
Abstract
Rickets is a disease of the growing child arising from alterations in calcium and phosphate homeostasis resulting in impaired apoptosis of hypertrophic chondrocytes in the growth plate. Its symptoms depend on the patients’ age, duration of disease, and underlying disorder. Common features include thickened wrists and ankles due to widened metaphyses, growth failure, bone pain, muscle weakness, waddling gait, and leg bowing. Affected infants often show delayed closure of the fontanelles, frontal bossing, and craniotabes. The diagnosis of rickets is based on the presence of these typical clinical symptoms and radiological findings on X-rays of the wrist or knee, showing metaphyseal fraying and widening of growth plates, in conjunction with elevated serum levels of alkaline phosphatase. Nutritional rickets due to vitamin D deficiency and/or dietary calcium deficiency is the most common cause of rickets. Currently, more than 20 acquired or hereditary causes of rickets are known. The latter are due to mutations in genes involved in vitamin D metabolism or action, renal phosphate reabsorption, or synthesis, or degradation of the phosphaturic hormone fibroblast growth factor 23 (FGF23). There is a substantial overlap in the clinical features between the various entities, requiring a thorough workup using biochemical analyses and, if necessary, genetic tests. Part I of this review focuses on the etiology, pathophysiology and clinical findings of rickets followed by the presentation of a diagnostic approach for correct diagnosis. Part II focuses on the management of rickets, including new therapeutic approaches based on recent clinical practice guidelines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
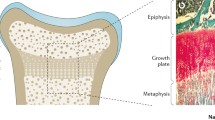
Rickets is a heterogeneous group of acquired and inherited diseases resulting in disturbances in calcium and/or phosphate heomeostasis and, thereby, affecting the growing skeleton. Rickets is characterized by impaired apoptosis of hypertrophic chondrocytes, resulting in widening of the growth plates in bones and is usually associated with osteomalacia (Fig. 1) [1]. The latter is characterized by an inadequate mineralization of the organic component of the bone matrix—the osteoid—by calcium salts, resulting in soft bone. Historically, rickets was classified as calcipenic or phosphopenic rickets. The latter is also called hypophosphatemic rickets. However, there is growing evidence that the ultimate cause of rickets is an insufficient availability of phosphate required for terminal differentiation and mineralization of growth plate chondrocytes [1,2,3,4].

reproduced from Carpenter et al. with permission [1]
Morphology of the growth plate in rickets. (a,b) Morphology of a healthy, human growth plate (physis). The growth plate is characterized by maturation of chondrocytes (cartilage cells) occurring progressively from the epiphysis to the metaphysis. The border between the metaphysis and the growth plate is marked by a provisional zone of calcification of the cartilage matrix (red–pink staining) which undergoes resorption and replacement with mineralized bone (turquoise staining). (c) A rachitic growth plate showing a marked increase in longitudinal width, marked by the persistence of the zone of hypertrophic chondrocytes with lost columnar arrangement. The growth plate abnormalities are the consequence of impaired chondrocyte apoptosis and impaired mineralization of the cartilage matrix surrounding the apoptotic chondrocytes. Apoptosis of hypertrophic chondrocytes is induced by extracellular phosphate via phosphorylation of mitogen-activated protein kinase (MAPK) pathway intermediates and downstream inhibition of the caspase-9-dependent mitochondrial apoptotic pathway. Thus, reduction in ambient phosphate availability to the chondrocyte, which is common to all forms of rickets, seems to be central to the impaired apoptosis. The ligand 1,25-dihydroxyvitamin D and its receptor may also be involved. Finally, expansion of the hypertrophic chondrocyte zone can be induced by impaired vascularization, influenced by vascular endothelial growth factor, which is regulated by MAPK pathway intermediates. Figure
Clinical presentation depends on age at onset, duration of disease, and underlying pathophysiology. Common features are thickened wrists and ankles due to widened metaphyses, growth failure, bone pain, muscle weakness, waddling gait, and leg bowing. Severe rickets in infancy may also include delayed closure of the fontanelles, parietal and frontal bossing and craniotabes (soft skull bones). Additional features may be present depending on the underlying disease, e.g., symptomatic hypocalcemia (seizures or tetany) in nutritional rickets—the most common cause of calcipenic rickets—and craniosynostosis, dental abscesses, and hearing loss in X-linked hypophosphatemia (XLH)—the most common cause of phosphopenic rickets. Impaired mineralization of the growth plate can be confirmed by typical radiography of the wrist or knee, showing metaphyseal fraying and widening of growth plates, in conjunction with elevated serum levels of the osteoblast marker alkaline phosphatase (ALP) (Figs. 2 and 3) [5, 6].

reproduced from Schnabel and Haffner with permission [109]
Clinical features of calcipenic rickets. (a) 18-month-old girl presenting with genu vara, and widening of the growth plates and metaphyseal fraying on X-rays, caused by nutritional rickets. (b, c) Two infants with widening of the wrist and rachitic rosary, respectively, due to nutritional rickets. (d) 14-year-old boy with genu valga due to nutritional rickets. (e) Infant with alopecia due to vitamin D-dependent rickets type 2A. Figure 2a, b, and e are

reproduced with permission from Haffner and Linglart [110]
Clinical features of phosphopenic rickets. (a) 2-year-old boy diagnosed with X-linked hypophosphatemia (XLH) at the age of 2 years, presenting with disproportionate short stature (–2.3 SD score), genu vara, and widening of growth plates and metaphyseal fraying on X-rays. (b) 3-year-old patient with XLH started on treatment with active vitamin D and phosphate at the age of 2 years showing disproportionate short stature (height, –2.4 SD score), frontal bossing, dolichocephalus and mild signs of rickets on X-ray. (c) Dental abscess on an apparently healthy tooth in a child with XLH. (d) 16-year-old boy with autosomal-recessive hypophosphatemic rickets type 2 (ARHR2) showing genu vara and mild ricketic signs on X-ray. Figure 3c
The discovery of new calcium and phosphate metabolism regulators, such as the phosphaturic hormone fibroblast growth factor 23 (FGF23), together with the elucidation of the underlying genetic defects in many hereditary forms of rickets and the availability of comprehensive genetic testing has improved our understanding of the underlying pathophysiology and revolutionized its diagnosis. Currently, more than 20 inherited and acquired causes of rickets are known, which show a considerable overlap in their clinical findings (Table 1) [1, 7]. Wrong or delayed diagnosis may result in inappropriate treatment of rickets and poor patient outcome. The diagnostic approach to rickets is primarily based on medical history, biochemical tests and radiography, which is followed, if necessary, by genetic tests. The latter is of especial importance in patients suspected to have a genetic form of rickets but no family history of the disease. In addition, targeted therapy has become available for some hereditary forms of rickets, such as XLH which requires correct diagnosis before treatment [8].
In part I of this review, we give an overview on the etiology, pathophysiology and clinical findings of rickets, followed by a diagnostic approach and discussion of important pitfalls in finding the correct diagnosis. Part II focuses on the treatment of rickets, including new therapeutic approaches based on recent clinical practice guidelines.
Calcium and phosphate homeostasis
Mineral homeostasis is maintained by complex interactions between organ systems—primarily the skeleton, intestine and the kidney. It is regulated by several hormones, including calciotropic and phosphate-regulating hormones, e.g., parathyroid hormone (PTH), vitamin D metabolites and FGF23 (Fig. 4). Adequate intake of calcium and phosphate is mandatory in order to maintain bone integrity and allow normal skeletal growth in children. Most of the calcium (> 99%) is stored in the skeleton, and less than 0.5% circulates in the blood, with approximately 50% being bound to albumin or globulin. The primary sources of dietary calcium in children are milk products. Typically, about 35% of dietary calcium is absorbed in the small intestine via transepithelial transport through the apical membrane calcium channel TRPV6, active extrusion across the basolateral membrane by PMCA1b and/or other mechanisms. Thereafter, calcium enters the extracellular fluid, and is deposited in bone, secreted back to the gut, or filtered and later partly reabsorbed via the TRPV5 channel within the kidneys. Children have a positive net balance of calcium which peaks in infancy and puberty, the periods of highest growth rates. Adequate availability of calcium is essential for hydroxyl-apatite [Ca5(PO4)3OH] formation in bone. Influx of ionized calcium (Ca++) regulates many biological processes, including neural transmission, muscle contraction, and protein secretion. Circulating calcium represents a central mediator between bone, parathyroid glands, the intestine and the kidneys and is kept constant, within close limits, by numerous hormonal signals.

