
Abstract
Background
Nephrocalcinosis (NC) is characterized by an excessive accumulation of calcium deposits in the kidneys. In children, it is often incidentally discovered with an uncertain prognosis.
Case-diagnosis/treatment
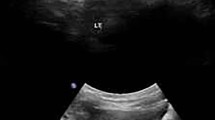
A 3-month-old girl suspected to have a milk protein allergy underwent an ultrasound that revealed increased echogenicity in the kidney pyramids suggestive of medullary NC. At the age of 18 months, imaging findings revealed not only hyperechogenicity in the medulla but also in the cortex. Over the course of a long follow-up, her kidneys maintained size within the upper limits but showed an increase by age 7. Genetic analysis identified PKHD1 variants, which required structural predictive tools to guide clinical diagnosis. Until the age of 7, her kidney function has remained intact; however, her prognosis is uncertain.
Conclusions
NC in newborns is a rare condition, but its incidence is rising. Recurrent urinary infections or kidney stones may lead to kidney failure. A proactive approach in sporadic NC enables an early diagnosis to orientate clinical supervision and facilitates counseling to support family planning decisions.

Similar content being viewed by others


Data availability
The data supporting the findings of this clinical case are available upon justified request. For inquiries regarding more information, interested parties may contact Paola Krall at paola.krall@uchile.cl. We are committed to transparency and facilitating further research endeavors.
References
Rodriguez Cuellar CI, Wang PZT, Freundlich M, Filler G (2020) Educational review: role of the pediatric nephrologists in the work-up and management of kidney stones. Pediatr Nephrol 35:383–397. https://doi.org/10.1007/s00467-018-4179-9
Letavernier E, Schwoehrer M, Livrozet M, Saint-Jacques C, Raymond L, Saraeva R, Haymann JP, Frochot V, Daudon M, Mesnard L (2021) Atypical clinical presentation of autosomal recessive polycystic kidney mimicking medullary sponge kidney disease. Kidney Int Rep 7:916–919. https://doi.org/10.1016/j.ekir.2021.11.035
Gunay-Aygun M, Turkbey BI, Bryant J, Daryanani KT, Gerstein MT, Piwnica-Worms K, Choyke P, Heller T, Gahl WA (2011) Hepatorenal findings in obligate heterozygotes for autosomal recessive polycystic kidney disease. Mol Genet Metab 104:677–681. https://doi.org/10.1016/j.ymgme.2011.09.001
Shan D, Rezonzew G, Mullen S, Roye R, Zhou J, Chumley P, Revell DZ, Challa A, Kim H, Lockhart ME, Schoeb TR, Croyle MJ, Kesterson RA, Yoder BK, Guay-Woodford LM, Mrug M (2019) Heterozygous Pkhd1C642* mice develop cystic liver disease and proximal tubule ectasia that mimics radiographic signs of medullary sponge kidney. Am J Physiol Renal Physiol 316:F463–F472. https://doi.org/10.1152/ajprenal.00181.2018
Cheungpasitporn W, Thongprayoon C, Brabec BA, Kittanamongkolchai W, Erickson SB (2016) Outcomes of living kidney donors with medullary sponge kidney. Clin Kidney J 9:866–870. https://doi.org/10.1093/ckj/sfv107
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Consent for publication
The patient’s legal guardians, represented by her parents, have provided consent to publish the clinical and genetic data.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Krall, P., Faundes, V., Gálvez, C. et al. A case of diffuse kidney hyperechogenicity in early childhood associated with biallelic PKHD1 variants. Pediatr Nephrol (2024). https://doi.org/10.1007/s00467-024-06348-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00467-024-06348-y