
Abstract
Background
Intravenous hypertonic saline is utilized commonly in critical care for treatment of acute or refractory elevations of intracranial pressure (ICP) in traumatic brain injury (TBI) patients. Though there is a clear understanding of the general physiological effects of a hypertonic saline solution over long periods of time, smaller epoch effects of hypertonic saline (HTS) have not been thoroughly analyzed. The aim of this study was to perform a direct evaluation of the high-frequency response of HTS on the cerebrovascular physiological responses in TBI.
Methods
We retrospectively reviewed our prospectively maintained adult TBI database for those with archived high-frequency cerebral physiology and available HTS treatment information. We evaluated different epochs of physiology around HTS bolus dosing, comparing pre- with post-HTS. We assessed for changes in slow fluctuations in ICP, pulse amplitude of ICP (AMP), cerebral perfusion pressure (CPP), mean arterial pressure (MAP), cerebrovascular reactivity (as measured through pressure reactivity index (PRx)), and cerebral compensatory reserve (correlation (R) between AMP (A) and ICP (P)). Comparisons of mean measures and percentage time above clinically relevant thresholds for the physiological parameters were compared pre- and post-HTS using descriptive statistics and Mann-Whitney U testing. We assessed for subgroups of physiological responses using latent profile analysis (LPA).
Results
Fifteen patients underwent 69 distinct bolus infusions of hypertonic saline. Apart from the well-documented decrease in ICP, there was also a reduction in AMP. The analysis of cerebrovascular reactivity response to HTS solution had two main effects. For patients with grossly impaired cerebrovascular reactivity pre-HTS (PRx > + 0.30), HTS bolus led to improved reactivity. However, for those with intact cerebrovascular reactivity pre-HTS (PRx < 0), HTS bolus demonstrated a trend towards more impaired reactivity. This indicates that HTS has different impacts, dependent on pre-bolus cerebrovascular status. There was no significant change in metrics of cerebral compensatory reserve. LPA failed to demonstrate any subgroups of physiological responses to HTS administration.
Conclusions
The direct decrease in ICP and AMP confirms that a bolus dose of a HTS solution is an effective therapeutic agent for intracranial hypertension. However, in patients with intact autoregulation, hypertonic saline may impair cerebral hemodynamics. These findings regarding cerebrovascular reactivity remain preliminary and require further investigation.
Similar content being viewed by others


Introduction
Hypertonic saline (HTS) solutions are used in the setting of moderate/severe traumatic brain injury (TBI) to treat instances of acute or refractory elevations in intracranial pressure (ICP) [9, 38]. There is substantial evidence to indicate that long periods of high ICP result in poor neurological outcomes in TBI [1, 14, 20, 23, 24], and that aggressive ICP and cerebral perfusion pressure (CPP)-directed management should be the mainstay of care for moderate/severe TBI. Though the impact of HTS on ICP response is well known, its impact on other aspects of cerebral physiology remains uncertain. Much of this uncertainty stems from the difficulty in obtaining high-frequency digital physiology, in concert with treatment information and timing. Most prior work has focused on mean hourly or peak hourly point measures of ICP and CPP, documenting the response to HTS administration [7]. The response of cerebrovascular reactivity and cerebral compensatory reserve measures to HTS administration remains under-investigated, with preliminary work demonstrating a decrease in ICP and a tendency to decrease pressure reactivity index (PRx), as well as a decrease in cerebral blood flow and cerebrovascular resistance [17].
For cerebrovascular reactivity, data supporting the strong association between impaired cerebrovascular reactivity and poor patient outcome in TBI is emerging [5, 15, 19, 39, 46, 50], as well as computed tomography (CT)-based peri-contusional edema progression [34, 35]. Furthermore, recent literature also supports limited to no impact of Brain Trauma Foundation (BTF) guideline-based therapies on cerebrovascular reactivity, including hyperosmolar/hypertonic agents [19, 22, 43, 47]. The main limitation of previous work in this area is the poor temporal resolution of the treatment data, relying on daily measures of treatment intensity levels. Thus, our true understanding of the impact of HTS on continuously assessed cerebrovascular reactivity, whether positive or negative, remains unknown.
Similarly, continuously assessed cerebral compensatory reserve, using the RAP (correlation (R) between pulse amplitude of ICP (A) and ICP (P)), has emerged as a potential surrogate metric for compliance [11, 31, 49, 50]. Recent data suggests association of high area under the RAP vs. time curve, with admission CT characteristics of diffuse intracranial injury, as well as poor global outcome at 6 months post-TBI [50]. However, there is limited data assessing its response to HTS, an agent designed to reduced ICP, and subsequently improve compliance. Knowledge of the responsiveness of this type of advanced cerebral physiological monitoring to HTS dosing may improve end-user uptake and provide some additional confidence in this metric’s ability to measure important aspects of intracranial physiology during injured states.
As such, the goal of this study was to assess the HTS response of slow fluctuations in cerebral physiology, cerebrovascular reactivity, and cerebral compensatory reserve, using archived high-frequency physiology data and treatment information stored in the Winnipeg Acute TBI Database.
