
Abstract
Altered intestinal microbial composition (dysbiosis) and metabolic products activate aggressive mucosal immune responses that mediate inflammatory bowel diseases (IBD). This dysbiosis impairs the function of regulatory immune cells, which normally promote mucosal homeostasis. Normalizing and maintaining regulatory immune cell function by correcting dysbiosis provides a promising approach to treat IBD patients. However, existing microbe-targeted therapies, including antibiotics, prebiotics, probiotics, and fecal microbial transplantation, provide variable outcomes that are not optimal for current clinical application. This review discusses recent progress in understanding the dysbiosis of IBD and the basis for therapeutic restoration of homeostatic immune function by manipulating an individual patient’s microbiota composition and function. We believe that identifying more precise therapeutic targets and developing appropriate rapid diagnostic tools will guide more effective and safer microbe-based induction and maintenance treatments for IBD patients that can be applied in a personalized manner.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
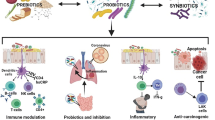
Hundreds of trillions of microorganisms, including bacteria, virus, fungi and archaea, reside in our distal intestines and mutually interact with co-evolved host immune cells in a beneficial reciprocal relationship that is influenced by host genetics and environmental factors, including the diet [1,2,3]. The microbiota evolved to colonize specialized ecological niches of the human gastrointestinal tract and to utilize variable diets, while the human mucosal immune system evolved to protect the host from harmful microbial pathogen exposures, yet prevent chronic intestinal inflammation [3, 4]. Enteric resident microbiota exists as a consortium that contains both putative proinflammatory and protective strains [5, 6]. A delicate balance between those functionally distinct populations is maintained in healthy individuals, while patients with inflammatory bowel diseases (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), harbor an altered gut microbial composition (dysbiosis) defined as increased potentially aggressive species in parallel with decreased anti-inflammatory groups [5,6,7,8]. Gut microbial diversity decreases and metabolic functions are altered in IBD patients, suggesting a loss of protective bacteria and their functions in IBD [9,10,11]. Prolonged dysbiotic conditions lead to dysfunction of the host immune system, which is considered the key mediator of the chronic inflammation of IBD [6, 11] (Fig. 1).

Dysbiosis-associated mucosal immune-dysfunction in IBD. Enteric infection, medications including antibiotics, NSAIDs and immunosuppressive drugs, diet, smoking and alcohol, psychological stress in susceptible genetic individuals cause microbial dysbiosis and metabolic changes. Prolonged dysbiotic conditions characterized by increased aggressive bacterial strains and decreased regulatory species lead to dysfunction of mucosal immune response. Aggressive microbial groups activate inflammatory response by inducing Th1/Th17-effector cells, while decreased regulatory species impair the induction and function of regulatory cells that include regulatory T cells (Treg), B cells (Breg), macrophages (MΦ), dendritic cells (DC) and innate lymphoid cells (ILCs). This imbalance of mucosal cytokine profiles in combination with defective barrier function sustains mucosal inflammation and can potentially lead to IBD in susceptible individuals
The activation, migration, proliferation, differentiation and maintenance of a variety of mucosal immune cells are directly regulated by resident microbiota. These activated immune cells cooperate to maintain intestinal homeostasis in normal hosts [4]. Inflammatory immune cells help eliminate invading pathogens by highly effective redundant innate and adaptive immune mechanisms. Microbiota boosts the innate immune response against pathogens by stimulating secretion of antimicrobial peptides and cytokines such as TNFα, IL-22 and IL-17, and activating the inflammasome for anti-pathogen defense [2]. On the other hand, regulatory immune cells including regulatory T cells [12,13,14,15], B cells [16,17,18,19], dendritic cells [20, 21], macrophages [22] and innate lymphoid cells (ILCs) [23] counteract excessive inflammatory reactions [2, 4, 5, 24,25,26]. The frequency and functions of these regulatory cells are impaired in IBD, but can potentially be stimulated by microbial manipulation to restore immune homeostasis to reverse and normalize dysregulated immune function and ameliorate mucosal inflammation [12, 27,28,29]. Thus, targeted induction and maintenance of regulatory immune cells by manipulating microbial profiles and functions offer a promising approach to treat IBD patients.
In this review, we discuss the characteristics of the dysbiosis associated with IBD to identify potential treatment targets as well as recent progress regarding therapeutic induction of regulatory immune cells by resident bacteria and their products. Understanding the detailed mechanisms of dysbiosis will open new insights into the pathogenesis of IBD and uncover new strategies to normalize regulatory immune cell functions by manipulating the microbiota as more physiologic and effective treatment options for IBD patients.
Dysbiosis-associated mucosal immune-dysfunction in IBD
It is still unclear whether dysbiosis is a cause or consequence of inflammation in IBD patients [30]. Mucosal inflammation can directly alter microbial composition by increasing oxygen concentrations and metabolic changes that might expand colitogenic aerobic or facultative oxygen-tolerant anaerobic species [31, 32]. In addition, inflammation stimulates macrophages to produce nitrate oxide (NO) and increase the bacterial groups that can synthesize NO. Some colitogenic bacterial species utilize NO to regulate their membrane electron transport and protect from oxidative stress, which are potentially beneficial for their survival [33]. For example, parasite infection with Toxoplasma gondii induces mucosal inflammation and marked bacterial dysbiosis, which might be due to upregulated nitrate synthesis that serves as a source for anaerobic respiration and supports overgrowth of colitogenic Enterobacteriaceae [34]. Likewise, we demonstrated that the relative composition of a defined group of human IBD-relevant bacterial strains evolved as colitis progressed in selectively colonized gnotobiotic Il10−/− mice in contrast to stable profiles of the same strains in identically colonized wild-type mice [35]. In parallel, experimental colonic inflammation alters luminal bacterial gene expression that potentially transforms certain resident bacterial species into a more colitogenic phenotype that can sustain inflammation [36,37,38].
