miller BL, Boxer AL (2018) CSF neurofilament light chain and phosphorylated tau 181 predict disease progression in PSP. Neurology 90(4):e273–e281
Rösler TW, Tayaranian Marvian A, Brendel M, Nykänen N-P, Höllerhage M, Schwarz SC, Hopfner F, Koeglsperger T, Respondek G, Schweyer K, Levin J, Villemagne VL, Barthel H, Sabri O, Müller U, Meissner WG, Kovacs GG, Höglinger GU (2019) Four-repeat tauopathies. Prog Neurobiol 180:101644
Rossi M, Candelise N, Baiardi S, Capellari S, Giannini G, Orrù CD, Antelmi E, Mammana A, Hughson AG, Calandra-Buonaura G, Ladogana A, Plazzi G, Cortelli P, Caughey B, Parchi P (2020) Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol 140(1):49–62
Russo MJ, Orru CD, Concha-Marambio L, Giaisi S, Groveman BR, Farris CM, Holguin B, Hughson AG, LaFontant D-E, Caspell-Garcia C, Coffey CS, Mollon J, Hutten SJ, Merchant K, Heym RG, Soto C, Caughey B, Kang UJ (2021) High diagnostic performance of independent alpha-synuclein seed amplification assays for detection of early Parkinson’s disease. Acta Neuropathol Commun 9(1):179
Safarpour D, Thibault DP, DeSanto CL, Boyd CM, Dorsey ER, Racette BA, Willis AW (2015) Nursing home and end-of-life care in Parkinson disease. Neurology 85(5):413–419
Saranza G, Villanueva EQ, Lang AE (2021) Preferences for communication about end-of-life care in atypical Parkinsonism. Movement Disord 36(9):2116–2125
Schrag A, Hommel ALAJ, Lorenzl S, Meissner WG, Odin P, Coelho M, Bloem BR, Dodel R (2020) The late stage of Parkinson’s -results of a large multinational study on motor and non-motor complications. Parkinsonism Relat Disord 75:91–96
Scott IA, Hilmer SN, Reeve E, Potter K, Le Couteur D, Rigby D, Gnjidic D, Del Mar CB, Roughead EE, Page A, Jansen J, Martin JH (2015) Reducing inappropriate polypharmacy: the process of deprescribing. JAMA Intern Med 175(5):827–834
Seeber AA, Pols AJ, Hijdra A, Grupstra HF, Willems DL, de Visser M (2019) Advance care planning in progressive neurological diseases: lessons from ALS. BMC Palliat Care 18(1):50
Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, Hu B, Schmeichel A, Singer W, Wu G, Tsai A-L, Shirani H, Nilsson KPR, Low PA, Soto C (2020) Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 578:273–277
Shi Y, Zhang W, Yang Y, Murzin AG, Falcon B, Kotecha A, van Beers M, Tarutani A, Kametani F, Garringer HJ, Vidal R, Hallinan GI, Lashley T, Saito Y, Murayama S, Yoshida M, Tanaka H, Kakita A, Ikeuchi T, Robinson AC, Mann DMA, Kovacs GG, Revesz T, Ghetti B, Hasegawa M, Goedert M, Scheres SHW (2021) Structure-based classification of tauopathies. Nature 598(7880):359–363
Simonet C, Schrag A, Lees AJ, Noyce AJ (2021) The motor prodromes of parkinson’s disease: from bedside observation to large-scale application. J Neurol 268(6):2099–2108
Singer W, Schmeichel AM, Shahnawaz M, Schmelzer JD, Sletten DM, Gehrking TL, Gehrking JA, Olson AD, Suarez MD, Misra PP, Soto C, Low PA (2021) Alpha-synuclein oligomers and neurofilament light chain predict phenoconversion of pure autonomic failure. Ann Neurol 89(6):1212–1220
Song M, Scheifele M, Barthel H, van Eimeren T, Beyer L, Marek K, Eckenweber F, Palleis C, Kaiser L, Finze A, Kern M, Nitschmann A, Biechele G, Katzdobler S, Bischof G, Hammes J, Jessen F, Saur D, Schroeter ML, Rumpf J-J, Rullmann M, Schildan A, Patt M, Neumaier B, Stephens AW, Rauchmann B-S, Perneczky R, Levin J, Classen J, Höglinger GU, Bartenstein P, Boening G, Ziegler S, Villemagne V, Drzezga A, Seibyl J, Sabri O, Brendel M (2021) Feasibility of short imaging protocols for 18FPI-2620 tau-PET in progressive supranuclear palsy. Eur J Nucl Med Mol Imaging 48(12):3872–3885