Materials and methods
Study design
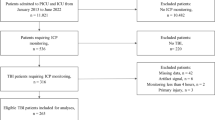
We retrospectively reviewed our prospectively maintained TBI database from the Winnipeg Acute TBI Laboratories at the University of Manitoba. We selected those patients with archived high-frequency digital physiology (ICP and ABP) and treatment data pertaining to HTS administration. All patients included in this database are age 17 or older, who have suffered moderate to severe TBI, requiring admission to the surgical intensive care unit (SICU) for invasive ICP monitoring. Patients have been sequentially entered into this database since January 2019, with no gaps or missed patients. Patients received treatment according to the Brain Trauma Foundation (BTF) guidelines [14] A total of 15 patients were identified with HTS data and high-frequency physiological recordings facilitating the assessment of cerebrovascular reactivity and compensatory reserve. There were 69 distinct episodes of bolus dose HTS administered, with all receiving the standard institutional bolus dose of 140 ml of 7.33% HTS solution for intermittent episodes of refractory ICP. All aspects of data collection for this database have been approved by the University of Manitoba research ethics board (H2017:181 and H2017:188), with approval for retrospective access for this project (H2020:118).
As mentioned, our patients received therapy according to the BTF guidelines. All patients with mass-occupying lesions are taken to the operating room on admission for evacuation ± primary decompression, depending on the individual situation. Our local tiered approach to ICP/CPP management is as follows, with a goal ICP of < 22 mmHg and CPP between 60 and 70 mmHg. First, elevation of the head of bed is 30° or higher (depending on tolerance) with assurance of no constraints on the neck to jugular venous return. Next, sedation is titrated to ensure comfort on the ventilator, followed by escalation in dosing titrated to ICP. We employ a combination of propofol, fentanyl, and midazolam as our baseline infusions. Spikes in ICP above 22 mmHg for 10 min, are tempered using either mannitol or HTS intermittent dosing. Our local institutional preference is for HTS. Failure of the above therapies is followed by ongoing sedative/hypertonic measures, with the addition of hypothermia therapy (via external cooling) or consideration for decompressive procedures. We do not employ barbiturate therapy in our patients. We rarely utilize hyperventilation therapy outside of temporizing measures to facilitate more definitive management (i.e., surgical intervention). We do not routinely employ cerebrospinal fluid diversion through external drainage to control ICP. We do not utilize cerebrovascular reactivity or compensatory reserve monitoring to direct therapies.
Patient data collection
As part of the ongoing prospective TBI database, all patient demographic, injury, and treatment information is recorded. For the purpose of this study, we extracted standard patient demographics (including age, sex, admission Glasgow Coma Score (GCS) total and motor, pupillary response, presence of pre-hospital hypoxia/hypotension, and admission computed tomography (CT) characteristics—including Marshall CT scores). All HTS dosing is recorded with a time stamp, allowing it to be linked to high-frequency recorded physiology.
All patients had ICP and arterial blood pressure (ABP) data prospectively recorded using intensive care monitoring plus (ICM+) software (Cambridge Enterprise Ltd, Cambridge, UK, http://icmplus.neurosurg.cam.ac.uk), with all signals recorded using the same software and digitized via an A/D converter (DT9804 or DT9826; Data Translation, Marlboro, MA), where appropriate; sampled at frequency of 100 Hz or higher. ICP was monitored using an intra-parenchymal strain gauge probe (Codman ICP Microsensor; Codman & Shurtleff Inc., Raynham, MA); ABP was obtained through arterial lines connected to a pressure transducer.
Signal processing
CPP was calculated as mean arterial pressure (MAP)-ICP. Pulse amplitude of ICP (AMP) was derived from the fundamental amplitude of the ICP waveform in the frequency domain, using Fourier analysis. A non-overlapping 10-s moving average filter was applied to the raw data to decimate the signals to 0.1 Hz, focusing on the frequency ranges associated with cerebral vasogenic activity [21, 27]. ICP, AMP, MAP, and CPP were subsequently output into 10-s by 10-s comma-separated value files for further analysis of the impact of HTS on cerebral physiology slow fluctuations.
Cerebrovascular reactivity was assessed through the derivation of the PRx. PRx was determined using standard means, by calculating the Pearson correlation coefficient between 30 consecutive 10-s measures of ICP and MAP, updated every minute [15]. Similarly, cerebral compensatory reserve was determined using the RAP index (correlation (R) between AMP (A) and ICP (P)), calculated following a similar fashion to PRx, but using AMP and ICP [11]. Data for PRx and RAP was output into minute-by-minute resolution comma-separated value files, for the analysis of the impact of HTS on both cerebrovascular reactivity and cerebral compensatory reserve.
Statistical analysis
All statistical analysis was performed using R statistical computing software (R Foundation for Statistical Computing (2020), Vienna, Austria, http://www.R-project.org/). Descriptive summary statistics for the patient population are provided in Table 1. Alpha for statistical significance was set at 0.05, with no correction for multiple comparisons, given this was an exploratory analysis. Error-bar plots were used to aid in the description of the data.
Initially, data surrounding each bolus injection was found and abstracted for 2 h before and after injecting, with incomplete data sets (i.e., missing ICP and ABP data) being removed. Then the data was aligned using the max peak ICP around the recorded injection time. If the ICP waveform had no distinct drop then this was deemed a non-responsive event, with the data stored separately for sub-group analysis. The cerebral physiology surrounding each HTS administration was manually inspected for ICP response. The criteria for a positive HTS response were (A) ICP had to drop by at least 2 mmHg during the initial 10 min post-HTS infusion and (B) ICP had to continue to drop by at least 5 mmHg from baseline during the following 30 min. Using these criteria, in total, there were 55 distinct episodes of responsive ICP to HTS and 14 distinct episodes that had non-responsive ICP to HTS. Examples of ICP response are demonstrated in Fig. 1.