On the other hand, dysbiosis can drive inflammatory immune responses. Dysbiosis was present in a significant portion of CD patients in a pediatric inception cohort at the time diagnosis [39] and multiple genetic polymorphisms associated with IBD, such as NOD2, ATG16L1, CARD9, CLEC7A, HLAs and mucin-related genes, influence the microbiome and its function [40, 41]. Genetically dysfunctional PPAR-γ signaling or elimination of PPAR-γ-producing bacteria with antibiotics promote a dysbiotic expansion of potentially colitogenic bacteria by reducing the bioavailability of respiratory electron acceptors [42]. In addition, exposure of antibiotics to IBD mothers during pregnancy increase risk of very early-onset IBD in their children, suggesting that antibiotic-mediated dysbiosis enhances the risk of mucosal inflammation in susceptible hosts [43]. In mouse studies, transfer of bacteria strains associated with IBD induces intestinal inflammation in susceptible gnotobiotic mice [35, 44] and fecal transplants from IBD donors to germ-free mice stimulate increased numbers of Th17 cells and inflammatory mediators compared with transfer of feces from healthy donors, which enhance numbers of inducible Tregs [45, 46]. These observations indicate that dysbiosis can have a causative role in inducing inflammation. Taken together, dysbiosis and intestinal inflammation appear to influence each other and synergistically perpetuate chronic immune activation that mediates IBD [47].
Since aggressive Th1 and Th17 immune responses directed against dysbiotic resident microbiota have a central role in the pathogenesis of CD [5, 6, 48], correcting dysbiosis can potentially alleviate gut inflammation and be an attractive therapeutic strategy for IBD. Effective microbial therapy depends on identifying the specific dominant microbial drivers of pathogenic effector immune responses. Multiple cohort studies in IBD patients and gnotobiotic animal experiments have identified specific bacterial families and species that influence dysbiosis-mediated immune dysfunction. Segmented filamentous bacteria (SFB) strongly activate Th17 responses in the small intestine of mice [49, 50], although SFB induces non-inflammatory homeostatic Th17 cells rather than infection-induced inflammatory Th17 [51]. Adherent-invasive Escherichia coli (AIEC) and Citrobacter rodentium induce colitogenic Th1 and Th17 responses in the colon [50, 52, 53] and oral Klebsiella pneumoniae strains activate Th1 cells and colitis when they ectopically colonize the colon [54].
Moreover, dysbiosis alters microbial metabolites in the intestine. For example, hydrogen sulfide, a dietary metabolite, is a toxin associated with progression of mucosal inflammation in UC by blocking butyrate metabolism in colonic epithelial cells [55]. Defective detoxification capacity may be involved in the pathogenesis of UC [55]. Trimethylamine-N-oxide (TMAO) is generated by enteric anaerobes through the digestion of dietary carnitine and phosphatidylcholine [56]. Western diets (enriched in fat, phosphatidylcholine, and l-carnitine) potentially promote mucosal inflammation through TMAO induction [57]. Both hydrogen sulfide and TMAO are increased in the feces of IBD patients [55, 56]. In contrast, multiple bacterial metabolites exert protective activities that stimulate mucosal homeostasis. For example, the bacterial metabolites short-chain fatty acids (SCFA), primarily butyrate and propionate, are the primary nutrients for colonic epithelial cells and stimulate regulatory T cells (Tregs) [58]. These protective metabolites are consistently decreased in IBD patients with dysbiosis [11]. Bacteroides-derived sphingolipids have defined protective roles in mucosal inflammation and are decreased in IBD subjects, although host-derived sphingolipids were identified as the most differentially abundant metabolites in stool from IBD patients [59]. Bile acids are also bacterial metabolites with pleotropic functions, including the regulation of metabolism and inflammation through interactions with both microbial and host receptors [60]. Ursodeoxycholic acid, a hydrophilic secondary bile acid, ameliorates mouse experimental colitis by expanding anti-inflammatory cluster XIVa Clostridium and Akkermansia muciniphila [61]. Dysbiosis in enteric bacteria changes bile acid receptor FXR expression, which is protective for experimental colitis models through inhibiting NF-kB signaling [62]. Conjugated bile acids activate sphingosine 1-phosphage receptor 2-mediated protective pathways [63]. Other protective bacterial metabolites, including indoles are decreased during dysbiosis [11, 64, 65], which subsequently decrease homeostatic immune cell and mucosal barrier functions. Indole metabolites stimulate IL-22 production by LP ILCs that mediate epithelial protection through AhR [66, 67].
Collectively, dysbiosis markedly impacts both inflammatory and regulatory responses of the host’s immune system. However, it is still unclear what types, degree and duration of dysbiosis are necessary to cause dysregulated mucosal immunity and IBD development and if and how rapidly correction of this dysbiosis can normalize homeostatic processes in IBD patients with genetic defects in immunoregulation and epithelial barrier function. It will be necessary to address these questions and more fully understand the IBD-specific dysbiosis to develop into new diagnostic tools and therapeutic targets for IBD.
Resident bacteria activate regulatory immune cells and signaling pathways
The regulatory cytokine IL-10, together with TGF-β and IL-35, is a key mediator in microbe-mediated gut homeostasis [68] and plays a pivotal role in the pathogenesis of IBD [69]. Multiple genome-wide association studies showed that genetic polymorphisms in the IL-10 signaling pathway are associated with worsening phenotype of UC and early onset of CD [70,71,72]. However, subcutaneous supplementation of recombinant IL-10 protein once daily did not prevent postoperative recurrence in the CD patients and raised safety concerns [73,74,75]. This negative outcome might be due to an improper selection of the patients (especially those with high mucosal IL-10 level due to IL-10 signaling-related gene polymorphisms), or inappropriate dose, timing of treatment and delivery method of IL-10 [73]. Since IL-10 is a short-life cytokine and functions locally, a more physiological and safer strategy may be to stimulate regulatory immune cells to secret adequate IL-10 at the intestinal mucosal site. For example, replacing missing or decreased regulatory resident microbes [12] or genetically engineered IL-10-secreting bacteria [76, 77] may be effective strategies, because they do not require frequent administration of high dose of IL-10 in the circulation and should have low toxicity profiles.