Regulation of calcium (A) phosphate (B) homeostasis. (A) The parathyroid gland senses extracellular calcium (Ca++) levels and secretes parathyroid hormone (PTH). PTH secretion is stimulated by low Ca++ and suppressed by high Ca++ plasma concentrations, respectively. PTH stimulates resorption of Ca++ from the bone, as well as renal Ca++ reabsorption. PTH also stimulates renal 1,25(OH)2D synthesis, and thereby enhances osteoclastic resorption of Ca++ from bone, as well as renal calcium reabsorption via TRPV5 and suppresses PTH synthesis. Circulating fibroblast growth factor 23 (FGF23) originates mainly from osteocytes. FGF23 suppresses both renal 1,25(OH)2D production and PTH. Both Ca++ and 1,25(OH)2D stimulate FGF23 production. The sites of defects of the different causes of hypocalcemic disorders (also called calcipenic rickets) are given in the purple boxes. This includes nutritional rickets due to vitamin D deficiency and/or impaired dietary calcium availability and genetic defects in vitamin D metabolism or action (vitamin D-dependent rickets (VDDR types 1–3)). Note: hypophosphatemia due to secondary hyperparathyroidism-associated renal phosphate wasting, rather than hypocalcemia ultimately causes rickets in calcipenic rickets. (B) FGF23 and PTH reduce renal tubular phosphate (Pi) reabsorption by reducing the apical expression of the sodium–phosphate cotransporters NaPi IIa and NaPi IIc. PTH stimulates, while FGF23 inhibits 1,25(OH)2D production. 1,25(OH)2D increases intestinal absorption of dietary Pi by enhancing NaPi IIb expression and stimulates FGF23 synthesis. PTH and FGF23 affect each other’s production through a negative feedback loop by as yet unknown mechanisms. The sites of defects of the different causes of hypophosphatemic disorders (“phosphopenic rickets”) are given in the purple boxes. This includes impaired dietary phosphate availability and genetic defects: XLH, X-linked hypophosphatemia; ARHR1/2/3, autosomal recessive hypophosphatemic rickets 1/2/3; FD/MAS, Fibrous dysplasia/McCune-Albright syndrome; HHRH, hereditary hypophosphatemic rickets with hypercalciuria; IIH, idiopathic hypercalcemia; ADHR, autosomal dominant hypophosphatemic rickets. *FGF23-protein resistant to degradation. Created with BioRender.com
Inorganic phosphate (Pi) is of major importance for the maintenance of cellular metabolism and skeletal mineralization. Pi contributes to approximately 0.6 and 1% of body weight in neonates and adults, respectively. The skeleton and teeth contain the vast majority (85%) of total body Pi in the form of hydroxyl-apatite. Approximately 14% is distributed within the intracellular compartment. Inorganic phosphate plays an important role in many cellular processes, e.g., energy metabolism (ATP), cell membrane function (phospholipids), cell signaling (phosphorylation by kinases), and DNA- or RNA-biosynthesis (phosphorylated nucleotides) [9]. The extracellular compartment contains only 1% of the total body Pi content, where it serves as an acid–base buffer in plasma and urine. As phosphate measurements in clinical practice are limited to serum and urine, only “the tip of the iceberg” is visible. Similarly to serum calcium, circulating Pi represents a central mediator between bone, parathyroid glands, the intestine and the kidneys, and is kept constant, within close limits, similarly to calcium by numerous hormonal signals (Fig. 4B).
After completion of skeletal growth, a steady state can be assumed where serum Pi concentration is kept constant by a balanced equilibrium between dietary phosphate absorption in the intestine (16 mg/kg per day), bone-turnover of phosphate (3 mg/kg per day) and urinary phosphate excretion (16 mg/kg per day), (Fig. 4B). By contrast, a positive Pi balance is required in children and adolescents to allow for normal skeletal growth. Therefore, adequate alimentary Pi is important and can be provided by protein-rich foods such as meat, milk and eggs. However, in contrast to calcium, phosphate is usually abundant in the Western diet, and a dietary phosphate deficiency is rarely observed. After intestinal absorption, Pi distributes in the extracellular compartment and equilibrates with the bone and intracellular compartment. Within the kidney, Pi is freely filtered across the capillaries of the glomerulus and then reabsorbed, mainly by the proximal renal tubules, depending on the actual needs of the body. The type II family of sodium-coupled phosphate-transporters, i.e., NaPi2a (encoded by SLC34A1), NaP2b (encoded by SLC34A2), and NaPi2c (encoded by SLC34A3) ensures the bulk of the intestinal and renal transepithelial Pi transport [10,11,12]. Approximately 90% of phosphate reabsorption within the kidney occurs via NaPi2a, which is localized at the brush border of renal proximal tubular cells. The rest is handled by NaPi2c within the proximal tubules of deep nephrons. Genetic defects of NaPi2A and NaPi2C result in renal phosphate wasting, causing hypophosphatemia and nephrocalcinosis, also known as idiopathic infantile hypercalcemia (IIH) and hypophosphatemic rickets with hypercalciuria (HHRH), respectively [13,14,15]. Hypercalcemia and hypercalciuria result from an enhanced 1,25(OH)2D synthesis. Absorption of dietary phosphate in the small intestine is carried out by NaPi2b which is localized in the brush border membrane of enterocytes and via passive paracellular diffusion [12].
Calcium and phosphate homeostasis regulators
The amount of intestinal calcium and phosphate absorption mainly depends on dietary calcium and Pi intake. Both are partially regulated by 1,25-dihydroxyvitamin D (1,25(OH)2D) [11, 12]. In addition, dietary Pi intake directly affects the amount of renal phosphate reabsorption. Renal phosphate handling is also regulated via phosphaturic hormones – parathyroid hormone (PTH) and FGF23 – both affecting the expression of NaPi2a and NaPi2c within the proximal tubular brush border membranes, in order to keep serum Pi within the normal range (Fig. 4B). Insulin-like growth factor 1 (IGF-1)—the main mediator of growth hormone—increases Pi reabsorption in the renal proximal tubules via upregulation of NaPi2a and NaPi2c, resulting in a positive Pi balance. Both, PTH and 1,25(OH)2D enhance calcium reabsorption in the distal renal tubules by upregulation/activation of the epithelial calcium channel TRPV5.
Parathyroid hormone synthesis and secretion is stimulated via the calcium-sensing receptor by low serum Ca++ and 1,25(OH)2D levels and elevated serum Pi concentrations, and is down-regulated by elevated serum Ca and 1,25(OH)2D concentrations and low Pi levels [16]. It inhibits NaPi-cotransport by enhancing the clearance of NaPi2a from the tubular epithelial brush border membrane and directly stimulates renal calcium reabsorption (vide supra). It also stimulates the synthesis of 1,25(OH)2D, resulting in increased intestinal Pi and Ca++ absorption. The total effect of these actions is an enhancement of circulating Ca++ levels and a reduction in circulating Pi levels (Fig. 4).
Fibroblast Growth Factor 23 is mainly synthesized in bone (osteocytes and osteoblasts) and teeth. Its synthesis is stimulated by Pi, Ca++, PTH, and vitamin D [17, 18]. FGF23 synthesis in bone is mainly regulated by two genes—phosphate-regulating neutral endopeptidase homolog (PHEX) and dentin matrix acidic phosphoprotein (DMP1)—by poorly understood mechanisms [19]. FGF23 requires binding to its co-receptor Klotho, a membrane-bound protein with ß-glucuronidase activity, to activate FGF-receptor 1 (FGFR1). Activation of FGFR1 results in inhibition of proximal tubular Pi reabsorption via triggering internalization and degradation of NaPi2a and NaPi2c, suppresses renal 1,25(OH)2D synthesis, and stimulates 1,25(OH)2D degradation. The total effect is a decrease in circulating Pi and 1,25(OH)2D concentrations (Fig. 4).
Vitamin D3 (cholecalciferol) is predominantly formed by sunlight (90%) (ultraviolet B in the 290–315 nm range) on the skin (deep layers of the epidermis) from 7-dehydrocholesterol (Fig. 5) [20]. The smaller portion of the daily vitamin D requirement is absorbed enterally (vitamin D2 from plant products, vitamin D3 from animal products). After binding to a specific protein (DBP), vitamin D2 and D3 are transported to the liver. In liver microsomes, hydroxylation occurs in 25-hydroxyvitamin D3 (25(OH)D (also known as calcidiol) via 25-hydroxylase (encoded by CYP2R1). After transport via vitamin D-binding protein (DBP) to the kidneys, a second hydroxylation occurs via 1α-hydroxylase (encoded by CYP27B1) to active vitamin D (1,25(OH)2D (also known as calcitriol). Whilst decreases in serum calcium and/or phosphate levels stimulate this hydroxylation, hypercalcemia, hyperphosphatemia, and elevated 1,25(OH)2D concentrations decrease it, and stimulate the conversion of 25OHD and 1,25(OH)2D via cytochrome P450 24A1 (encoded by CYP24A1) to biologically less effective 24,25(OH)2D and 1,24,25(OH2)D in the renal tubules. Vitamin D metabolites can also be inactivated by cytochrome P450 3A4 (encoded by CYP3A4) which is highly expressed in the liver.

Vitamin D homeostasis and hereditary causes of impaired VDDR function. The overall metabolic control of vitamin D homeostasis is shown. Synthesis of calciferols via sunlight exposure or dietary intake of vitamin D2 and D3 is given on the left-hand side. The genes involved in the different forms of VDRR are indicated in italics in the rounded boxes. Created with BioRender.com
Calcitriol and its precursors execute their genomic effects via the intracellular nuclear receptor termed VDR. Their non-genomic actions are mediated by binding to a presumptive plasma membrane receptor. Thereby, calcitriol can directly stimulate renal tubular calcium reabsorption via upregulation of the expression of the epithelial calcium channel TRPV5 and increase intestinal calcium and phosphate absorption via stimulation of the epithelial calcium channel TRPV6 and phosphate transporter NaPi2b, respectively [21,22,23]. This results in a positive calcium and phosphate net balance. Furthermore, 1,25(OH)2D stimulates osteoblasts to increase cytokine synthesis and, thereby, osteoclastogenesis and bone resorption. Compared to vitamin D3, 25(OH)D is metabolically tenfold, and 1,25(OH)2D 1,000-fold more potent.