Examples of responsive vs. non-responsive ICP to HTS. The pressure waveforms of responsive vs. non-responsive ICP to HTS. The shaded areas indicate the two 20-min windows with a 10-min delay used to evaluate the change due to HTS. AMP, amplitude pulse of ICP; CPP, cerebral perfusion pressure; HTS, hypertonic saline; ICP, intracranial pressure; MAP, mean arterial blood pressure; Mins/mins, minutes; mmHg, millimeters of mercury; PRx, pressure reactivity index
For each HTS injection, two distinct comparisons were conducted for the recorded physiology pre-/post-HTS. First, the time over threshold before and after bolus infusion was compared, using the following thresholds: (A) ICP above 22 mmHg [14], (B) AMP above 2 mmHg (though 1–10 mmHg was tested) [29], (C) CPP below 60 mmHg [18], (D) PRx above + 0.3 [4, 48], and (E) RAP above + 0.4 [50]. These thresholds were chosen as they have been quoted in TBI literature to be associated with worse global outcome. The window lengths of data to compare varied from 5 to 60 min with a delay varied from 5–60 min to evaluate response; the final window time and delay used for all the final evaluations in this analysis can be seen in Fig. 1 (10-min delay between two 20-min windows). For each window, the time over the indicated threshold was found for each bolus injection. Finally, a Mann-Whitney U test was performed between the two windows of data. The other variations in data windows, and gaps, failed to lead to any significant differences in the relationships described in the “Results” section. As such, we focused on the above defined data windows (i.e., 20 min pre-HTS, 10 min gap allowing for HTS infusion, and 20 min post-HTS) when presenting the results. Second, we compared the grand mean physiological value across the time windows using Mann-Whitney U test.
Furthermore, we wished to see if there was a difference in physiological response to HTS for those patients with deranged cerebrovascular reactivity pre-HTS (i.e., mean PRx for 30 min before HTS was above + 0.3) [10], clearly intact cerebrovascular reactivity pre-HTS (i.e., mean PRx for 30 min before HTS was below 0) [10]. We subsequently performed the previously stated analyses (time over threshold and grand mean value over window with a Mann-Whitney U test for each) with the same 20-min windows and 10-min delay.
Finally, for the MAP, ICP, CPP, and AMP slow-wave data (i.e., 10-s data), latent profile analysis (LPA) [36] was performed to identify any key features/classes of responses to HTS. In order to perform an accurate analysis on the data, values that contained null values were interpolated using linear regression of its nearest 5 neighbors to substitute for the missing data value [26]. The now fully filled data sets were then scaled so that each individual bolus infusion data set had a maximum variance of 1. Lastly, the filled and scaled data was then averaged over 5 min to reduce the dimensionality for LPA, then an LPA was performed to identify any sub-group of HTS responses for MAP, ICP, AMP, and CPP that the waveforms may contain. LPA failed to identify sub-groups of responses to HTS for any of the physiology assessed, and thus details of this negative analysis are not presented. Finally, we compared the influence of HTS on ICP and its association with other measured physiology or cerebral reactivity. The delta ICP for each bolus infusion of HTS was determined as the difference between the ICP mean value of two 30-min windows, one immediately before HTS infusion and one 30 min after HTS infusion. The mean/time over threshold was found over a variety of time windows immediately before HTS infusion (10 to 120 min). Using all HTS bolus infusions, mean/time over threshold vs. delta ICP were used to find a Pearson correlation and p value.
Results
Patient characteristics
Table 1 provides a table with the core patient characteristics. In total, there were 15 patients with 69 separate episodes of HTS administration analyzed. The median age was 35 (IQR, 23–54), with 11 being male. The median admission GCS motor sub-score was 5 (IQR, 4–5), with 10 and 2 having pre-hospital episodes of hypoxia and hypotension, respectively. Finally, Marshall CT grades on admission were 4 patients with class III, 6 with IV, and 5 with V.
Physiological response
HTS had no significant effect on slow fluctuations in MAP. There was, however, a significant response in slow fluctuations in ICP, CPP, and AMP. Table 2 demonstrates the response that HTS induced on these waveforms. In Table 2, the delay was 10 min and the time window 20 min, though as mentioned above, altering the time windows/delay failed to change the described relationships. ICP and AMP consistently demonstrated a decrease in magnitude, while CPP had an increase. As can be interfered from Fig. 1, an increase in the delay improved the significance up to about 40 min post-HTS, likewise an increase in the time window also increases the significance in ICP, AMP, and CPP responses. The 10-min delay, with a 20-min time window, demonstrated the most immediate significant response to cerebral physiology post-HTS infusion.
Despite the significant decrease in ICP and AMP demonstrated in 55 treatment episodes, in some patients, the bolus dose of HTS resulted in a non-significant response of ICP, as Fig. 1 illustrates. This lack of response in both ICP and AMP was similar in all infusion examples with a non-responsive ICP waveform (n = 14). In Supplementary Appendix A, more examples of responsive and non-responsive waveforms are shown.
Cerebrovascular reactivity status pre-HTS
To evaluate HTS on cerebrovascular reactivity, we separated the responsive ICP data into three different categories, impaired (mean PRx pre-HTS above + 0.3; n = 15), intact (mean PRx pre-HTS below 0; n = 17) or in the “grey” zone (mean PRx pre-HTS between 0 and + 0.3; n = 23). Table 2 demonstrates impaired and intact cerebrovascular reactivity responses to HTS. For a more intuitive description of the effects of PRx and HTS on cerebral response, there are examples in the Supplementary Appendix B using the error bar plots of each physiological measure and its effect to HTS, both for impaired and intact cerebrovascular reactivity across the entire population. In these error bar plots, ICP, AMP, MAP, and CPP all demonstrated the same physiological response as discussed previously. As can be seen in Table 2, when PRx is initially high, HTS caused a decrease in PRx mean and time over threshold. RAP failed to demonstrate a significant difference, regardless of the data window utilized for comparison.