Among the regulatory immune cells, regulatory T cells (Treg), consisting of thymus-derived Treg (tTreg) and inducible Treg (iTreg), have been most thoroughly studied in the pathogenesis of IBD [13,14,15]. iTreg are induced by normal resident bacteria but several studies showed that specific bacterial species are capable of inducing and maintaining iTreg. Human-derived 17 strains of Clostridium species induce Foxp3+CD4+Treg through IL-10, TGF-β1, butyrate and inducible T cell costimulatory (ICOS), and prevent mucosal inflammation in several murine colitis models [12, 78]. Polysaccharides from B. fragilis [79, 80] and protein components and supernatants of F. prausnitzii [81, 82] induce IL-10-producing mucosal Treg in mice. Recent studies show that a microbiota-activated iTreg population co-expressing RORγt+Foxp3+CD4+ (RORγt+Treg) possesses strong anti-inflammatory functions in the intestine [45, 83,84,85]. Normal resident bacteria as a whole are capable of inducing this population [19], but which bacterial strains or metabolites most efficiently activate RORγt+Tregs remain unknown. Britton et al. showed that transplant of feces from healthy human subjects induced higher concentrations of colonic lamina propria (LP) iTreg than did transfer of stools from IBD patients, which preferentially activated RORγt+ Th17 cells [45]. Bacterial metabolites can also induce Treg and intestinal homeostasis. Gut resident microbiota ferment dietary fiber to develop SCFAs that are essential in maintaining mucosal homeostasis. In particular, butyrate produced by the Firmicutes family members Ruminococcaceae, Lachnospiraceae, Erysipelotrichaceae or Clostridiaceae, activate and maintain intestinal Foxp3+ regulatory T cells. Treg induction by butyrate is mediated through activating G protein-coupled receptors (GPCRs), such as GPR41, GPR43, and GPR109A, and through inhibiting histone deacetylase synthesis [86, 87]. Another SCFA, propionate, also increases colonic Treg numbers by signaling via Ffar2 on Tregs and alleviates experimental colitis [58, 88]. Tryptophan is a major precursor of microbiota-derived AHR agonists such as various indoles that regulate chronic intestinal inflammation by promoting IL-10-producing T regulatory-1 (Tr1) cell induction and increasing the frequency of CD103+CD11b− regulatory DCs [64,65,66].
Regulatory B cells (Breg) exert a prominent role in mucosal homeostasis by directly inhibiting inflammatory responses through the regulatory cytokines IL-10, TGF-β and IL-35 that enhance Treg function and expansion, and inhibit effector APC function [16,17,18,19, 89, 90]. We have shown that IL-10-producing immunoregulatory Breg are induced by resident bacteria through the TLR2/MyD88, Akt and PI3Kinase signaling pathway [19]. Identifying the resident microbiota families and species that preferentially activate Bregs would be helpful to develop new therapeutic reagents for IBD management. Based on our studies, it is likely that different bacterial populations, bacterial components and metabolites activate Bregs and Tregs, although some overlap may exist. For example, TLRs 2 and 9 ligands activate IL-10 production by B cells but not T cells [19].
Dendritic cells (DCs) classically act as APC and CX3CR1intermediateCD70+CD11b+ DCs in mouse or CD14+CD163low DCs in humans can activate inflammatory Th17 cells [91, 92]. In contrast, DCs expressing CD103 have tolerogenic activities, promote iTreg differentiation, and inhibit intestinal Th1/Th17 immune responses by producing TGF-β, retinoic acid, AhR ligands and carbonic anhydrate I epitope peptide [26, 91, 93,94,95]. Resident bacteria also stimulate CX3CR1+CD103−CD11b+ DCs to augment the proliferation of Treg cells in an IFN-β and TLR4-dependent manner in mice [96].
Macrophages exhibit plasticity in their activation phenotype under different cytokine conditions [97, 98]. CD11chighCCR2+CX3CR1+ macrophages or M1 macrophages triggered by polarization signals from IFN-γ and microbial stimulation are proinflammatory [99]. Dysbiosis in IBD preferentially alters the dominant phenotype of intestinal LP macrophages into proinflammatory cells that exaggerate mucosal inflammation [100, 101]. Alternatively, CD11c−CCR2−CX3CR1− macrophages or M2 macrophages induced by IL-4, IL-13, IL-10 or TGF-β show an anti-inflammatory profile in experimental colitis and are decreased in the colons of patients with active IBD [26, 99,100,101]. M2 macrophages are preferentially induced by a mixture of probiotics (Vivomixx) containing four strains of Lactobacilli, three strains of Bifidobacteria, and Streptococcus thermophiles [102]. GPBAR1, the primary and secondary bile acid receptor, on macrophages regulates the M1/M2 phenotype and alleviates murine colitis [103].
ILCs are innate cells that are induced by bacterial stimulation, including indoles that serve as AhR ligands, and influence intestinal homeostasis [104, 105]. Regulatory ILCs, defined as Lin−CD45+CD127+IL-10+ ILCs that are mainly located in the small intestinal LP can suppress the activation of proinflammatory ILC1s and ILC3s and confer protection from innate intestinal inflammation through secreting IL-10 and TGF-β1 [23]. However, further studies will be required to determine which type of bacterial stimulation specifically induces regulatory ILCs.
Reversing dysbiosis as a nontoxic treatment of IBD
Recent progress in IBD treatments including the expansion of biological agents has provided rapid clinical remission and improved the quality of life in many IBD patients [106, 107]. However, those potent immunosuppressive therapies are not always effective, are quite expensive and potentially induce serious side effects [106, 107]. Recent reports reveal that the overall treatment efficacy, safety and cost-effectiveness of biologic therapies are not as striking as expected. For example, anti-TNF agents rapidly suppress inflammation and induce remission but did not change the long-term course in certain subsets of pediatric CD patients [108]. Murthy et al. showed that anti-TNF therapy did not reduce the frequency of hospitalization and surgical treatments in CD patients [109]. Therefore, more physiological approaches to induce and sustain remissions with limited toxicity and high cost-effectiveness are needed. Based on the current knowledge in the pathogenesis of IBD, manipulating the microbiota is considered as one of the rational treatment strategies for IBD [6, 28, 110, 111]. However, existing microbiota-targeting therapies including antibiotics, prebiotics, probiotics, and fecal microbial transplantation (FMT) demonstrate inconsistent results and the overall outcomes are not satisfactory in clinical practice [6, 28, 112].