Pathophysiology of rickets
It was previously assumed that rickets primarily resulted from disturbances of calcium and vitamin D metabolism and was classified into two major groups, based on the leading biochemical alteration, into calcipenic and phosphopenic rickets. However, recent data provides strong evidence that the real culprit is insufficient availability of phosphate which is a prerequisite for terminal differentiation of growth plate chondrocytes [2,3,4]. Endochondral bone growth is characterized by an orderly transformation of growth plate chondrocytes into bone [24]. Cartilage cells of the "resting zone", adjacent to the epiphysis, undergo maturation to chondrocytes which are organized in columns aligned along the longitudinal axis and hypertrophy with time. Thereafter, terminally differentiated hypertrophic chondrocytes are replaced via vascular invasion, apoptosis, mineralization and invasion of osteoclasts, resulting in primary cancellous bone. Studies in various animal models of rickets show that reduced concentrations of extracellular phosphate impair caspase-9-mediated apoptosis of hypertrophic chondrocytes, causing the characteristic rachitic changes (Fig. 1) [2, 4]. This led to the concept that hypophosphatemia is the denominator of all forms of rickets. Indeed, both forms of rickets—calcipenic and phosphopenic—are associated with low serum Pi levels. Therefore, the underlying causes of hypophosphatemia are the basis of the diagnostic algorithm given in Fig. 6 which is described below. However, the historical terms calcipenic and phosphopenic rickets will still be used in this review and are integrated in the algorithm for didactic reasons.

Algorithm for the evaluation of a child presenting with rickets. The differential diagnoses are based on the mechanisms leading to hypophosphatemia, i.e., high parathyroid hormone (PTH) activity (calcipenic rickets), inadequate intestinal phosphate absorption, or renal phosphate wasting (phosphopenic rickets). The latter may be due to either primary tubular defects or high serum FGF23 concentratons. Further details are given in Table 1; FGF23, fibroblast growth factor 23. Created with BioRender.com
Calcipenic rickets
Calcipenic rickets results from impaired calcium availability, which is either due to reduced calcium intake and/or vitamin D deficiency (nutritional rickets) or impaired action of 1,25(OH)2D (vitamin D-dependent rickets) (VDRR)) [1, 25, 26]. In general, the reduced availability of calcium results in a tendency toward lower serum Ca which stimulates PTH synthesis, in order to maintain normal serum Ca++ levels by enhancing renal 1,25(OH)2D synthesis and, thereby, intestinal calcium absorption. In addition, PTH causes internalization, and subsequent degradation, of NaPi2a and NaPi2C in the proximal tubule, leading to renal phosphate wasting and consecutive hypophosphatemia. The latter results in rickets and/or osteomalacia. When PTH-driven demineralization of the skeleton cannot further compensate calcium deprivation, patients may present with symptomatic hypocalcemia, i.e., seizures and tetany and heart failure due to dilated cardiomyopathy in infants (Fig. 7).

reproduced from Uday and Högler with permission [29]
Pathophysiology of nutritional rickets, due to vitamin D deficiency and/or dietary calcium deficiency. Both etiologies result in calcium deprivation and hyperparathyroidism occurs in an attempt to maintain normal serum calcium levels. In the long run, calcium deprivation and phosphate loss result in hypocalcaemic and hypophosphataemic complications. Figure
Nutritional rickets, also called “the English disease,” is still the most common cause of rickets globally [1, 27]. Its prevalence declined during the twentieth century, but recent reports indicate that it is still prevalent, even in so-called developed countries, due to changes in lifestyle (decreased sunlight exposure, which is needed for activation of vitamin D) and nutrition (e.g., vegan diet) and lack of or inconsistent vitamin D prophylaxis during the first year of life. Many risk factors for nutritional rickets have been identified (Table 2). Finally, severely ill children may present with malnutrition and vitamin D deficiency secondary to chronic disease [28,29,30].
Clinical presentation largely depends on the age of the patient and the severity and duration of the underlying deficiency [28,29,30]. Symptomatic hypocalcemia peaks in infancy and puberty, due to the high calcium demands during these periods of rapid growth. Young infants may show irritability and poor feeding and sometimes apnea and stridor. In addition, craniotabes (soft skull) and large fontanelle may be present. Radiological signs of rickets may be mild or even lacking, as prolonged periods of elevated PTH levels are required before they manifest in early infancy. Older infants usually present with the typical features of rickets, including failure to thrive, delayed development, hypotonia, enlarged wrists/ankles, rachitic rosary (enlarged costochondral junctions of the rib) and hypocalcemic seizures. In addition, tachycardia, tachypnea, hepatomegaly and edema suggest the presence of heart failure, due to cardiomyopathy, which is associated with increased risk of death. In children, additional features such as abnormal dentition, leg bowing, and fractures may occur. Adolescents may manifest with symptomatic hypocalcemia, bone pain, muscle weakness, waddling gait, leg bowing and fractures. Symptoms may mimic myopathies, often resulting in delayed diagnosis (Table 3).
Vitamin D-dependent rickets (VDDR) is a group of rare, inherited defects of vitamin D metabolism (failure to synthesize 25(OH)D, or 1,25(OH)2D) or impaired vitamin D receptor signaling), resulting in end-organ resistance to 1,25(OH)2D (Fig. 5). To date, 5 types of VDDR have been described [25, 31, 32]. VDDR shares many clinical and biochemical features with nutritional rickets (Tables 1 and 3). Patients with VDDR1A usually become symptomatic in early infancy, presenting with typical skeletal signs of rickets, failure to thrive, hypotonia, irritability, tetany or seizures, and, in cases of late diagnosis, fractures [33]. Patients with VDDR1B show a similar phenotype which may sometimes be much milder and often improves with age [34]. Patients with VDDR2A/B develop severe hypocalcemia in infancy. About half of VDDR2A, but less so with VDDR2B patients develop alopecia, failure to grow eyelashes and eyebrows due to disrupted VDR, as a functional VDR is required for normal hair follicle cycling (Fig. 2e) [35]. VDDR3 is extremely rare and associated with features similar to VDDR1 [31].
Phosphopenic rickets
Phosphopenic rickets, also called hypophosphatemic rickets, can either be due to dietary phosphate deficiency or impaired bioavailability, FGF23-mediated renal phosphate wasting or to primary or acquired renal tubular phosphate wasting (Fig. 4B, Table 1) [3, 7, 36]. X-linked hypophosphatemia is the most frequent cause of hypophosphatemic rickets and responsible for approximately 80% of all cases. There is considerable overlap between the clinical features of XLH and other forms of hypophosphatemic rickets.
Dietary phosphate deficiency or impaired availability may cause rickets, especially in high-risk populations. Phosphate-deficient diets are rare, and nutritional rickets is usually mainly due to impaired calcium intake or vitamin D deficiency [1]. However, very-low-birthweight infants, especially when they are breast-fed and no phosphate supplements are given, may present with rickets due to their high phosphate requirements [37]. In addition, a special elemental or hypoallergenic formula diet may be associated with impaired phosphate bioavailability, under certain clinical circumstances [38, 39]. Gastrointestinal surgery or short bowel syndrome may also result in low phosphate uptake. Finally, excessive use of phosphate binders or too much restriction of phosphate intake, especially in formula-fed infants suffering from chronic kidney disease, must be considered.
FGF23-mediated renal phosphate wasting is a group of disorders characterized by increased synthesis of FGF23 or reduced degradation of intact FGF23. X-linked hypophosphatemia is a dominant inherited disorder with a prevalence of approximately 4–5/100,000 children and caused by mutations in PHEX, underlying in about 90% of familial cases and 70% of sporadic cases of hypophosphatemic rickets [40, 41]. The mechanism by which pathogenic variants in the PHEX gene lead to increased circulating FGF23 concentrations is still largely unknown [19]. PHEX encodes a membrane-bound endopeptidase and is primarily expressed in bone (osteoblasts and osteocytes), and teeth (odontoblasts) [42] and is thought to affect the expression of FGF23 rather than its degradation [19, 43]. PHEX may regulate serum FGF23 by indirect cleavage of proprotein convertases such as subtilisin/kexin-type 2 (PC2). In addition, PHEX malfunction results in increased skeletal synthesis of osteopontin and acid serine aspartate-rich-MEPE-associated protein (ASARM) peptide, both of which also contribute to impaired bone mineralization in XLH [19, 44]. Thus, XLH is due to a complex osteoblast/odontoblast defect which may explain its special clinical features.
Clinical symptoms of XLH usually develop around the age of walking and resemble those of other forms of rickets including growth failure, thickened wrists and ankles due to widened metaphyses, bone deformities, a waddling gait and a large forehead. Characteristically, additional features are disproportionate short stature (short legs, and preserved trunk length), dental abscesses, craniosynostosis, leading to a dolichocephalic shape of the head, and sensorineural hearing loss [7, 45,46,47]. However, the latter features may also be noted in other forms of hereditary FGF23-mediated hypophosphatemia (vide infra). Tooth eruption is often delayed. Mineralization of dentine is markedly impaired resulting in spontaneous endodontic abscesses and early decay of lacteal and permanent teeth [48]. Type 1 Chiari malformation and syringomyelia are rare, mostly asymptomatic complications of XLH, but may result in headaches and neck pain [45]. Diagnosis may be delayed until adulthood, especially in patients with a negative family history and mild clinical phenotype. Adults with XLH often develop periodontitis, alveolar bone and tooth loss, enthesopathies, (ossification of the entheses, e.g., at the Achilles tendon), osteoarthritis (e.g., of the hip), and pseudofractures (characterized by cortical infraction surrounded by a thickened periosteum on X-ray) which may result in pain, reduced quality of life, and/or immobility [7, 49].