However, when cerebrovascular reactivity was intact pre-HTS, HTS bolus had the tendency to increase PRx. Though again, the change in window and delay caused a change in significance. As with the intact cerebrovascular reactivity group, the response in RAP was not significant on any time window of delay tested. Similar analysis for those HTS administration episodes where pre-HTS PRx was in the “grey” zone of 0 to + 0.30 can be found in Supplementary Appendix C.
Discussion
Through this analysis of the impact of HTS on high-frequency cerebrovascular physiology, some direct responses of note were revealed. First, there is a significant and reliable decrease in AMP with ICP with HTS bolus dosing, corroborating previous literature on the topic [11, 17, 49]. The two waveforms are heavily correlated as demonstrated by the persistently positive RAP (i.e., correlation between ICP and AMP) in all bolus infusions in which ICP responded to HTS (n = 55). This conclusion supports the idea that AMP may be effective at identifying true ICP (also termed compensatory reserve weighted ICP), though much further work in this area is required [25, 30, 37]. Also in line with the ICP reduction is the significant improvement in CPP seen post-HTS dosing. Of note, LPA analysis failed to uncover any sub-groups of responders with different temporal profiles in cerebral physiological response to HTS administration, including the response of ICP, AMP, MAP, and CPP.
Second, a small number of instances (n = 14) existed where there was no response in ICP to HTS administration. Based on inspection of the cerebral physiology pre-HTS in these cases, a clear common physiological pattern in these instances did not exist. However, there are some non-responsive episodes where the PRx was high (impaired reactivity) [3, 15, 39, 50], potentially reflecting a system where the homeostatic mechanisms are deranged beyond the point of salvage or response to therapeutics. Given the small number of these types of events seen in our data set, much larger multi-center cohorts of data with treatment information are required to explore reasons for responsive and non-responsive periods.
Third, cerebrovascular reactivity appears to improve with HTS administration, in the short term (up to 60 min post-HTS). These HTS results have been described previously in one study [17], with the inter-dependence of ICP and cerebrovascular reactivity alluded to in other retrospective series [4, 10, 40, 47]. However, this improvement in cerebrovascular reactivity tended to only occur when there was gross derangement in reactivity pre-HTS (i.e., PRx > + 0.30 for 30 min pre-HTS). Those treatment instances where cerebrovascular reactivity remained intact pre-HTS displayed a trend towards more positive PRx values post-HTS. This has not been previously described and carries importance moving forward for ongoing studies into cerebrovascular reactivity monitoring and individualized CPP/ICP thresholds based on PRx. Furthermore, it carries implications for prophylactic dosing of HTS in moderate/severe TBI. It is becoming more apparent that the time spent with abnormal cerebrovascular reactivity is associated with poorer 6-month outcome in moderate/severe TBI [20, 24, 45, 48]. As such, if particular treatments lead to worse cerebrovascular reactivity, this may impact patient outcomes. Evaluating the cause for the rise in PRx post-HTS has left us with more questions than answers. Given that HTS reduced ICP, one natural thought as to the cause for the rise in PRx would be a subsequent rise in MAP/CPP towards the upper limit of regulation (ULR) [18]. However, as seen in Supplementary Appendix B, at a population level, this is not the case. In the cohort of “intact” reactivity prior to HTS, we can see that HTS administration failed to lead to a significant increase in either MAP or CPP, yet did lead to an increase in PRx. With that said, evaluating each response at an individual level, there were some cases where HTS administration did lead to MAP/CPP increases that in theory could have approached the ULR (see Supplementary Appendix A—example of HTS response). Worsening of cerebrovascular reactivity after HTS administration has been described in aneurysmal subarachnoid hemorrhage patients, using transcranial Doppler (TCD) mean flow index (Mx; correlation between mean flow velocity from the middle cerebral artery and CPP) [41]. This prior study in subarachnoid hemorrhage demonstrated significant ICP reductions with increased MAP, CPP, and CBF (using Xenon CT). However, there was a significant hemispheric discrepancy in Mx pre-HTS, with the ipsilateral hemisphere (ipsilateral to hemorrhage/aneurysm) displaying “impaired” reactivity pre-HTS (+ 0.20 ± 0.92), and the contralateral hemisphere displaying relatively “intact” reactivity pre-HTS (+ 0.01 ± 0.95). With HTS administration, the ipsilateral hemisphere did not show any change in Mx; however, the contralateral hemisphere had worsening cerebrovascular reactivity with HTS administration (increase of + 0.22 ± 0.28, p < 0.01). These previous findings, in conjunction with our results in this TBI cohort, suggest that HTS may induce impaired cerebrovascular reactivity if reactivity is intact prior to HTS administration. It must be emphasized that our findings are based on a small number of patients and treatments but raise the question of a disparity in cerebrovascular reactivity response to HTS bolus dosing. Although not directly confirmed, the only other previous study that analyzed the effects of HTS on cerebrovascular reactivity in TBI may also have demonstrated this phenomenon though all of these episodes were amalgamated into one group [17]. Finally, the episodes where PRx was in a grey zone (i.e., 0 to + 0.30) prior to HTS were analyzed in Supplementary Appendix C. These episodes of PRx were not the main focus of the study, as we wanted to do a sub-group analysis based on clearly intact vs. clearly impaired cerebrovascular reactivity. Our analysis of this grey zone sub-group demonstrated that there was no significant impact of HTS on recorded cerebrovascular reactivity or compensatory reserve. This grey zone for PRx remains unclear in the literature, and may represent a transitional phase for cerebrovascular reactivity, as PRx is unlikely a binary function and likely represents a spectrum of vascular function. Further investigation into this disparity of response to HTS is required in order to fully understand the impact of HTS on cerebrovascular reactivity in TBI.