Single antibiotic therapies provide modest effects to certain group of CD patients [113,114,115,116]. Oral metronidazole and ciprofloxacin are effective for anal lesions and delay of postoperative recurrence in CD [113, 115]. Rifaximin has a favorable safety profile because of the minimal systemic absorption, but does not yet have validated efficacy [114]. Broad spectrum antibiotics also can be effective in active IBD [113, 115, 117] and combination of antibiotics targeting Fusobacterium varium improved the outcome of UC patients [118]. Despite some favorable clinical effects, the long-term use of broad spectrum antibiotics potentially eliminates beneficial resident microbiota, induces antibiotic-resident species and creates a different type of dysbiosis by decreasing bacterial diversity [119].
Currently available probiotics potentially modulate dysbiosis in IBD, but their effects are transient and limited in most IBD subsets [6, 111, 112]. The possible reasons are: (1) single bacterial strains or combinations of traditional probiotics are not designed to replace the microbial species that are depleted in IBD patients and are unlikely to be effective given the broad heterogeneity in the microbial profile of individual IBD patients [9]. The microbiome profile of each IBD patient is different and defined IBD-specific dysbiosis is not present in all patients. (2) Most existing probiotics find colonization resistance in the host intestine, so that they do not colonize and are, therefore, present for a limited period, even after prolonged administration [120]. Baseline personalized host and mucosal microbial features are associated with probiotics persistence [121]. (3) The treatment timing is important to achieve the best effect of probiotics. (4) The proper delivery methods of live bacteria should be considered [122]. The most common species of current probiotics are Lactobacillus and Bifidobacterium, which have limited effects on IBD [112, 123]. Several clinical trials demonstrated that combination probiotics VSL#3 (containing 8 live bacterial species), E. coli Nissle, B. bifidum + L. acidophilus, Lactobacillus GG were effective in UC patients [112]. However, as mentioned above, the overall outcome of these probiotics is not fully satisfactory and new candidates for more effective colonizing probiotics that include combinations of protective resident strains (live biotherapeutic products, LBPs) are emerging [122].
Prebiotics, non-digestible carbohydrates that are metabolized by resident bacteria, can improve the composition and metabolic function of beneficial resident intestinal bacterial species [124]. Inulin, fructo-oligosaccharide, galacto-oligosaccharide, and lactulose are commonly used as prebiotics [124, 125]. Those prebiotics can increase the synthesis of SCFA, which improve barrier function, enhance regulatory immune responses and prevent pathobiont invasion by reducing pH levels in the intestine [125, 126]. In clinical practice, prebiotics can provide beneficial effect in IBD treatment, but their effects are modest with inconsistent results [28, 116, 125]. More mechanistic studies underlying the interactions among prebiotics, IBD-related dysbiosis and regulatory immune cells are required.
FMT is established as a standard treatment for recurrent Clostridium difficile infection, while the efficacy of FMT on IBD is still controversial [112, 127, 128]. Initial clinical studies show that FMT is effective at inducing remission in a small subset of UC patients with variable results in different studies; this variability might be due to different experimental designs including donor selection, delivery methods, pre-transplant preparation, frequency and timing of administration, as well as suitable controls. Recently, a randomized placebo-controlled trial demonstrated that multidonor intensive FMT with repeat administration 5 times/week for 6 weeks improved the efficacy in treating active UC [129]. Achieving remission by FMT was associated with increased microbial diversity with enrichment of Eubacterium hallii and Roseburia inulivorans, and increased levels of SCFA biosynthesis and secondary bile acids in the patients’ stool [130]. These types of deep analyses of microbial and host predictors of success vs failure will identify the characteristics of optimal donors and recipients to guide future application of this approach to treating IBD patients. However, importantly, FMT potentially can cause life-threatening side effects. The Food and Drug Administration (FDA) recently issued an alert after 2 patients died after FMT due to multi-antibiotics-resistant bacterial infection. These issues should be carefully considered in clinical practice.
The use of various combinations of specific intestinal-protective microbial strains or their metabolites may be safer and potentially more effective than whole FMT. This approach is more likely to achieve regulatory approval and be amenable to treating individual patients by matching replacement therapies in a rational targeted fashion based on the individual’s profile of fecal bacteria and metabolites (personalized therapy). The next generation of LBP or microbial products may include Faecalibacterium prausnitzii, which has a high capacity to induce IL-10-producing Treg [81, 82], Clostridium species (17 strains of clusters IV and XIVa, C. butyricum) [12, 78, 131] and Bacteroides species (thetaiotaomicron [132], uniformis [133], ovatus [134], and fragilis [79, 80]). Many other novel LBP formulations are being developed to replace protective bacterial species or homeostatic microbial products that are decreased in IBD patients.
Future treatment options in IBD
Due to the increasing demand of microbe-based therapeutics, multiple preclinical and clinical trials are currently underway or planned to find better treatment approach in IBD [111]. Several novel strategies that extend beyond traditional antimicrobial and LBP approaches are briefly mentioned. These include targeting specific pathobionts and modifying bacterial functions by genetic engineering or pharmacologic approaches. Strategies to directly modulate specific pathobionts include preventing AIEC mucosal attachment by blocking fimH [135], depletion of pathobionts with bacteriophages [136], CRISPER-CAS editing to produce specific bacteriocines [137] and replacing ecologic niches with competing commensals [6, 111]. Despite remaining safety and environmental concerns, genetically modified bacteria such as anti-TNF nanobody-producing Lactobacillus species [138, IL-35 producing E. coli [139], and IL-10-, IL-27-, HO-1-secreting Lactococcus species could more efficiently alleviate mucosal inflammation by promoting a homeostatic immunologic profile, including Treg induction [90, 111, 140]. Moreover, a 16-kDa protein of helminths produced in E. coli protects against DSS-induced colitis by inhibiting PPAR-α signaling [141] and the formulation of several strains with beneficial other additives have been proposed [111]. Precision editing of the gut microbiota by tungstate ameliorates experimental gut inflammation through preventing the dysbiotic expansion of Enterobacteriaceae [142]. Blocking intestinal bacterial enzymatic functions may also improve intestinal homeostasis and improve efficacy and decrease toxicity of conventional and developing IBD therapies, as have been accomplished for cancer therapies (Pharmacomicrobiomics) [143].