Autosomal-dominant hypophosphatemic rickets (ADHR) is a rare disorder, due to activating pathogenic variants of the FGF23 gene. The resulting mutated protein is resistant to proteolytic cleavage, leading to enhanced FGF23 serum concentrations and, thereby, clinical consequences as in other forms of FGF23 excess [50]. Due to its incomplete penetrance, ADHR patients show a highly variable phenotype and patients usually become symptomatic after childhood [51]. Leading complications due to osteomalacia in adult patients include weakness, fatigue, bone pain and pseudofractures and tumor-induced osteomalacia (TIO) is the most important differential diagnosis (vide infra). Iron status is an important regulator of FGF23 metabolic pathways, and iron deficiency predisposes ADHR patients to become clinically symptomatic [52].
Autosomal-recessive forms of hypophosphatemic rickets (ARHR1 and 2) are either caused by pathogenic variants in the dentine matrix acidic phosphoprotein 1 (DMP1) gene or in the ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) gene, which are both expressed in bone and teeth [53,54,55,56]. The mechanism of how ENPP1 and DMP1 gene mutations result in elevated FGF23 serum levels is largely unclear [19]. Clinical symptoms resemble those seen in XLH. DMP1 protein plays an important role in the development of bone, cartilage and teeth, whereas ENPP1 is a regulator of bone mineralization and tissue calcification via formation of inorganic pyrophosphate (PPi). ENPP1 mutations result in an unbalanced ratio of phosphate and PPi and increased mineral accumulation in tissue and bone, and impaired bone development. Lorenz-Depiereux et al. were the first to describe inactivating mutations in the ENPP1 gene in 4 families with hypophosphataemic rickets [55]. In ARHR2, in contrast to ENPP1-related GACI, a significantly stronger hypophosphatemia is found, which possibly represents a “protective mechanism” against arterial calcification. ENPP1 mutations were initially described in cases of infantile arterial calcification [57]. There is a high phenotypic heterogeneity and clinical presentation may differ widely in family members showing the same biallelic ENPP1 mutations, including enthesopathy and primary hyperparathyroidism [58].
Raine syndrome is a very rare disease caused by mutations in FAM20C, a major regulator of osteoblasts and osteocytes and stimulator of FGF23 cleavage [59]. Patients present generalized osteosclerosis, and a wide spectrum of other features, including craniofacial anomalies (hypoplasia of the nose/midface), seizures, sensorineural hearing loss, delayed motor development, large fontanelle and abnormal enamel formation. However, the dysmorphic features may be subtle in some patients.
Tumor-induced osteomalacia (TIO) is due to overproduction of FGF23 and other phosphaturic factors by benign slow-growing mesenchymal tumors [60]. TIO has rarely been described in children but is an important differential diagnosis in adults presenting with bone pain, muscle weakness, fatigue and/or pseudofractures in conjunction with hypophosphatemia and high levels of circulating FGF23.
Fibrous dysplasia (FD) is due to activating mutations in GNAS observed in McCune-Albright syndrome (MAS), which is characterized by FD of bone, café-au-lait spots, and precocious puberty, resulting in increased FGF23-synthesis in bone [61]. Hypophosphatemia is usually mild and associated with phosphate-wasting, but may result in bone fractures and bone pain.
Finally, cutaneous disorders such as epidermal nevi (EN), congenital melanocytic nevi (CMN) and Schimmelpenning–Feuerstein–Mims syndrome are rare causes of FGF23-excess. Patients show multiple segmental skin and bone lesions, due to activating somatic mutations of HRAS or NRAS, which are also responsible for the enhanced FGF23-synthesis in bone [62].
Hypophosphatemic disorders with normal or suppressed FGF23-activity include HHRH, which is due to mutations in SLC34A3 encoding for NaPi2C [14, 63, 64]. HHRH usually shows increased 1,25(OH)2D serum concentrations which promote hypercalciuria, low PTH serum levels, nephrocalcinosis and/or nephrolithiasis. However, clinical symptoms and changes in biochemical parameters may be subtle. The clue is to thoroughly assess the urinary calcium–creatinine ratio in all patients with hypophosphatemic rickets, which is usually normal in XLH but elevated in HHRH.
Hypophosphatemia and nephrocalcinosis is due to mutations in the SLC34A1 gene encoding for NaPi2a. Clinical presentations vary widely including idiopathic infantile hypercalcemia (IIH), nephrolithiasis, and rickets associated with renal phosphate wasting [14, 65].
Osteoglophonic dysplasia is due to activating mutations in FGFR1 (Fibroblast growth factor receptor 1) [66, 67]. Clinical symptoms include craniosynostosis, facial dysmorphism, delayed eruption of teeth and growth impairment, which may be associated with hypophosphatemic rickets due to enhanced bone FGF23-synthesis.
Renal Fanconi syndrome is another important cause of renal phosphate wasting, due to a generalized proximal tubular dysfunction [68]. Clinical consequences result mainly from polyuria, hypophosphatemia and metabolic acidosis, i.e., malnutrition, rickets and growth failure. Nephropathic cystinosis represents its most common hereditary form [69]. Incomplete Fanconi syndrome is noted in Dent disease 1, which is due to inactivating mutations in the CLCN5 gene encoding for CIC-5 expressed in proximal renal tubules, resulting in low molecular weight proteinuria associated with hypercalciuria, nephrolithiasis, and often, renal phosphate wasting and impaired urinary acidification, resulting in rickets [70].
Iatrogenic phosphate wasting may be caused by exposure to certain chemotherapies such as cisplatin and ifosfamide, and other drugs (e.g., tenofovir, protease inhibitors) [71].
General diagnostic approach
The diagnosis of rickets is based on typical clinical symptoms (e.g., widened wrists, frontal bossing, leg deformities, waddling gait, muscle weakness, and growth failure) and radiological findings (e.g., metaphyseal fraying and widening of growth plates) in the presence of elevated serum ALP levels (Fig. 6). The biochemical workup is based on assessment of serum phosphate, (ionized) calcium, creatinine, bicarbonate, ALP, PTH, 25(OH)D, 1,25(OH)2D, plasma FGF23 (if available) and an assessment of the renal tubular function (Table 4) [1, 7, 25].
The differential diagnosis of rickets can be straightforward, e.g., an infant presenting with typical clinical symptoms and a history of low calcium intake and lack of vitamin D prophylaxis suggests nutritional rickets, but can be tricky in a child with a rare case of hereditary rickets, especially when presenting with mild symptoms and concomitant decreased vitamin D levels, which is not uncommon. As outlined above, hypophosphatemia is the denominator of all forms of rickets, except metabolic acidosis and CKD-associated rickets, which must be excluded before using the diagnostic algorithm shown in Fig. 6. This approach was originally proposed by Penido and Alon [3] and is based on the underlying mechanisms of hypophosphatemia, i.e., i) elevated levels of the phosphaturic hormone PTH, ii) inadequate intestinal phosphate absorption, and iii) renal phosphate wasting. The latter can either be due to increased activity of the phosphaturic hormone FGF23 or primary/acquired tubular defects. Further details of the underlying entities are given in Table 1. The use of the diagnostic algorithm based on patient history, clinical presentation, laboratory and imaging studies, as well as its pitfalls, are outlined in the following section.
Medical history
A detailed medical history, including dietary intake and medication, as well as family history, is essential in order to establish the mode of inheritance. The recommended dietary calcium intake in order to prevent rickets amounts to 200 and 260 mg/day in infants 0–6 and 6–12 months of age, respectively [28]. Thereafter, an intake above 500 mg/day is recommended, and an intake of less than 300 mg is considered to be deficient (Table 5). However, there is no clear cut-off at which low calcium intake results in nutritional rickets, as this is often due to a combination of low calcium intake and vitamin D deficiency, which are both associated with the various risk factors given in Table 2, which should also be considered [26, 28,29,30]. Breast milk-fed infants of vitamin D deficient mothers are most prone to hypocalcemic complications such as poor feeding, irritablitiy, and seizures if unsupplemented. Older patients may complain of limb pain, muscle spasms, and fatigue (Table 3).
An elemental or hypoallergenic formula diet or parental nutrition may cause hypophosphatemic rickets [38, 39]. A history of gastrointestinal surgery may point to impaired dietary phosphate availability. Certain drugs, including valproate, cisplatin, ifosfamide, gentamycin, and excessive use of phosphate binders, may cause renal Fanconi syndrome or interfere with calcium/vitamin D metabolism or phosphate intake [68, 71]. In cases of symptoms such as polyuria, polydipsia, and failure to thrive, a workup for Fanconi syndrome should be initiated.
If a family member is affected, they should be investigated for disproportionate short stature, leg deformities, abnormalities of skull shape (frontal bossing), history of previous orthopedic operations, dental abscesses, periodontal disease, permanent tooth and hearing loss, suggesting XLH. These patients may also suffer from “bone pain” due to osteoarthritis, enthesopathy and pseudofractures. Due to its X-dominant inheritance, 50% of the offspring of affected females and all daughters, but not sons, of affected males will be affected. However, about 30% of XLH cases are due to de novo mutations. An autosomal, dominant inheritance suggests ADHR (Table 1), whereas an X-linked recessive inheritance suggests Lowe syndrome or Dent disease, often presenting with Fanconi syndrome. In a patient with unaffected parents, especially if they are consanguineous or, in the case of an affected sibling, several autosomal recessive disorders should be considered, including VDDR types 1A, 1B, 2A, and 2B, ARHR types 1 and 2, HHRH, hypophosphatemia, nephrocalcinosis, and nephropathic cystinosis.