Fourth, the impact of HTS on continuously assessed cerebral compensatory reserve (i.e., RAP) was underwhelming in our cohort. Despite the improvement in AMP and ICP with HTS administration, RAP itself (i.e., a surrogate for phase shift between AMP and ICP), failed to demonstrate a significant difference comparing values pre- with post-HTS. There appeared to be a trend towards a decrease in RAP when PRx was impaired pre-HTS, however this was not significant. These findings are in slight contradiction with those found with barbiturate therapy in TBI [28]. The prior work on RAP and its response to barbiturate therapy in TBI did demonstrate a reduction in RAP, AMP, and ICP slope with thiopental coma. Thus, RAP does appear to demonstrate a temporal responsiveness to BTF-directed therapeutic interventions. It must be acknowledged that HTS and barbiturate therapies have different mechanisms of action, which may account for the different responses seen in our paper compared with the barbiturate literature. HTS primarily functions based on improved rheology and osmotic properties, while barbiturates function based on metabolic suppression. Similarly, the temporal response pattern of RAP is difficult to assess, as changes in brain edema may take extended time periods to occur, and may depend on individual genetics [16, 44]. We are currently left with assessing mean RAP or time above threshold, though integrated area under RAP vs. time curves have been utilized [50]. Given the negative parabolic nature of the relationship between RAP and ICP, low or negative value RAP can mean both preserved and severely impaired compensatory reserve, depending on the situation [11]. Thus, the interpretation of RAP is complex. We were interested in performing a sub-group analysis of those with grossly impaired pre-HTS mean RAP (i.e., > + 0.4) vs. those with pre-HTS RAP < + 0.4, however, all of our instances of HTS administration had a mean RAP value above + 0.4. Thus, such an analysis was not possible. Other RAP thresholds were not investigated, as the selection of other higher RAP values would have been arbitrary. Such investigation into RAP thresholding requires dedicated larger multi-center data sets and is beyond the scope of this paper. Unfortunately, given the relatively new nature of RAP in TBI monitoring, we are still in stages of infancy regarding its application and analysis. Thus, the role for continuous RAP monitoring remains unclear at this time, with much further investigation required in TBI multi-modal monitoring. As such, the negative results regarding RAP found in this study should be interpreted with caution.
Finally, we were unable to find any pre-HTS physiological variables which were predictive of the ICP change seen as a result of HTS administration. As mentioned in the “Materials and methods” section, we performed LPA in order to assess for any latent class/groupings of response to HTS administration, evaluating all physiological variables. This analysis failed to demonstrate any sub-classes of HTS administrations that were different from the others. Similarly, we also assessed the correlation between pre-HTS physiology, using data windows ranging from 10 min up to 2 h pre-HTS, and compared them with the delta-ICP resulting from HTS administration. As this analysis was not the focus of the manuscript, it can be found in Supplementary Appendix D. However, this correlation testing also failed to demonstrate moderate or strong correlations between pre-HTS cerebral physiology and the delta-ICP response. Predictors of ICP response to the various BTF guideline-based therapeutics are crucial, as they would allow for clinical end-users to stratify patients pre-treatment into suspected responders vs. non-responders, which may facilitate alternative treatments or increase in aggressiveness of therapies applied. Much further work, using high-frequency physiological responses, is required in this regard, and will be the focus of ongoing work from European [2, 6, 33] and Canadian [8] collaborative research groups.
Limitations
Despite the interesting results described above, there are some limitations which deserve highlighting. First, this study is based on small patient cohort with unique data consisting of high-frequency digital physiology and available time-linked HTS administration data. Thus, despite having 69 episodes of HTS administration, the described results should remain exploratory at this time. With only 15 patients with 69 HTS administration events, our data set is small, yet relatively unique globally with high-frequency physiology linked with treatment information. Similar methodologies for the exploration of the relationships between TBI therapeutics and continuous physiological responses have been employed by other centers globally [12, 13, 17, 32, 33, 41, 42]. As these types of data sets are rare, we are left with exploring such treatment-related impacts on small patient numbers. This type of analysis, evaluating individual treatment events in a small number of patients, serve only as a preliminary exploratory probe into such relationships, and are merely hypothesis generating, requiring much further evaluation using larger multi-center data sets. As such, the results described within should be interpreted with caution. Second, we focused on basic statistics and descriptive analysis for the HTS response. Future work in this area will require time-series techniques, evaluating the multi-variate relationships pre- and post-treatment. Third, we evaluated various time windows of physiological data pre- and post-HTS administration, in order to exclude the impact of data window length on the relationships seen. Caution must be taken when evaluating windows for data averaging that are less than 30 min in duration. Particularly, evaluating derived correlation indices, such as those for cerebrovascular reactivity (PRx) and compensatory reserve (RAP), such caution is required in the interpretation of data that is averaged using less than 30 min of recordings. Shorter data windows may not reduce the signal-to-noise ratio sufficiently, particularly in the case of metrics of compensatory reserve which may take substantially longer to respond to intervention, compared with other high-frequency physiology measures such as ICP. Fourth, all patients in this study received active treatment according to the BTF guidelines. As such, the response to HTS seen may not truly reflect the isolated response to HTS but the combination of therapies received during the HTS dosing. Furthermore, aggressiveness of therapy changed throughout these patients stay, based on ICP/CPP responses and serial cranial imaging. As such, we cannot confidently say that the physiological responses seen during HTS administration were solely due to HTS. This further highlights the need for large multi-center data sets to evaluate the impact of all BTF based therapeutic strategies on cerebrovascular reactivity and compensatory reserve. Finally, the small number of episodes where ICP was non-responsive to HTS dosing remain puzzling. The results from this sub-group require further investigation, as there were only 14 episodes where this occurred.