Fully understanding the interactions between microbiota and the host immune system, in concert with environmental and genetic factors unique to each individual, is necessary to target the most effective therapies for each patient. Personalized diagnostic profiles will require identifying an individual’s metabolic functions and dominant microbial antigens by shotgun metagenomic and metabolomic profiling, in concert with host microbial transcriptomic and genetic profiling. Microbiota reciprocally interacts with each other and the diet to provide immunological signals to host and the same microbe sometimes behaves differently in different individuals [1, 11, 144]. Host genetic and nutritional factors will need to be considered in an integrated and personalized manner to increase the effectiveness and efficacy of microbiota-based therapies [111, 145]. Selection of optimal approaches and therapeutic targets based on analysis of an individual’s microbiota pattern will be important to replace missing or dysfunctional bacterial components. We believe that a combined strategy to promote homeostatic immune responses, improve mucosal barrier function and restore eubiosis by targeting dominant pathobionts and replacing missing protective species or their functions by manipulating the bacterial microbiota and diet may be best. This integrated approach should provide a more physiologic, safer and more cost-effective means to sustained remission of IBD than the current lifelong treatments with immunosuppressives. It is our belief that this approach will be more effective as maintenance therapies once induction of disease remission has been accomplished by traditional therapies, but then toxic induction regimens can be withdrawn to decrease toxicity.
Conclusions and a path to improve personalized treatment
Human IBD includes genetically and clinically heterozygous patient subpopulations with very unique intestinal bacterial compositions and functions that help determine immune responses and disease outcomes. Therefore, we believe that it will be feasible to evaluate the microbe/immune profiles by rapid diagnostic tests of microbiota functional and mucosal immune profiles to direct highly effective and safe treatments in a personalized manner (Fig. 2). Restoring impaired regulatory immune cell activity by correcting dysbiosis and defective microbial metabolic functions is a novel and highly promising therapeutic approach to managing IBD in a more physiologic, safer and sustained manner. Unveiling the mechanisms underlying specific defective bacteria–host interactions in each IBD patient will enable precision editing of microbiota and their function with maximum effectiveness and efficiency.

Current and proposed treatment strategies in microbe-based treatment for IBD. Currently, we diagnose and treat IBD patients based on clinical parameters including fecal calprotectin, serum CRP level, disease activity index (DAI) and endoscopic findings. These clinical observations do not provide insight into the degree of mucosal dysbiosis or impaired regulatory immune response in IBD patients. Therefore, empiric microbe-based therapies are used, such as existing probiotics, prebiotics, antibiotics, fecal microbial transplantation in addition to standard of care anti-TNF agents or immunomodulators (IM). Since these empiric treatments have a limited efficacy in current clinical practice, we propose a more rational and scientific approach based on the fecal microbial and mucosal immune profiles in each IBD patient determined by rapid diagnosis tests. These fecal metabolic profiles and mucosal immune cytokine expression levels allow us to provide more effective and lower toxic microbe-based treatments based on various combinations of protective bacterial strains (LBP live biotherapeutic products) that are then applied in a customized way to restore microbial homeostasis based on dysbiosis in an individual patient. This approach can potentially provide cost-effective, nontoxic treatment and higher quality of life for IBD patients. HC healthy control, Pt IBD patient
References
Rothschild D, Weissbrod O, Barkan E, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–5.
Cheng H-Y, Ning M-X, Chen D-K, et al. Interactions between the gut microbiota and the host innate immune response against pathogens. Front Immunol. 2019;10:607.
Dominguez-Bello MG, Godoy-Vitorino F, Knight R, et al. Role of the microbiome in human development. Gut. 2019;68:1108–14.
Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–41.
Nagao-Kitamoto H, Kamada N. Host-microbial cross-talk in Inflammatory Bowel Disease. Immun Netw. 2017;17:1.
Sartor RB, Wu GD. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology. 2017;152:327–339.e4.
Harris KG, Chang EB. The intestinal microbiota in the pathogenesis of inflammatory bowel diseases: new insights into complex disease. Clin Sci (Lond.). 2018;132:2013–28.
Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–99.
Integrative HMP. (iHMP) Research network consortium. The integrative human microbiome project. Nature. 2019;569:641–8.
Kriss M, Hazleton KZ, Nusbacher NM, et al. Low diversity gut microbiota dysbiosis: drivers, functional implications and recovery. Curr Opin Microbiol. 2018;44:34–40.
Lloyd-Price J, Arze C, Ananthakrishnan AN, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–62.
Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–6.
Roers A, Siewe L, Strittmatter E, et al. T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J Exp Med. 2004;200:1289–97.
Groux H, Powrie F. Regulatory T cells and inflammatory bowel disease. Immunol Today. 1999;20:442–6.
Brockmann L, Soukou S, Steglich B, et al. Molecular and functional heterogeneity of IL-10-producing CD4 + T cells. Nat Commun. 2018;9:5457.
Mishima Y, Liu B, Hansen JJ, et al. Resident bacteria-stimulated IL-10-secreting B cells ameliorate T cell-mediated colitis by inducing Tr-1 cells that require IL-27-signaling. Cell Mol Gastroenterol Hepatol. 2015;1:295–310.
Mishima Y, Ishihara S, Aziz MM, et al. Decreased production of interleukin-10 and transforming growth factor-β in Toll-like receptor-activated intestinal B cells in SAMP1/Yit mice. Immunology. 2010;131:473–87.
Mizoguchi A, Mizoguchi E, Takedatsu H, et al. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 2002;16:219–30.
Mishima Y, Oka A, Liu B, et al. Microbiota maintain colonic homeostasis by activating TLR2/MyD88/PI3 K signaling in IL-10-producing regulatory B cells. J Clin Invest. 2019;130:pii: 93820.
Liu B, Tonkonogy SL, Sartor RB. Antigen-presenting cell production of IL-10 inhibits T-helper 1 and 17 cell responses and suppresses colitis in mice. Gastroenterology. 2011;141(653–62):662.e1–4.
Rutella S, Locatelli F. Intestinal dendritic cells in the pathogenesis of inflammatory bowel disease. World J Gastroenterol. 2011;17:3761–75.
Shouval DS, Biswas A, Goettel JA, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–19.
Wang S, Xia P, Chen Y, et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell. 2017;171(201–216):e18.
Giuffrida P, Cococcia S, Delliponti M, et al. Controlling gut inflammation by restoring anti-inflammatory pathways in inflammatory Bowel disease. Cells. 2019;8:397.