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388(6645):839–840
Stewart T, Liu C, Ginghina C, Cain KC, Auinger P, Cholerton B, Shi M, Zhang J (2014) Cerebrospinal fluid α-synuclein predicts cognitive decline in Parkinson disease progression in the DATATOP cohort. Am J Pathol 184(4):966–975
Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, Keutzer J, Mistry PK, Chandra SS (2017) Glucosylsphingosine promotes α-synuclein pathology in mutant GBA-Associated Parkinson’s disease. J Neurosci 37(40):9617–9631
Tennigkeit J, Feige T, Haak M, Hellqvist C, Seven ÜS, Kalbe E, Schwarz J, Warnecke T, Tönges L, Eggers C, Loewenbrück KF (2020) Structured care and self-management education for persons with parkinson’s disease: why the first does not go without the second-systematic review, experiences and implementation concepts from Sweden and Germany. J Clin Med 9(9):2787
Tessitore A, Marano P, Modugno N, Pontieri FE, Tambasco N, Canesi M, Latorre A, Lopiano L, Sensi M, Quatrale R, Solla P, Defazio G, Melzi G, Costanzo AM, Gualberti G, Di Luzio PU, Antonini A (2018) Caregiver burden and its related factors in advanced Parkinson’s disease: data from the PREDICT study. J Neurol 265(5):1124–1137
Trapp S, MacKenzie J, Gonzalez-Arredondo S, Rodriguez-Agudelo Y, Arango-Lasprilla JC (2019) Mediating role of caregiver burden among family caregivers of patients with Parkinson’s disease in Mexico. Int J Psychiatry Med 54(3):203–216
Truong D-JJ, Phlairaharn T, Eßwein B, Gruber C, Tümen D, Baligács E, Armbrust N, Vaccaro FL, Lederer E-M, Beck EM, Geilenkeuser J, Göppert S, Krumwiede L, Grätz C, Raffl G, Schwarz D, Zirngibl M, Živanić M, Beyer M, Körner JD, Santl T, Evsyukov V, Strauß T, Schwarz SC, Höglinger GU, Heutink P, Doll S, Conrad M, Giesert F, Wurst W, Westmeyer GG (2021) Non-invasive and high-throughput interrogation of exon-specific isoform expression. Nat Cell Biol 23(6):652–663
Türker F, Cook EK, Margolis SS (2021) The proteasome and its role in the nervous system. Cell Chem Biol 28(7):903–917
Uemura N, Uemura MT, Luk KC, Lee VM-Y, Trojanowski JQ (2020) Cell-to-cell transmission of tau and α-Synuclein. Trends Mol Med 26(10):936–952
van Rooden SM, Heiser WJ, Kok JN, Verbaan D, van Hilten JJ, Marinus J (2010) The identification of Parkinson’s disease subtypes using cluster analysis: a systematic review. Movement Disord 25(8):969–978
Villemagne VL, Doré V, Burnham SC, Masters CL, Rowe CC (2018) Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat Rev Neurol 14(4):225–236
Viscidi E, Litvan I, Dam T, Juneja M, Li L, Krzywy H, Eaton S, Hall S, Kupferman J, Höglinger GU (2021) Clinical features of patients with progressive supranuclear palsy in an US insurance claims database. Front Neurol 12:571800
Wagner J, Krauss S, Shi S, Ryazanov S, Steffen J, Miklitz C, Leonov A, Kleinknecht A, Göricke B, Weishaupt JH, Weckbecker D, Reiner AM, Zinth W, Levin J, Ehninger D, Remy S, Kretzschmar HA, Griesinger C, Giese A, Fuhrmann M (2015) Reducing tau aggregates with anle138b delays disease progression in a mouse model of tauopathies. Acta Neuropathol 130(5):619–631
Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F, Prix C, Pan-Montojo F, Bertsch U, Mitteregger-Kretzschmar G, Geissen M, Eiden M, Leidel F, Hirschberger T, Deeg AA, Krauth JJ, Zinth W, Tavan P, Pilger J, Zweckstetter M, Frank T, Bähr M, Weishaupt JH, Uhr M, Urlaub H, Teichmann U, Samwer M, Bötzel K, Groschup M, Kretzschmar H, Griesinger C, Giese A (2013) Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol 125(6):795–813