Future directions
As this is only the second study evaluating the impact of hypertonic saline dosing on cerebrovascular reactivity, and compensatory reserve, there exists the need for much further work. Such work will require larger multi-center datasets, with treatment data temporally linked. Such work is the focus of the Canadian High-Resolution TBI (CAHR-TBI) Research Collaborative. Furthermore, as mentioned above, future investigation would benefit from time-series analytical methodologies, using multi-variate vector autoregressive integrative moving average (VARIMA) models, impulse response function analysis, and Granger causality testing both pre- and post-HTS. Such complex work is the focus of ongoing efforts in our laboratory. In addition, despite the lack of significant results in this work, LPA is a technique to profile and identify key features within systems that may help differentiate distinct responses to an event. To leverage this more effectively, an increase in sample size may identify more individualized physiological responses. With larger datasets, we may find sub-groups of responders to HTS. Finally, the addition of other continuous multi-modal monitoring would allow us to comment on the impacts of HTS on cerebral blood flow velocity, cerebral oxygen saturations and extracellular oxygen diffusion, cerebral blood flow, and cerebral metabolic response in the extracellular space. It is only through such complex datasets that we will truly gain an understanding of the impact of current TBI therapeutics on cerebral physiology, allowing us to move closer to personalized medicine in moderate/severe TBI care.
Conclusions
HTS bolus dosing leads to a consistent decrease in ICP and AMP, with an improvement in CPP. Cerebrovascular reactivity appears to improve with HTS dosing, but only in those with grossly impaired reactivity pre-HTS. There is a trend towards worse cerebrovascular reactivity after HTS administration in those patients with intact reactivity pre-HTS. Future investigation is required to fully demonstrate the underlying effects.
References
Adams H, Donnelly J, Czosnyka M, Kolias AG, Helmy A, Menon DK, Smielewski P, Hutchinson PJ (2017) Temporal profile of intracranial pressure and cerebrovascular reactivity in severe traumatic brain injury and association with fatal outcome: an observational study. PLoS Med 14(7):e1002353
AIR M, Menon DK, Steyerberg EW, Citerio G, Lecky F, Manley GT, Hill S, Legrand V, Sorgner A, CENTER-TBI Participants and Investigators (2015) Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury (CENTER-TBI): a prospective longitudinal observational study. Neurosurgery 76(1):67–80
Avezaat CJJ, van Eijndhoven JHM (1986) The role of the pulsatile pressure variations in intracranial pressure monitoring. Neurosurg Rev 9(1–2):113–120
Balestreri M, Czosnyka M, Steiner LA, Hiler M, Schmidt EA, Matta B, Menon D, Hutchinson P, Pickard JD (2005) Association between outcome, cerebral pressure reactivity and slow ICP waves following head injury. Acta Neurochir Suppl 95:25–28
Bennis FC, Teeuwen B, Zeiler FA, Elting JW, van der Naalt J, Bonizzi P, Delhaas T, Aries MJ (2020) Improving prediction of favourable outcome after 6 months in patients with severe traumatic brain injury using physiological cerebral parameters in a multivariable logistic regression model. Neurocrit Care. https://doi.org/10.1007/s12028-020-00930-6
Beqiri E, Smielewski P, Robba C et al (2019) Feasibility of individualised severe traumatic brain injury management using an automated assessment of optimal cerebral perfusion pressure: the COGiTATE phase II study protocol. BMJ Open. https://doi.org/10.1136/bmjopen-2019-030727
Berger-Pelleiter E, Émond M, Lauzier F, Shields J-F, Turgeon AF (2016) Hypertonic saline in severe traumatic brain injury: a systematic review and meta-analysis of randomized controlled trials. CJEM 18(2):112–120
Bernard F, Gallagher C, Griesdale D, Kramer A, Sekhon M, Zeiler FA (2020) The Canadian high-resolution traumatic brain injury (CAHR-TBI) research collaborative. Can J Neurol Sci J Can Sci Neurol 1–6
Boone MD, Oren-Grinberg A, Robinson TM, Chen CC, Kasper EM (2015) Mannitol or hypertonic saline in the setting of traumatic brain injury: what have we learned? Surg Neurol Int. https://doi.org/10.4103/2152-7806.170248
Budohoski KP, Czosnyka M, de Riva N, Smielewski P, Pickard JD, Menon DK, Kirkpatrick PJ, Lavinio A (2012) The relationship between cerebral blood flow autoregulation and cerebrovascular pressure reactivity after traumatic brain injury. Neurosurgery 71(3):652–661
Calviello L, Donnelly J, Cardim D, Robba C, Zeiler FA, Smielewski P, Czosnyka M (2018) Compensatory-reserve-weighted intracranial pressure and its association with outcome after traumatic brain injury. Neurocrit Care 28(2):212–220
Calviello LA, Zeiler FA, Donnelly J, Uryga A, de Riva N, Smielewski P, Czosnyka M (2019) Estimation of pulsatile cerebral arterial blood volume based on transcranial doppler signals. Med Eng Phys 74:23–32
Cardim D, Schmidt B, Robba C, Donnelly J, Puppo C, Czosnyka M, Smielewski P (2017) Transcranial Doppler monitoring of intracranial pressure plateau waves. Neurocrit Care 26(3):330–338