Onali S, Favale A, Fantini MC. The resolution of intestinal inflammation: the Peace–Keeper’s Perspective. Cells. 2019;8:344.
Sun M, He C, Cong Y, Liu Z. Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol. 2015;8:969–78.
Ciullini Mannurita S, Gambineri E. Novel molecular defects associated with very early-onset inflammatory bowel. Curr Opin Allergy Clin Immunol. 2017;17:317–24.
Knox NC, Forbes JD, Van Domselaar G, et al. The gut microbiome as a target for ibd treatment: are we there yet? Curr Treat Options Gastroenterol. 2019;17:115–26.
Nishida A, Inoue R, Inatomi O, et al. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol. 2018;11:1–10.
Ni J, Wu GD, Albenberg L, et al. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14:573–84.
Lewis JD, Chen EZ, Baldassano RN, et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric crohn’s disease. Cell Host Microbe. 2015;18:489–500.
Albenberg L, Esipova TV, Judge CP, et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology. 2014;147(1055–63):e8.
Fang FC, Vázquez-Torres A. Reactive nitrogen species in host-bacterial interactions. Curr Opin Immunol. 2019;60:96–102.
Wang S, El-Fahmawi A, Christian DA, et al. Infection-induced intestinal dysbiosis is mediated by macrophage activation and nitrate production. MBio. 2019;10:pii: e00935-19.
Eun CS, Mishima Y, Wohlgemuth S, et al. Induction of bacterial antigen-specific colitis by a simplified human microbiota consortium in gnotobiotic interleukin-10-/- mice. Infect Immun. 2014;82:2239–46.
Hansen JJ, Huang Y, Peterson DA, et al. The colitis-associated transcriptional profile of commensal Bacteroides thetaiotaomicron enhances adaptive immune responses to a bacterial antigen. PLoS One. 2012;7:e42645.
Hansen JJ. Immune responses to intestinal microbes in inflammatory Bowel diseases. Curr Allergy Asthma Rep. 2015;15:61.
Lengfelder I, Sava IG, Hansen JJ, et al. Complex bacterial consortia reprogram the colitogenic activity of enterococcus faecalis in a gnotobiotic mouse model of chronic. Immun Med Colitis Front Immunol. 2019;10:1420.
Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–92.
Knights D, Silverberg MS, Weersma RK, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med. 2014;6:107.
Lamas B, Richard ML, Leducq V, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med. 2016;22:598–605.
Byndloss MX, Olsan EE, Rivera-Chávez F, et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science. 2017;357:570–5.
Örtqvist AK, Lundholm C, Halfvarson J, et al. Fetal and early life antibiotics exposure and very early onset inflammatory bowel disease: a population-based study. Gut. 2019;68:218–25.
Schaubeck M, Clavel T, Calasan J, et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut. 2016;65:225–37.
Britton GJ, Contijoch EJ, Mogno I, et al. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORγt + Regulatory T Cells and Exacerbate Colitis in Mice. Immunity. 2019;50(212–224):e4.
Nagao-Kitamoto H, Shreiner AB, Gillilland MG, et al. Functional characterization of inflammatory bowel disease-associated gut dysbiosis in gnotobiotic mice. Cell Mol Gastroenterol Hepatol. 2016;2:468–81.
Round JL, Palm NW. Causal effects of the microbiota on immune-mediated diseases. Sci Immunol. 2018;3:eaao1603.
Ahluwalia B, Moraes L, Magnusson MK, et al. Immunopathogenesis of inflammatory bowel disease and mechanisms of biological therapies. Scand J Gastroenterol. 2018;53:379–89.
Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–98.
Atarashi K, Tanoue T, Ando M, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell. 2015;163:367–80.
Omenetti S, Bussi C, Metidji A, et al. The intestine harbors functionally distinct homeostatic tissue-resident and inflammatory Th17 Cells. Immunity. 2019;51:77–89.e6.
Viladomiu M, Kivolowitz C, Abdulhamid A, et al. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci Transl Med. 2017;9:pii: eaaf9655.
Kim SC, Tonkonogy SL, Albright CA, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906.
Atarashi K, Suda W, Luo C, et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science. 2017;358:359–65.
Teigen LM, Geng Z, Sadowsky MJ, et al. Dietary factors in sulfur metabolism and pathogenesis of ulcerative colitis. Nutrients. 2019;11:931.
Wilson A, Teft WA, Morse BL, et al. Trimethylamine-N-oxide: a novel biomarker for the identification of inflammatory bowel disease. Dig Dis Sci. 2015;60:3620–30.
Tilg H, Moschen AR. Food, immunity, and the microbiome. Gastroenterology. 2015;148:1107–19.
Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–73.
Brown EM, Ke X, Hitchcock D, et al. Bacteroides-derived sphingolipids are critical for maintaining intestinal homeostasis and symbiosis. Cell Host Microbe. 2019;25(668–680):e7.
Chen ML, Takeda K, Sundrud MS. Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunol. 2019;12:851–61.
Van den Bossche L, Hindryckx P, Devisscher L, et al. Ursodeoxycholic acid and its taurine- or glycine-conjugated species reduce colitogenic dysbiosis and equally suppress experimental colitis in mice. Appl Environ Microbiol Am Soc Microbiol. 2017;83:e02766-16.
Ding L, Yang L, Wang Z, et al. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm Sin B. 2015;5:135–44.
Nagahashi M, Takabe K, Liu R, et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology. 2015;61:1216–26.
Aoki R, Aoki-Yoshida A, Suzuki C, et al. Indole-3-pyruvic acid, an aryl hydrocarbon receptor activator, suppresses experimental colitis in mice. J Immunol. 2018;201:3683–93.
Naganuma M, Sugimoto S, Mitsuyama K, et al. Efficacy of indigo naturalis in a multicenter randomized controlled trial of patients with ulcerative colitis. Gastroenterology. 2018;154:935–47.
Zelante T, Iannitti RG, Cunha C, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39:372–85.
Monteleone I, Rizzo A, Sarra M, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141(237–48):248.e1.
Levast B, Li Z, Madrenas J. The role of IL-10 in microbiome-associated immune modulation and disease tolerance. Cytokine. 2015;75:291–301.