Physical investigation
A detailed examination, including assessment of height and sitting height, in order to detect a disproportionate short stature (short legs and normal trunk length), presence of limb deformities (genu varum and genu valgum), and thickened wrists and ankles (widened metaphysis), and enlarged costochondral junctions of the rib (rachitic rosary)) should be undertaken. The degree of limb deformities may be assessed by measurement of the intermalleolar and intercondylar distances [72]. A waddling gait may be related to muscle weakness and/or coxa vara. Tibial intorsion may present with an in-toed gait. The head should be assessed for abnormal shape with frontal bossing and dolichocephaly, craniotabes and large fontanelle in infants. A detailed neurological examination and fundoscopy should be undertaken in patients with signs suggesting craniosynostosis/Chiary type 1 malformations, including dolichocephaly, persistent headache and/or ataxia.
It must be pointed out that there is a considerable overlap of the clinical features of the various causes of calcipenic and phosphopenic rickets, making it usually impossible to render a specific diagnosis solely based on clinical examination. In general, clinical presentation may be more severe in patients with VDDR. Neuromuscular irritability and tetany during provocation tests (Chvostek and Trousseau signs) suggests hypocalcemia, which is noted in calcipenic but not in hypophosphatemic rickets (Fig. 7). Characteristic clinical features pointing to certain causes of rickets are given in Table 3.
Laboratory investigations
Serum phosphate
Serum Pi levels are decreased in most forms of rickets, and tend to be even more decreased in patients with phosphopenic rickets compared to calcipenic rickets, as the designations already suggest. In patients with calcipenic rickets, serum phosphate levels may initially remain in the lower normal range until PTH-related renal phosphate loss has resulted in demineralized bone, which makes maintenance of normal serum phosphate levels impossible. Serum Pi levels must always be interpreted together with other biochemical parameters, including PTH values and parameters of urinary phosphate handling (vide infra), which are strongly age-dependent, with highest values during the first months of life and a continuous decrease until adulthood (Table 5). Establishing a diagnosis of hypophosphatemia in patients with inherited forms of hypophosphatemic rickets, such as XLH, is hampered in early infancy as serum phosphate levels often remain in the lower normal range during the first 4–6 months [3]. It must be stressed that published pediatric reference values differ considerably and it is recommended that diagnosis of hypophosphatemia is based on locally established pediatric reference values, if available [73]. In Table 5, we present one widely used data set. Repeated measurements may be required to confirm hypophosphatemia and it should be taken into account that pediatric patients may show lower serum phosphate concentrations compared to healthy pediatric volunteers, which hampers the interpretation of reduced serum phosphate levels in these patients. Finally, due to the diurnal fluctuations of serum phosphate levels with lowest levels in the morning, fasting state, and highest levels in the afternoon and evening, it is recommended to establish the diagnosis of hypophosphatemia in the morning fasting state [7, 36].
Renal phosphate handling
Determination of tubular maximum reabsorption of Pi per glomerular filtration rate (TmP/GFR) using a second morning spot urine and serum sample taken at the same time allows for proof of renal phosphate wasting [74,75,76,77]. Both circulating levels of Pi and TmP/GFR are highest in infants and young children, and constantly decline thereafter until adulthood. Three methods (two formula and one nomogram-based) are available to estimate TmP/GFR and it is important to employ the appropriate reference data using the same methodology [77]. A formula proposed by Brodehl et al. to calculate TmP/GFR in children and adolescents, which is reliable in the fasting and non-fasting state, age-related reference values, and a link to an online calculator are presented in Table 5. The nomogram from Walton and Bijvoet was originally established in adults [78]. Its use is not recommended in children, as it results in a slight overestimation of TmP/GFR when used in this population, as compared to the formula provided by Brodehl et al. [77]. Finally, the formula and respective reference values provided by Payne et al. may be employed [79]. The calculation of fractional tubular reabsorption of phosphate (TRP) is not reliable for assessing renal phosphate handling, as it does not account for the amount of filtered phosphate. In the setting of very low serum phosphate concentrations, the remaining capacity of the proximal renal tubules may still be enough to maintain a normal TRP, whereas TmP/GFR is already clearly reduced.
It is necessary to mention that the assessment of TmP/GFR does not allow for discrimination between calcipenic or phosphopenic rickets, as in both entities renal phosphate wasting, and thus reduced TmP/GFR is present, either due to increased PTH (calcipenic rickets), or FGF23 activity or inherited/ acquired impairment of tubular phosphate transporters (phosphopenic rickets) (Table 1).
There is one important pitfall when evaluating renal phosphate wasting by calculating TmP/GFR. In rare cases, when hypophosphatemia is due to decreased phosphate bioavailability or insufficient phosphate intake from the gut, TmP/GFR values may be “falsely” reduced (Table 1) [3]. This dilemma becomes clear when looking at the formulae given in Table 5. As TmP/GFR amounts to serum phosphate concentration minus a sum based on the ratios of urinary and serum phosphate and creatinine concentrations, it is per se always equal to or less than the serum phosphate concentration. Consequently, estimated TmP/GFR may be reduced in cases of dietary phosphate deficiency or impaired bioavailability, resulting in very low serum phosphate levels, even when correcting for filtered amounts of Pi. This holds true for all available methods [3, 74, 78, 79]. The clue to identifying this pitfall is a very low urine phosphate concentration, which can easily be assessed by calculating the urinary phosphate to creatinine ratio (U/P/Crea). In case of low U/P/Crea, TmP/GFR should only be assessed after raising serum and urine phosphate levels via phosphate supplementation [3]. This also explains why urinary phosphate levels (U/P/Crea) and not TmP/GFR was introduced into the diagnostic algorithm given in Fig. 6. Pediatric reference values for U/P/Crea are given in Table 5.
Urine analyses
A urine dipstick allows for identification of glucosuria and proteinuria, suggesting renal Fanconi syndrome. Urinary calcium to creatinine ratio (UCa/Crea) should be assessed using a spot urine sample, related to age-specific reference values (Table 4), and should be confirmed by repeated measurement or a 24 h urine sample, in cases of abnormal values [7]. A urinary calcium excretion above 4 mg/kg per day indicates hypercalciuria. Patients with calcipenic rickets usually present with low UCa/Crea, due to impaired vitamin D metabolism. Patients with FGF23-mediated renal phosphate wasting usually also show low UCa/Crea values due to diminished 1,25 vitamin D synthesis, whereas most patients with primary renal tubular phosphate wasting show hypercalciuria due to elevated 1,25(OH)2D synthesis (e.g., HHRH, IIH, cystinosis, Dent disease), which can be associated with nephrocalcinosis/nephrolithiasis (Table 1). Therefore, a patient with a presumed diagnosis of XLH presenting with hypercalciuria should never be started on conventional treatment (phosphate supplements and active vitamin D) or burosumab—an FGF23 antibody—as this may promote progressive nephrocalcinosis. Instead, a diagnostic workup for other causes of rickets, as outlined above, should be undertaken. Finally, urine amino acids and low molecular weight proteins should be evaluated to detect Fanconi syndrome.
Serum calcium
Serum calcium levels are only minimally age-dependent, with higher levels in young children (Table 5). Ionized calcium, or—if not available—albumin-corrected calcium levels should be used in patients with hypoalbuminemia. Patients with calcipenic rickets usually present with low calcium levels, but serum levels may be in the lower normal range in the early stages, when PTH-driven calcium release from bone can compensate for calcium deficiency. By contrast, serum calcium levels in treatment-naïve patients with phosphopenic rickets are usually in the normal range (Table 1).
Serum alkaline phosphatase
Serum ALP levels serve as a marker of osteoblast activity and are generally elevated in all forms of rickets [1, 80]. In children, 80–90% of total ALP has a bone origin. Therefore, total ALP may be used instead of bone-specific ALP in pediatric patients after exclusion of liver disease, which includes the evaluation of liver enzymes [7]. ALP levels show a tetraphasic course, with highest serum concentrations in infancy and puberty and troughs at mid-childhood and post-puberty [81]. Therefore, ALP levels must always be interpreted in comparison with age- and sex-related normative values. Unfortunately, these differ markedly depending on the assay used. In Table 5, we show recent international pediatric reference level values, which may be used when local normal values are not available. Pediatric reference values for bone-specific ALP are also available [82].
Elevated serum ALP levels confirm the diagnosis of rickets in patients with clinical and radiological signs of rickets. By contrast, normal/reduced ALP levels are seen in Blount’s disease, metaphyseal dysplasia, and hypophosphatasia, which may mimic rickets.
ALP levels are usually highly elevated in treatment-naïve patients with calcipenic rickets (up to tenfold upper normal limit (UNL) or more), and moderately elevated (1 to 3 times UNL) in those with phosphopenic rickets (Table 1). A study comparing age and sex-related z-scores for total ALP levels in treatment-naïve patients with different types of rickets revealed mean values of 11.2 ± 2.6 (VDRR), 7.1 ± 3.8 (nutritional rickets), and 4.2 ± 1.6 (hypophosphatemic rickets), respectively (p < 0.001 between groups) [80]. However, there was a considerable overlap between groups. Finally, circulating ALP is a useful marker in disease monitoring.
Parathyroid hormone
Major stimuli for the synthesis of PTH in the parathyroid glands are hypocalcemia, low 1,25(OH)2D levels, and hyperphosphatemia, whereas hypercalcemia and hypophosphatemia suppress PTH levels (Fig. 4) [16]. PTH levels are markedly increased in untreated patients with calcipenic rickets in order to maintain normal serum calcium levels (Table 1). By contrast, PTH levels are usually in the normal range in patients with phosphopenic rickets, but can be slightly elevated in patients with FGF23-driven phosphopenic rickets, as FGF23 suppresses 1,25(OH)2D levels, which in turn stimulates PTH secretion in the parathyroid glands. PTH levels are often suppressed in patients with HHRH, due to 1,25(OH)2D excess. Finally, PTH levels are expected to be markedly elevated in children presenting with rickets due to CKD, which is usually associated with elevated serum phosphate and creatinine levels. Monitoring of PTH is an excellent marker for the response to therapy in patients with calcipenic rickets.