Carney N, Totten AM, O’Reilly C et al (2017) Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery 80(1):6–15
Czosnyka M, Smielewski P, Kirkpatrick P, Laing RJ, Menon D, Pickard JD (1997) Continuous assessment of the cerebral vasomotor reactivity in head injury. Neurosurgery 41(1):11–19
Dardiotis E, Paterakis K, Tsivgoulis G et al (2014) AQP4 tag single nucleotide polymorphisms in patients with traumatic brain injury. J Neurotrauma 31(23):1920–1926
Dias C, Silva MJ, Pereira E, Silva S, Cerejo A, Smielewski P, Rocha AP, Gaio AR, Paiva J-A, Czosnyka M (2014) Post-traumatic multimodal brain monitoring: response to hypertonic saline. J Neurotrauma 31(22):1872–1880
Donnelly J, Czosnyka M, Adams H et al (2017) Individualizing thresholds of cerebral perfusion pressure using estimated limits of autoregulation. Crit Care Med 45(9):1464–1471
Donnelly J, Czosnyka M, Adams H et al (2019) Twenty-five years of intracranial pressure monitoring after severe traumatic brain injury: a retrospective, single-center analysis. Neurosurgery 85(1):E75–E82
Flechet M, Meyfroidt G, Piper I et al (2018) Visualizing cerebrovascular autoregulation insults and their association with outcome in adult and paediatric traumatic brain injury. In: Heldt T (ed) Intracranial Press. Neuromonitoring XVI. Springer International Publishing, Cham, pp 291–295
Fraser CD, Brady KM, Rhee CJ, Easley RB, Kibler K, Smielewski P, Czosnyka M, Kaczka DW, Andropoulos DB, Rusin C (2013) The frequency response of cerebral autoregulation. J Appl Physiol 115(1):52–56
Froese L, Batson C, Gomez A, Dian J, Zeiler FA (2020) The limited impact of current therapeutic interventions on cerebrovascular reactivity in traumatic brain injury: a narrative overview. Neurocrit Care. https://doi.org/10.1007/s12028-020-01003-4
Güiza F, Depreitere B, Piper I et al (2015) Visualizing the pressure and time burden of intracranial hypertension in adult and paediatric traumatic brain injury. Intensive Care Med 41(6):1067–1076
Güiza F, Meyfroidt G, Piper I et al (2017) Cerebral perfusion pressure insults and associations with outcome in adult traumatic brain injury. J Neurotrauma 34(16):2425–2431
Hall A, O’Kane R (2016) The best marker for guiding the clinical management of patients with raised intracranial pressure—the RAP index or the mean pulse amplitude? Acta Neurochir (Wien) 158(10):1997–2009
Horton NJ, Lipsitz SR (2001) Multiple imputation in practice. Am Stat 55(3):244–254
Howells T, Johnson U, McKelvey T, Enblad P (2015) An optimal frequency range for assessing the pressure reactivity index in patients with traumatic brain injury. J Clin Monit Comput 29(1):97–105
Howells T, Lewén A, Sköld MK, Ronne-Engström E, Enblad P (2012) An evaluation of three measures of intracranial compliance in traumatic brain injury patients. Intensive Care Med 38(6):1061–1068
International Symposium on Intracranial Pressure and Brain Monitoring, Manley GT (eds) (2008) Intracranial pressure and brain monitoring XIII: mechanisms and treatment; [XIIIth International Symposium on Intracranial Pressure and Brain Monitoring, July 22 - 26, 2007, San Francisco, CA]. Springer, Wien
Ji Z, Zhu L, Yang X, Jiang L (2011) Single wave extraction in continuous intracranial pressure signal with lifting wavelet transformation and discrimination rules. EURASIP J Adv Signal Process 2011(1):43
Kim D-J, Czosnyka Z, Keong N, Radolovich DK, Smielewski P, Sutcliffe MPF, Pickard JD, Czosnyka M (2009) Index of cerebrospinal compensatory reserve in hydrocephalus. Neurosurgery 64(3):494–501 discussion 501-502
Kirkpatrick PJ, Smielewski P, Piechnik S, Pickard JD, Czosnyka M (1996) Early effects of mannitol in patients with head injuries assessed using bedside multimodality monitoring. Neurosurgery 39(4):714–721
Klein SP, Fieuws S, Meyfroidt G, Depreitere B (2020) Effects of norepinephrine, propofol and hemoglobin concentration on dynamic measurements of cerebrovascular reactivity in acute brain injury. J Neurotrauma. https://doi.org/10.1089/neu.2020.7160
Mathieu F, Zeiler FA, Ercole A et al (2020) Relationship between measures of cerebrovascular reactivity and intracranial lesion progression in acute traumatic brain injury patients: a CENTER-TBI study. J Neurotrauma. https://doi.org/10.1089/neu.2019.6814
Mathieu F, Zeiler FA, Whitehouse DP, Das T, Ercole A, Smielewski P, Hutchinson PJ, Czosnyka M, Newcombe VFJ, Menon DK (2020) Relationship between measures of cerebrovascular reactivity and intracranial lesion progression in acute TBI patients: an exploratory analysis. Neurocrit Care 32(2):373–382
Oberski D (2016) Mixture models: latent profile and latent class analysis. In: Robertson J, Kaptein M (eds) Mod. Stat. Methods HCI. Springer International Publishing, Cham, pp 275–287
Passi N, Degnan AJ, Levy LM (2013) MR imaging of papilledema and visual pathways: effects of increased intracranial pressure and pathophysiologic mechanisms. AJNR Am J Neuroradiol 34(5):919–924
Rangel-Castillo L, Gopinath S, Robertson CS (2008) Management of intracranial hypertension. Neurol Clin 26(2):521–541
Sorrentino E, Diedler J, Kasprowicz M et al (2012) Critical thresholds for cerebrovascular reactivity after traumatic brain injury. Neurocrit Care 16(2):258–266