Shouval DS, Ouahed J, Biswas A, et al. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv Immunol. 2014;122:177–210.
Franke A, Balschun T, Karlsen TH, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–23.
Amre DK, Mack DR, Morgan K, et al. Interleukin 10 (IL-10) gene variants and susceptibility for paediatric onset Crohn’s disease. Aliment Pharmacol Ther. 2009;29:1025–31.
Glocker EEO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–45.
Marlow GJ, van Gent D, Ferguson LR. Why interleukin-10 supplementation does not work in Crohn’s disease patients. World J Gastroenterol. 2013;19:3931–41.
Schreiber S, Fedorak RN, Nielsen OH, et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Gastroenterology. 2000;119:1461–72.
Colombel J-F, Rutgeerts P, Malchow H, et al. Interleukin 10 (Tenovil) in the prevention of postoperative recurrence of Crohn’s disease. Gut. 2001;49:42–6.
Steidler L, Hans W, Schotte L, et al. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science. 2000;289:1352–5.
Braat H, Rottiers P, Hommes DW, et al. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin Gastroenterol Hepatol. 2006;4:754–9.
Atarashi K, Tanoue T, Shima T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–41.
Round JL, Lee SM, Li J, et al. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–7.
Furusawa Y, Obata Y, Hase K. Commensal microbiota regulates T cell fate decision in the gut. Semin Immunopathol. 2015;37:17–25.
Quévrain E, Maubert MA, Michon C, et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut. 2016;65:415–25.
Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci. 2008;105:16731–6.
Yang B-H, Hagemann S, Mamareli P, et al. Foxp3(+) T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal. Immunol. 2016;9:444–57.
Lochner M, Peduto L, Cherrier M, et al. In vivo equilibrium of proinflammatory IL-17 + and regulatory IL-10 + Foxp3 + RORgamma t + T cells. J Exp Med. 2008;205:1381–93.
Sefik E, Geva-Zatorsky N, Oh S, et al. Mucosal immunology. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science. 2015;349:993–7.
Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–50.
Parada Venegas D, De la Fuente MK, Landskron G, et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019;10:277.
Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19:29–41.
Ray A, Wang L, Dittel BN. IL-10-independent regulatory B-cell subsets and mechanisms of action. Int Immunol. 2015;27:531–6.
Hanson ML, Hixon JA, Li W, et al. Oral delivery of IL-27 recombinant bacteria attenuates immune colitis in mice. Gastroenterology. 2014;1:210–221.e13.
Sichien D, Lambrecht BN, Guilliams M, et al. Development of conventional dendritic cells: from common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal. Immunol. 2017;10:831–44.
Kayama H, Nishimura J, Takeda K. Regulation of intestinal homeostasis by innate immune cells. Immun Netw. 2013;13:227–34.
Yagi S, Abe M, Yamashita M, et al. Carbonic anhydrate I epitope peptide improves inflammation in a murine model of inflammatory Bowel disease. Inflamm Bowel Dis. 2016;22:1835–46.
Scott CL, Aumeunier AM, Mowat AM. Intestinal CD103 + dendritic cells: master regulators of tolerance? Trends Immunol. 2011;32:412–9.
Bain CC, Montgomery J, Scott CL, et al. TGFβR signalling controls CD103 + CD11b + dendritic cell development in the intestine. Nat Commun. 2017;8:620.
Nakahashi-Oda C, Udayanga KGS, Nakamura Y, et al. Apoptotic epithelial cells control the abundance of Treg cells at barrier surfaces. Nat Immunol. 2016;17:441–50.
Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–55.
Mowat AM, Scott CL, Bain CC. Barrier-tissue macrophages: functional adaptation to environmental challenges. Nat Med. 2017;23:1258–70.
Orecchioni M, Ghosheh Y, Pramod AB, et al. Macrophage polarization: different gene signatures in M1(LPS +) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol. 2019;10:1084.
Na YR, Stakenborg M, Seok SH, et al. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019;16:531–43.
Bernardo D, Marin AC, Fernández-Tomé S, et al. Human intestinal pro-inflammatory CD11chighCCR101 + CX3CR101 + macrophages, but not their tolerogenic CD11c-CCR101-CX3CR101-ounterparts, are expanded in inflammatory bowel disease. Mucosal Immunol. 2018;11:1114–26.
Biagioli M, Capobianco D, Carino A, et al. Divergent effectiveness of multispecies probiotic preparations on intestinal microbiota structure depends on metabolic properties. Nutrients. 2019;11:pii: E325.
Biagioli M, Carino A, Cipriani S, et al. The bile acid receptor GPBAR1 regulates the M1/M2 phenotype of intestinal macrophages and activation of GPBAR1 rescues mice from murine colitis. J Immunol. 2017;199:718–33.
Vivier E, Artis D, Colonna M, et al. Innate lymphoid cells: 10 years On. Cell. 2018;174:1054–66.
Klose CSN, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. 2016;17:765–74.
Ford AC, Sandborn WJ, Khan KJ, et al. Efficacy of biological therapies in inflammatory bowel disease: systematic review and meta-analysis. Am J Gastroenterol. 2011;106:644–59.
Abraham C, Dulai PS, Vermeire S, et al. Lessons learned from trials targeting cytokine pathways in patients with inflammatory bowel diseases. Gastroenterology. 2017;152:374–388.e4.
Kugathasan S, Denson LA, Walters TD, et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. Lancet (Lond Engl.). 2017;389:1710–8.
Murthy SK, Begum J, Benchimol EI, et al. Introduction of anti-TNF therapy has not yielded expected declines in hospitalisation and intestinal resection rates in inflammatory bowel diseases: a population-based interrupted time series study. Gut. 2019.https://doi.org/10.1136/gutjnl-2019-318440.
Sartor RB. Therapeutic correction of bacterial dysbiosis discovered by molecular techniques. Proc Natl Acad Sci USA. 2008;105:16413–4.
Cohen LJ, Cho JH, Gevers D, et al. Genetic factors and the intestinal microbiome guide development of microbe-based therapies for inflammatory bowel diseases. Gastroenterology. 2019;156:2174–89.
Basso PJ, Câmara NOS, Sales-Campos H. Microbial-based therapies in the treatment of inflammatory bowel disease—an overview of human studies. Front Pharmacol. 2018;9:1571.