Vitamin D levels
Serum levels of 25(OH)D are a reliable marker of vitamin D status in a child. Vitamin D sufficiency is defined by 25(OH)D levels above 50 nmol/l, insufficiency by levels between 30 and 50 nmol/l, and deficiency by levels below 30 nmol/l (Table 5) [28]. Serum 25(OH)D levels are usually normal in patients with phosphopenic rickets. However, vitamin D insufficiency is frequently observed in the general population. In addition, normal 25(OH)D levels do not exclude nutritional rickets as nutritional rickets may also be related to reduced dietary calcium intake. In addition, reduced 25(OH)D levels are also observed in VDDR types 1B and 3 (Table 1). Therefore, 25(OH)D levels must be carefully interpreted together with a history of dietary intake (vide supra) and other biochemical parameters including 1,25(OH)2D levels (vide infra).
If the proposed diagnostic algorithm is followed, 25(OH)D values should be assessed in patients presenting with calcipenic rickets, as suggested by elevated PTH levels (Fig. 6). According to a global consensus recommendation on prevention and management of nutritional rickets, a serum 25(OH)D level below 30 nmol/L (12 ng/ml) supports the diagnosis of nutritional rickets due to vitamin D deficiency [28]. The duration of vitamin D deficiency, dietary calcium intake (vide supra), the patient’s growth rate and other risk factors (Table 2) must also be taken into account. If a patient with the presumed diagnosis of nutritional rickets does not respond to treatment with native vitamin D and calcium the diagnosis must be reconsidered, including other causes of low 25(OH)D levels (VDDR types 1B and 3).
Serum levels of 1,25(OH)2D vary largely depending on the methodology used, and local pediatric reference values may not be available. Pediatric reference values using a fully automated chemiluminescence immunoassay were published by the Canadian Laboratory Initiative in Pediatric Reference Intervals (CALIPER) [83]. Serum levels of 1,25(OH)2D were age-dependent with higher levels in infancy and stable levels after the age of 3 years, which is comparable to those in adults (Table 5).
Increased 1,25(OH)2D levels in patients with calcipenic rickets suggest a diagnosis of calcium deficiency or VDR defects, i.e., VDDR types 2A and 2B, whereas reduced 1,25(OH)2D levels suggests VDDR types 1A, 1B and 3. Diagnosis of the latter two is further supported by low 25(OH)D levels (Table 1, Fig. 5).
In patients with FGF23-mediated rickets, 1,25(OH)2D levels are usually low or inappropriately normal in the setting of hypophosphatemia. By contrast, 1,25(OH)2D levels are usually increased in patients with an inherited renal tubular transporter (NaPi2a and 2C) defect. Patients with nephropathic cystinosis may show normal or reduced 1,25(OH)2D levels, depending on the CKD stage (Table 1) [84].
Fibroblast growth factor 23
The measurement of FGF23 levels is helpful in the diagnostic workup of phosphopenic rickets in order to differentiate between FGF23-mediated and other forms (Fig. 6, Table 1) [3, 7, 36, 85, 86]. Several ELISA assays for biological, active, intact FGF23 (Immunotopics, San Clemente, CA, USA; Kainos, Tokyo, Japan; Millipore, Bedford, MA), an automated, intact method (Diasorin liaison, Saluggia, Italy) and c-terminal FGF23 (Immunotopics; Biomedica ELISA, Wien, Austria; Quidel) are available. Use of plasma EDTA is recommended for most assays. FGF23-concentrations decline when centrifugation is delayed (> 1 h) [87]. Therefore, prompt centrifugation is recommended to avoid falsely normal levels. Age-related reference values for intact [84] and c-terminal FGF23 [82] are available, but vary considerably according to the assay used. It is therefore recommended that the results in an individual patient should always be compared with the reference value obtained by the same assay. Pi intake and vitamin D therapy strongly enhance serum FGF23 levels. FGF23 levels are 2–threefold higher in XLH patients receiving conventional treatment with oral phosphate and active vitamin D, compared to untreated patients [88, 89]. Therefore, FGF23 levels are most informative in untreated patients. In patients have already started on conventional treatment, fasting Pi and FGF23 samples should ideally be collected 1 or 2 weeks after discontinuation of treatment. FGF23 levels are also not informative in patients on burosumab treatment, as this interferes with the FGF23 assay. Indeed, because burosumab binds FGF23 in the circulation, this may result in markedly elevated intact and c-terminal FGF23 concentrations, irrespective of the assay used [90]. Finally, high normal FGF23 levels in the setting of hypophosphatemia should be interpreted as inappropriately normal and therefore do not exclude FGF23-driven hypophosphatemia [7].
Endo et al. evaluated the diagnostic value of intact FGF23 levels, using the Kainos assay in two studies in pediatric and adult Japanese patients with FGF23-mediated hypophosphatemia, including TIO, XLH, ARHR, ADHR, McCune-Albright syndrome/fibrous dysplasia in comparison to patients with non-FGF23-mediated forms of rickets (e.g., nutritional rickets, VDDR, Fanconi syndrome) [91, 92]. FGF23 levels were above the UNL (50 pg/ml) in most patients of the former group and were undetectable or below 23.9 pg/ml in the latter group. Mean FGF23 levels were significantly higher in TIO patients compared to those with FGF23-mediated rickets due to genetic defects, but varied widely in both groups. The combination of hypophosphatemia and FGF23 levels above 30 pg/ml allows for identification of all patients with TIO and genetic hypophosphatemia and excludes all patients with other forms of rickets. From these results, they proposed a cut-off level of 30 pg/ml to confirm FGF23-mediated hypophosphatemia in children and adults when using the Kainos assay [85].
As the degree of elevated FGF23 does not allow for discrimination between the various forms of FGF23-mediated rickets, final diagnosis may require additional genetic testing, especially in cases of negative family history. Bearing in mind the uncertainties of FGF23-assessment, one may also decide upon genetic testing if results are readily available. However, in cases of negative genetic testing for inherited forms of hypophosphatemic rickets, non-genetic forms such as TIO must be considered.
Apparative diagnostics
Radiographs
Radiological changes in rickets are best assessed on radiography at the growth plates of rapidly growing bones, i.e., the distal ulna, metaphyses around the knees and ankles. Early signs include increased height of the physis, widening of the epiphyseal plate and loss of definition in the zone of provisional calcification at the epiphyseal/metaphyseal interface (Figs. 2 and 3) [1, 7]. Later on, a progressive disorganization of the growth plate with cupping, splaying, formation of cortical spurs and stippling, as well as a delay in the appearance of the epiphyseal bone centers is noted. Deformities of the weight-bearing long bones may be present. Finally, Milkman pseudofractures, characterized by pathological fractures and Looser's zones, narrow radiolucent lines 2 to 5 mm in width with sclerotic borders, may be present in adolescents and adults suffering from XLH.
The severity of rickets in XLH patients was monitored in clinical trials using the Rickets Severity Score (RSS), a quantitative scale originally created for assessment of nutritional rickets by Tom Thacher [93, 94]. RSS is based on the degree of metaphyseal fraying and concavity, and the proportion of the growth plate affected at the wrist and knee. It is important to note that radiography of XLH patients shows the metaphyseal signs seen in common rickets, but lacks signs of bone resorption and does not usually show radiolucency. This explains why the Thacher score in patients with XLH is usually graded below 3 on a scale of 10 [93]. Unfortunately, it requires simultaneous radiography of the wrist and knee and is therefore associated with higher X-ray exposure, compared to routine radiography of one joint, which hampers its use in clinical practice.
Kidney ultrasound
The presence of nephrocalcinosis or nephrolithiasis in a patient prior to initiation of therapy, suggests diseases associated with hypercalciuria such as HHRH (NaPi 2C defects), Dent disease 1(CLCN5 defect) or hypophosphatemia and nephrocalcinosis (NaPi2a defects). Nephrocalcinosis may also be noted in children with nephropathic cystinosis and was observed in 30–70% of XLH patients on long-term conventional treatment. Diagnosis of FGF23-mediated phosphopenic rickets, including XLH, should always be questioned in a treatment-naïve patient showing nephrocalcinosis.
Cardiovascular investigations
Infants with nutritional rickets should undergo a heart echo- and electrocardiogram evaluation, in order to detect dilated cardiomyopathy (heart failure, arrhythmia) [29]. Left ventricular hypertrophy and/or hypertension have rarely been reported in XLH patients on long-term conventional treatment. Assessment for these changes may not be required at the initial diagnosis workup [7].
Diagnostic workup for TIO
Children manifesting with hypophosphatemic rickets after early childhood, associated with elevated FGF23 levels, negative family history, and/or negative genetic tests for hereditary causes should be candidates for a TIO diagnostic workup [60]. The biochemical changes resemble those seen in XLH. Localization of these, usually, very small and slow-growing tumors is challenging. They may be located anywhere in soft tissue or the skeleton and are more likely seen in craniofacial bones or extremities. A palpable mass can rarely be identified. Therefore, full-body imaging techniques, including 68GA-DOTA-based PET/CT scans and Technecium 99 m octreotide with single-photon emission computed tomography (octro-SPECT), are suggested for tumor localization, which should be followed by histological confirmation. A comprehensive review on the optimal diagnostic approach was recently provided by Florenzano et al. [60].