Wenning GK, Stankovic I, Vignatelli L, Fanciulli A, Calandra-Buonaura G, Seppi K, Palma J-A, Meissner WG, Krismer F, Berg D, Cortelli P, Freeman R, Halliday G, Höglinger G, Lang A, Ling H, Litvan I, Low P, Miki Y, Panicker J, Pellecchia MT, Quinn N, Sakakibara R, Stamelou M, Tolosa E, Tsuji S, Warner T, Poewe W, Kaufmann H (2022) The movement disorder society criteria for the diagnosis of multiple system atrophy. Movement Disord. https://doi.org/10.1002/mds.29005
Winslow AR, Rubinsztein DC (2011) The Parkinson disease protein α-synuclein inhibits autophagy. Autophagy 7(4):429–431
Zarit SH, Todd PA, Zarit JM (1986) Subjective burden of husbands and wives as caregivers: a longitudinal study. Gerontologist 26(3):260–266
Zella MAS, Bartig D, Herrmann L, Respondek G, Höglinger G, Gold R, Woitalla D, Krogias C, Tönges L (2020) Hospitalization rates and comorbidities in patients with progressive supranuclear palsy in Germany from 2010 to 2017. J Clin Med 9(8):2454
Zhang H, Iranzo A, Högl B, Arnulf I, Ferini-Strambi L, Manni R, Miyamoto T, Oertel WH, Dauvilliers Y, Ju Y-E, Puligheddu M, Sonka K, Pelletier A, Montplaisir JY, Stefani A, Ibrahim A, Frauscher B, Leu-Semenescu S, Zucconi M, Terzaghi M, Miyamoto M, Janzen A, Figorilli M, Fantini ML, Postuma RB (2022a) Risk factors for phenoconversion in rapid eye movement sleep behavior disorder. Ann Neurol 91(3):404–416
Zhang L, Cao B, Hou Y, Gu X, Wei Q, Ou R, Zhao B, Luo C, Shang H (2022b) Neurofilament light chain predicts disease severity and progression in multiple system atrophy. Movement Disord 37(2):421–426
Zhang S, Liu Y-Q, Jia C, Lim Y-J, Feng G, Xu E, Long H, Kimura Y, Tao Y, Zhao C, Wang C, Liu Z, Hu J-J, Ma M-R, Liu Z, Jiang L, Li D, Wang R, Dawson VL, Dawson TM, Li Y-M, Mao X, Liu C (2021) Mechanistic basis for receptor-mediated pathological α-synuclein fibril cell-to-cell transmission in Parkinson’s disease. Proc Natl Acad Sci USA 118(26):e2011196118
Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, Vidal R, Garringer HJ, Shi Y, Ikeuchi T, Murayama S, Ghetti B, Hasegawa M, Goedert M, Scheres SHW (2020) Novel tau filament fold in corticobasal degeneration. Nature 580(7802):283–287
Zhong M, Evans A, Peppard R, Velakoulis D (2013) Validity and reliability of the PDCB: a tool for the assessment of caregiver burden in Parkinson’s disease. Int Psychogeriatr 25(9):1437–1441
Acknowledgements
We thank all the patients and colleagues stimulating our knowledge and understanding of these diseases in various ways.
Funding
Open Access funding enabled and organized by Projekt DEAL. This article had no specific funding.
Author information
Authors and Affiliations
Contributions
MH and MK wrote the first draft of this manuscript. GUH made substantial contributions to the writing and language editing of the final draft and enhanced the quality of the manuscript by revising it critically for important intellectual content. All authors approved to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to report.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Höllerhage, M., Klietz, M. & Höglinger, G.U. Disease modification in Parkinsonism: obstacles and ways forward. J Neural Transm 129, 1133–1153 (2022). https://doi.org/10.1007/s00702-022-02520-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-022-02520-6