Khan KJ, Ullman TA, Ford AC, et al. Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Am J Gastroenterol. 2011;106:661–73.
Sartor RB. Review article: the potential mechanisms of action of rifaximin in the management of inflammatory bowel diseases. Aliment Pharmacol Ther. 2016;43(Suppl 1):27–36.
Nitzan O, Elias M, Peretz A, et al. Role of antibiotics for treatment of inflammatory bowel disease. World J Gastroenterol. 2016;22:1078–87.
Zuo T, Ng SC. The Gut microbiota in the pathogenesis and therapeutics of inflammatory bowel disease. Front Microbiol. 2018;9:2247.
Turner D, Levine A, Kolho K-L, et al. Combination of oral antibiotics may be effective in severe pediatric ulcerative colitis: a preliminary report. J Crohns Colitis. 2014;8:1464–70.
Ohkusa T, Kato K, Terao S, et al. Newly developed antibiotic combination therapy for ulcerative colitis: a double-blind placebo-controlled multicenter trial. Am J Gastroenterol. 2010;105:1820–9.
Vangay P, Ward T, Gerber JS, et al. Antibiotics, pediatric dysbiosis, and disease. Cell Host Microbe. 2015;17:553–64.
Gionchetti P, Rizzello F, Venturi A, et al. Oral bacteriotherapy as maintenance treatment in patients with chronic pouchitis: a double-blind, placebo-controlled trial. Gastroenterology. 2000;119:305–9.
Zmora N, Zilberman-Schapira G, Suez J, et al. Personalized Gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell. 2018;174(1388–1405):e21.
O’Toole PW, Marchesi JR, Hill C. Next-generation probiotics: the spectrum from probiotics to live biotherapeutics. Nat Microbiol. 2017;2:17057.
Sartor RB. Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: antibiotics, probiotics, and prebiotics. Gastroenterology. 2004;126:1620–33.
Gibson GR, Hutkins R, Sanders ME, et al. Expert consensus document: the International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat Rev Gastroenterol Hepatol. 2017;14:491–502.
Eom T, Kim YS, Choi CH, et al. Current understanding of microbiota- and dietary-therapies for treating inflammatory bowel disease. J Microbiol. 2018;56:189–98.
Shokryazdan P, Faseleh Jahromi M, Navidshad B, et al. Effects of prebiotics on immune system and cytokine expression. Med Microbiol Immunol. 2017;206:1–9.
Kassam Z, Lee CH, Yuan Y, et al. Fecal Microbiota Transplantation for Clostridium difficile Infection: systematic review and meta-analysis. Am J Gastroenterol. 2013;108:500–8.
Vaughn BP, Rank KM, Khoruts A. Fecal Microbiota Transplantation: current status in treatment of GI and liver disease. Clin Gastroenterol Hepatol. 2019;17:353–61.
Paramsothy S, Kamm MA, Kaakoush NO, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet (Lond Engl.). 2017;389:1218–28.
Paramsothy S, Nielsen S, Kamm MA, et al. Specific bacteria and metabolites associated with response to fecal microbiota transplantation in patients with ulcerative colitis. Gastroenterology. 2019;156:1440–1454.e2.
Kanai T, Mikami Y, Hayashi A. A breakthrough in probiotics: clostridium butyricum regulates gut homeostasis and anti-inflammatory response in inflammatory bowel disease. J Gastroenterol. 2015;50:928–39.
Delday M, Mulder I, Logan ET, et al. Bacteroides thetaiotaomicron Ameliorates Colon Inflammation in preclinical models of Crohn’s disease. Inflamm Bowel Dis. 2019;25:85–96.
Takahashi K, Nishida A, Fujimoto T, et al. Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in Crohn’s disease. Digestion. 2016;93:59–65.
Ihekweazu FD, Fofanova TY, Queliza K, et al. Bacteroides ovatus ATCC 8483 monotherapy is superior to traditional fecal transplant and multi-strain bacteriotherapy in a murine colitis model. Gut Microb. 2019;10:504–20.
Sivignon A, Bouckaert J, Bernard J, et al. The potential of FimH as a novel therapeutic target for the treatment of Crohn’s disease. Expert Opin Ther Targets. 2017;21:837–47.
Galtier M, De Sordi L, Sivignon A, et al. Bacteriophages targeting adherent invasive Escherichia coli strains as a promising new treatment for Crohn’s disease. J. Crohn’s Colitis. 2017;11:840–7.
Bikard D, Euler CW, Jiang W, et al. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol. 2014;32:1146–50.
Vandenbroucke K, de Haard H, Beirnaert E, et al. Orally administered L. lactis secreting an anti-TNF Nanobody demonstrate efficacy in chronic colitis. Mucosal Immunol. 2010;3:49–56.
Zhang B, Liu Y, Lan X, et al. Oral Escherichia coli expressing IL-35 meliorates experimental colitis in mice. J Transl Med. 2018;16:71.
Shigemori S, Shimosato T. Applications of genetically modified immunobiotics with high immunoregulatory capacity for treatment of inflammatory Bowel diseases. Front Immunol. 2017;8:22.
Wang L, Xie H, Xu L, et al. rSj16 protects against DSS-induced colitis by inhibiting the PPAR-α signaling pathway. Theranostics. 2017;7:3446–60.
Zhu W, Winter MG, Byndloss MX, et al. Precision editing of the gut microbiota ameliorates colitis. Nature. 2018;553:208–11.
Wallace BD, Wang H, Lane KT, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831–5.
Gilbert JA, Blaser MJ, Caporaso JG, et al. Current understanding of the human microbiome. Nat Med. 2018;24:392–400.
Zmora N, Suez J, Elinav E. You are what you eat: diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol. 2019;16:35–56.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Mishima has no financial conflicts of interest to declare. Dr. Sartor receives research preclinical grant support from Janssen, Gusto Global, Vedanta, Seres and BiomX and is on Advisory Boards for Danone/Yakult, Second Genome, BiomX, Biomica and Qu Pharmaceuticals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mishima, Y., Sartor, R.B. Manipulating resident microbiota to enhance regulatory immune function to treat inflammatory bowel diseases. J Gastroenterol 55, 4–14 (2020). https://doi.org/10.1007/s00535-019-01618-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-019-01618-1