Genetic studies
The diagnostic measures outlined above may not allow for confirmation of the exact underlying cause in patients with inherited forms of rickets, especially in cases of negative family history or unusual clinical presentation. In addition, burosumab, a humanized FGF23 antibody, has only been approved in patients with XLH and TIO, but not in other forms of FGF23-driven phosphopenic rickets. Therefore, genetic confirmation of the diagnosis is generally recommended, unless a non-genetic cause of rickets can be proven, or in cases of a positive family history and clear clinical presentation (Fig. 6) [1, 7]. PHEX mutations can be found in 87% of familial cases and 72% of sporadic cases of clinically and laboratory classified XLH [40, 95].
Key summary points
-
The diagnosis of rickets is based on the presence of typical clinical symptoms and radiological findings on X-rays of the wrist or knee, showing widening of the growth plates and metaphyseal fraying and in conjunction with elevated serum levels of alkaline phosphatase.
-
Nutritional rickets, due to vitamin D deficiency and/or dietary calcium deficiency, is the most common cause of rickets.
-
Hereditary causes of rickets are due to mutations in genes involved in vitamin D metabolism or action, renal phosphate reabsorption, or synthesis or degradation of the phosphaturic hormone fibroblast growth factor 23.
-
There is a substantial overlap of the clinical features between the various entities, requiring a thorough workup by biochemical analyses and, if necessary, genetic tests.
Multiple choice questions
-
1.
Which of the following statements is true?
Clinical signs of rickets are:
-
a)
Blue sclerae and an increased fracture rate.
-
b)
Early childhood caries.
-
c)
Progressive deformities of the lower extremities.
-
d)
Shortened upper extremities.
-
e)
Premature closure of cranial sutures.
-
a)
-
2.
Which of the following statements is true?
-
a)
The daily vitamin D requirement is mainly met from animal products.
-
b)
Vitamin D promotes the renal excretion of calcium and phosphate.
-
c)
The most metabolically effective vitamin D metabolite is 25-hydroxy vitamin D.
-
d)
The liver and kidneys are important sites for vitamin D metabolite synthesis.
-
e)
In Europe, endogenous vitamin D synthesis is strongly reduced from April to September.
-
a)
-
3.
Which of the following statements is true?
-
a)
Calcipenic rickets is usually associated with normal serum phosphate and parathyroid hormone levels.
-
b)
1,25(OH)2D levels are reduced in nutritional rickets.
-
c)
Increased renal calcium excretion is a common finding in calcipenic rickets
-
d)
Alkaline phosphatase levels are usually less increased in calcipenic rickets compared to hypophosphatemic rickets.
-
e)
Reduced calcium intake and/or low 25-OHD levels may cause calcipenic rickets.
-
a)
-
4.
Which of the following statements is true?
-
a)
Hypophosphatemic rickets results, in most cases, from a diminished phosphate intake.
-
b)
Hypophosphatemic rickets can be caused by elevated levels of fibroblast growth factor 23 or primary renal phosphate wasting.
-
c)
Hypophosphatemic rickets mainly affects adolescents rather than young children.
-
d)
In contrast to calcipenic rickets, hypophosphatemic rickets is associated with impaired apoptosis of hypertrophic chondrocytes, resulting in widening of the growth plates in bones.
-
e)
Hypophosphatemic rickets is usually associated with increased levels of parathyroid hormone, resulting in decreased serum phosphate concentrations.
-
a)
-
5.
Which of the following statements is true?
-
a)
Hypophosphatemic rickets is most commonly inherited as an X-linked dominant disorder.
-
b)
Hypophophatemic rickets is most commonly inherited as an autosomal dominant disorder.
-
c)
In XLH, PHEX gene mutations can be detected in approx. 50 percent of cases.
-
d)
In the case of a father affected by XLH, the daughters and sons have a 50% risk of XLH.
-
e)
Hypophosphatemic rickets is most commonly inherited as an autosomal recessive disorder.
-
a)
Change history
18 August 2022
The missing funding note has been inserted.
References
Carpenter TO, Shaw NJ, Portale AA, Ward LM, Abrams SA, Pettifor JM (2017) Rickets Nat Rev Dis Primers 3:17101
Sabbagh Y, Carpenter TO, Demay MB (2005) Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci U S A 102:9637–9642
Penido MG, Alon US (2013) Hypophosphatemic rickets due to perturbations in renal tubular function. Pediatr Nephrol 29:361–373
Tiosano D, Hochberg Z (2009) Hypophosphatemia: The common denominator of all rickets. J Bone Miner Metab 27:392–401
Shore RM, Chesney RW (2013) Rickets: Part I. Pediatr Radiol 43:140–151
Shore RM, Chesney RW (2013) Rickets: Part II. Pediatr Radiol 43:152–172
Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, Wicart P, Bockenhauer D, Santos F, Levtchenko E, Harvengt P, Kirchhoff M, Di Rocco F, Chaussain C, Brandi ML, Savendahl L, Briot K, Kamenicky P, Rejnmark L, Linglart A (2019) Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol 15:435–455
Emma F, Haffner D (2018) FGF23 blockade coming to clinical practice. Kidney Int 94:846–848
Manghat P, Sodi R, Swaminathan R (2014) Phosphate homeostasis and disorders. Ann Clin Biochem 51:631–656
Biber J, Hernando N, Forster I (2013) Phosphate transporters and their function. Annu Rev Physiol 75:535–550
Bergwitz C, Jüppner H (2010) Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med 61:91–104
Levi M, Gratton E, Forster IC, Hernando N, Wagner CA, Biber J, Sorribas V, Murer H (2019) Mechanisms of phosphate transport. Nat Rev Nephrol 15:482–500
Jaureguiberry G, Carpenter TO, Forman S, Juppner H, Bergwitz C (2008) A novel missense mutation in SLC34A3 that causes hereditary hypophosphatemic rickets with hypercalciuria in humans identifies threonine 137 as an important determinant of sodium-phosphate cotransport in NaPi-IIc. Am J Physiol Renal Physiol 295:371
Segawa H, Kaneko I, Takahashi A, Kuwahata M, Ito M, Ohkido I, Tatsumi S, Miyamoto K (2002) Growth-related renal type II na/pi cotransporter. J Biol Chem 277:19665–19672
Gorvin CM (2021) Genetic causes of neonatal and infantile hypercalcaemia. Pediatr Nephrol. https://doi.org/10.1007/s00467-021-05082-z
Goltzman D, Mannstadt M, Marcocci C (2018) Physiology of the calcium-parathyroid hormone-vitamin D axis. Front Horm Res 50:1–13
Ho BB, Bergwitz C (2021) FGF23 signalling and physiology. J Mol Endocrinol 66:R23–R32
Bär L, Stournaras C, Lang F, Föller M (2019) Regulation of fibroblast growth factor 23 (FGF23) in health and disease. FEBS Lett 593:1879–1900
Beck-Nielsen SS, Mughal Z, Haffner D, Nilsson O, Levtchenko E, Ariceta G, de Lucas CC, Schnabel D, Jandhyala R, Mäkitie O (2019) FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J Rare Dis 14:58
Dusso AS, Brown AJ, Slatopolsky E (2005) Vitamin D. Am J Physiol Renal Physiol 289:8
Christakos S, Veldurthy V, Patel N, Wei R (2017) Intestinal regulation of calcium: Vitamin D and bone physiology. Adv Exp Med Biol 1033:3–12
Ishizawa M, Akagi D, Yamamoto J, Makishima M (2017) 1α,25-dihydroxyvitamin D(3) enhances TRPV6 transcription through p38 MAPK activation and GADD45 expression. J Steroid Biochem Mol Biol 172:55–61
Hoenderop JG, Müller D, Van Der Kemp AW, Hartog A, Suzuki M, Ishibashi K, Imai M, Sweep F, Willems PH, Van Os CH, Bindels RJ (2001) Calcitriol controls the epithelial calcium channel in kidney. J Am Soc Nephrol 12:1342–1349
Kobayashi H, Saito T, Tanaka S (2014) Mineralization of cartilage in growth plate. Clin Calcium 24:177–184
Levine MA (2020) Diagnosis and management of vitamin D dependent rickets. Front Pediatr 8:315
Bouillon R (2021) Nutritional rickets: Calcium or vitamin D deficiency? Am J Clin Nutr 114:3–4
Wheeler BJ, Snoddy AME, Munns C, Simm P, Siafarikas A, Jefferies C (2019) A brief history of nutritional rickets. Front Endocrinol (Lausanne) 10:795
Munns CF, Shaw N, Kiely M, Specker BL, Thacher TD, Ozono K, Michigami T, Tiosano D, Mughal MZ, Mäkitie O, Ramos-Abad L, Ward L, DiMeglio LA, Atapattu N, Cassinelli H, Braegger C, Pettifor JM, Seth A, Idris HW, Bhatia V, Fu J, Goldberg G, Sävendahl L, Khadgawat R, Pludowski P, Maddock J, Hyppönen E, Oduwole A, Frew E, Aguiar M, Tulchinsky T, Butler G, Högler W (2016) Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab 101:394–415
Uday S, Högler W (2020) Nutritional rickets osteomalacia: A practical approach to management. Indian J Med Res 152:356–367