Steiner L, Czosnyka M, Piechnik S, Smielewski P, Chatfield D, Menon D, Pickard J (2002) Continuous monitoring of cerebrovascular pressure reactivity allows determination of optimal cerebral perfusion pressure in patients with traumatic brain injury. Crit Care Med 30(4):733–738
Tseng M-Y, Al-Rawi PG, Czosnyka M, Hutchinson PJ, Richards H, Pickard JD, Kirkpatrick PJ (2007) Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 107(2):274–282
Tymko MM, Donnelly J, Smielewski P, Zeiler FA, Sykora M, Haubrich C, Nasr N, Czosnyka M (2019) Changes in cardiac autonomic activity during intracranial pressure plateau waves in patients with traumatic brain injury. Clin Auton Res 29(1):123–126
Weersink CSA, Aries MJH, Dias C et al (2015) Clinical and physiological events that contribute to the success rate of finding “optimal” cerebral perfusion pressure in severe brain trauma patients. Crit Care Med 43(9):1952–1963
Yin J, Zhang H, Chen H, Lv Q, Jin X (2018) Hypertonic saline alleviates brain edema after traumatic brain injury via downregulation of aquaporin 4 in rats. Med Sci Monit Int Med J Exp Clin Res 24:1863–1870
Zeiler FA, Beqiri E, Cabeleira M, Hutchinson PJ, Stocchetti N, Menon DK, Czosnyka M, Smielewski P, Ercole A, Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury (CENTER-TBI) High-Resolution ICU (HR ICU) Sub-Study Participants and Investigators (2020) Brain tissue oxygen and cerebrovascular reactivity in traumatic brain injury: a collaborative european neurotrauma effectiveness research in traumatic brain injury exploratory analysis of insult burden. J Neurotrauma. https://doi.org/10.1089/neu.2020.7024
Zeiler FA, Donnelly J, Smielewski P, Menon DK, Hutchinson PJ, Czosnyka M (2018) Critical thresholds of intracranial pressure-derived continuous cerebrovascular reactivity indices for outcome prediction in noncraniectomized patients with traumatic brain injury. J Neurotrauma 35(10):1107–1115
Zeiler FA, Ercole A, Beqiri E et al (2019) Cerebrovascular reactivity is not associated with therapeutic intensity in adult traumatic brain injury: a CENTER-TBI analysis. Acta Neurochir (Wien) 161(9):1955–1964
Zeiler FA, Ercole A, Beqiri E et al (2019) Association between cerebrovascular reactivity monitoring and mortality is preserved when adjusting for baseline admission characteristics in adult traumatic brain injury: a CENTER-TBI study. J Neurotrauma. https://doi.org/10.1089/neu.2019.6808
Zeiler FA, Ercole A, Cabeleira M et al (2019) Compensatory-reserve-weighted intracranial pressure versus intracranial pressure for outcome association in adult traumatic brain injury: a CENTER-TBI validation study. Acta Neurochir (Wien) 161(7):1275–1284
Zeiler FA, Kim D-J, Cabeleira M, Calviello L, Smielewski P, Czosnyka M (2018) Impaired cerebral compensatory reserve is associated with admission imaging characteristics of diffuse insult in traumatic brain injury. Acta Neurochir (Wien) 160(12):2277–2287
Funding
This work was supported directly through the Manitoba Public Insurance (MPI) Neuroscience/TBI Research Endowment providing infrastructures and disposable support; the Canada Foundation for Innovation (CFI) (Project #: 38583), Research Manitoba (Grant #: 3906), University of Manitoba - Department of Surgery GFT Research Grant providing graduate student salary support; and the University of Manitoba Office of Research Services (ORS) - University Research Grant Program (URGP) providing hardware support. The sponsor had no role in the design or conduct of this research. FAZ receives research support from the Manitoba Public Insurance (MPI) Neuroscience/TBI Research Endowment, the Health Sciences Centre Foundation Winnipeg, the United States National Institutes of Health (NIH) through the National Institute of Neurological Disorders and Stroke (NINDS), the Canadian Institutes of Health Research (CIHR), the Canada Foundation for Innovation (CFI), Research Manitoba, the University of Manitoba VPRI Research Investment Fund (RIF), the University of Manitoba Centre on Aging, and the University of Manitoba Rudy Falk Clinician-Scientist Professorship. LF is supported by the University of Manitoba - Department of Surgery GFT Research Grant and the University of Manitoba Office of Research Services (ORS) - University Research Grant Program (URGP). CB is supported by the Centre on Aging Fellowship at the University of Manitoba. AG is supported by the University of Manitoba Clinician Investigator Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee (University of Manitoba Research Ethics Board; H2017:1818, H2017:188, H2020:118) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. For this type of study, formal consent is not required. No identifying participant information is present in this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Brain trauma
Rights and permissions
About this article

Cite this article
Froese, L., Dian, J., Batson, C. et al. The impact of hypertonic saline on cerebrovascular reactivity and compensatory reserve in traumatic brain injury: an exploratory analysis. Acta Neurochir 162, 2683–2693 (2020). https://doi.org/10.1007/s00701-020-04579-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-020-04579-0