Roth DE, Abrams SA, Aloia J, Bergeron G, Bourassa MW, Brown KH, Calvo MS, Cashman KD, Combs G, De-Regil LM, Jefferds ME, Jones KS, Kapner H, Martineau AR, Neufeld LM, Schleicher RL, Thacher TD, Whiting SJ (2018) Global prevalence and disease burden of vitamin D deficiency: A roadmap for action in low- and middle-income countries. Ann N Y Acad Sci 1430:44–79
Roizen JD, Li D, O’Lear L, Javaid MK, Shaw NJ, Ebeling PR, Nguyen HH, Rodda CP, Thummel KE, Thacher TD, Hakonarson H, Levine MA (2018) CYP3A4 mutation causes vitamin D-dependent rickets type 3. J Clin Invest 128:1913–1918
Thacher TD, Fischer PR, Singh RJ, Roizen J, Levine MA (2015) CYP2R1 mutations impair generation of 25-hydroxyvitamin D and cause an atypical form of vitamin D deficiency. J Clin Endocrinol Metab 100:1005
Ozden A, Doneray H (2021) The genetics and clinical manifestations of patients with vitamin D dependent rickets type 1A. J Pediatr Endocrinol Metab 34:781–789
Molin A, Wiedemann A, Demers N, Kaufmann M, Do Cao J, Mainard L, Dousset B, Journeau P, Abeguile G, Coudray N, Mittre H, Richard N, Weryha G, Sorlin A, Jones G, Kottler ML, Feillet F (2017) Vitamin D-dependent rickets type 1B (25-hydroxylase deficiency): A rare condition or a misdiagnosed condition? J Bone Miner Res 32:1893–1899
Demir K, Zou M, Al-Rijjal RA, BinEssa H, Acar S, Durmaz E, Çatlı G, Al-Enezi AF, Alzahrani AS, Meyer BF, Shi Y (2020) Novel vdr mutations in patients with vitamin D-dependent rickets type 2a: A mild disease phenotype caused by a novel canonical splice-site mutation. Endocr Pract 26:72–81
Florenzano P, Cipriani C, Roszko KL, Fukumoto S, Collins MT, Minisola S, Pepe J (2020) Approach to patients with hypophosphataemia. Lancet Diabetes Endocrinol 8:163–174
Chacham S, Pasi R, Chegondi M, Ahmad N, Mohanty SB (2020) Metabolic bone disease in premature neonates: An unmet challenge. J Clin Res Pediatr Endocrinol 12:332–339
Uday S, Sakka S, Davies JH, Randell T, Arya V, Brain C, Tighe M, Allgrove J, Arundel P, Pryce R, Högler W, Shaw NJ (2019) Elemental formula associated hypophosphataemic rickets. Clin Nutr 38:2246–2250
Castro S, Velasco Suárez C, Vieites A, Bergadá I, Cassinelli H (2021) Rickets associated to the use of elemental formula: A case report. Arch Argent Pediatr 119:e49–e53
Gaucher C, Walrant-Debray O, Nguyen TM, Esterle L, Garabedian M, Jehan F (2009) PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum Genet 125:401–411
(1995) A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. the HYP consortium. Nat Genet 11:130–136
Du L, Desbarats M, Viel J, Glorieux FH, Cawthorn C, Ecarot B (1996) cDNA cloning of the murine pex gene implicated in X-linked hypophosphatemia and evidence for expression in bone. Genomics 36:22–28
Gattineni J, Baum M (2010) Regulation of phosphate transport by fibroblast growth factor 23 (FGF23): Implications for disorders of phosphate metabolism. Pediatr Nephrol 25:591–601
Barros NM, Hoac B, Neves RL, Addison WN, Assis DM, Murshed M, Carmona AK, McKee MD (2013) Proteolytic processing of osteopontin by PHEX and accumulation of osteopontin fragments in hyp mouse bone, the murine model of X-linked hypophosphatemia. J Bone Miner Res 28:688–699
Rothenbuhler A, Fadel N, Debza Y, Bacchetta J, Diallo MT, Adamsbaum C, Linglart A, Di Rocco F (2019) High incidence of cranial synostosis and chiari I malformation in children with X-linked hypophosphatemic rickets (XLHR). J Bone Miner Res 34:490–496
Coyac BR, Falgayrac G, Penel G, Schmitt A, Schinke T, Linglart A, McKee MD, Chaussain C, Bardet C (2018) Impaired mineral quality in dentin in X-linked hypophosphatemia. Connect Tissue Res 59:91–96
Zivicnjak M, Schnabel D, Billing H, Staude H, Filler G, Querfeld U, Schumacher M, Pyper A, Schroder C, Bramswig J, Haffner D, Hypophosphatemic Rickets Study Group of Arbeitsgemeinschaft fur Padiatrische Endokrinologie and Gesellschaft fur Padiatrische Nephrologie (2011) Age-related stature and linear body segments in children with X-linked hypophosphatemic rickets. Pediatr Nephrol 26:223–231
Biosse Duplan M, Coyac BR, Bardet C, Zadikian C, Rothenbuhler A, Kamenicky P, Briot K, Linglart A, Chaussain C (2017) Phosphate and vitamin D prevent periodontitis in X-linked hypophosphatemia. J Dent Res 96:388–395
Seefried L, Smyth M, Keen R, Harvengt P (2021) Burden of disease associated with X-linked hypophosphataemia in adults: A systematic literature review. Osteoporos Int 32:7–22
Gribaa M, Younes M, Bouyacoub Y, Korbaa W, Ben Charfeddine I, Touzi M, Adala L, Mamay O, Bergaoui N, Saad A (2010) An autosomal dominant hypophosphatemic rickets phenotype in a tunisian family caused by a new FGF23 missense mutation. J Bone Miner Metab 28:111–115
Econs MJ, McEnery PT (1997) Autosomal dominant hypophosphatemic rickets/osteomalacia: Clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab 82:674–681
Liu C, Li X, Zhao Z, Chi Y, Cui L, Zhang Q, Ping F, Chai X, Jiang Y, Wang O, Li M, Xing X, Xia W (2021) Iron deficiency plays essential roles in the trigger, treatment, and prognosis of autosomal dominant hypophosphatemic rickets. Osteoporos Int 32:737–745
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Juppner H, Strom TM (2006) DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet 38:1248–1250
Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM (2010) Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet 86:267–272
Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J, Parvari R (2010) Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet 86:273–278
Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nurnberg P (2003) Mutations in ENPP1 are associated with “idiopathic” infantile arterial calcification. Nat Genet 34:379–381
Kotwal A, Ferrer A, Kumar R, Singh RJ, Murthy V, Schultz-Rogers L, Zimmermann M, Lanpher B, Zimmerman K, Stabach PR, Klee E, Braddock DT, Wermers RA (2020) Clinical and biochemical phenotypes in a family with ENPP1 mutations. J Bone Miner Res 35:662–670
Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, Bjerknes R (2013) Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res 28:1378–1385
Florenzano P, Hartley IR, Jimenez M, Roszko K, Gafni RI, Collins MT (2021) Tumor-induced osteomalacia. Calcif Tissue Int 108:128–142
de Castro LF, Ovejero D, Boyce AM (2020) DIAGNOSIS OF ENDOCRINE DISEASE: Mosaic disorders of FGF23 excess: Fibrous dysplasia/McCune-albright syndrome and cutaneous skeletal hypophosphatemia syndrome. Eur J Endocrinol 182:R83–R99
Lim YH, Ovejero D, Sugarman JS, Deklotz CM, Maruri A, Eichenfield LF, Kelley PK, Jüppner H, Gottschalk M, Tifft CJ, Gafni RI, Boyce AM, Cowen EW, Bhattacharyya N, Guthrie LC, Gahl WA, Golas G, Loring EC, Overton JD, Mane SM, Lifton RP, Levy ML, Collins MT, Choate KA (2014) Multilineage somatic activating mutations in HRAS and NRAS cause mosaic cutaneous and skeletal lesions, elevated FGF23 and hypophosphatemia. Hum Mol Genet 23:397–407
Tieder M, Modai D, Samuel R, Arie R, Halabe A, Bab I, Gabizon D, Liberman UA (1985) Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med 312:611–617
Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, Schlingmann KP, Janner M, Biggin A, Lazier J, Gessner M, Chrysis D, Tuchman S, Baluarte HJ, Levine MA, Tiosano D, Insogna K, Hanley DA, Carpenter TO, Ichikawa S, Hoppe B, Konrad M, Sävendahl L, Munns CF, Lee H, Jüppner H, Bergwitz C (2014) Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J Am Soc Nephrol 25:2366–2375
Ma Y, Lv H, Wang J, Tan J (2020) Heterozygous mutation of SLC34A1 in patients with hypophosphatemic kidney stones and osteoporosis: A case report. J Int Med Res 48:300060519896146
Marzin P, Baujat G, Gensburger D, Huber C, Bole C, Panuel M, Finidori G, De la Dure M, Cormier-Daire V (2020) Heterozygous FGFR1 mutation may be responsible for an incomplete form of osteoglophonic dysplasia, characterized only by radiolucent bone lesions and teeth retentions. Eur J Med Genet 63:103729
White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu X, Shaw NJ, McLellan NJ, McKeown C, Fitzpatrick D, Yu K, Ornitz DM, Econs MJ (2005) Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet 76:361–367
Foreman JW (2019) Fanconi syndrome. Pediatr Clin North Am 66:159–167
Emma F, Nesterova G, Langman C, Labbe A, Cherqui S, Goodyer P, Janssen MC, Greco M, Topaloglu R, Elenberg E, Dohil R, Trauner D, Antignac C, Cochat P, Kaskel F, Servais A, Wuhl E, Niaudet P, Van't Hoff W, Gahl W, Levtchenko E (2014) Nephropathic cystinosis: An international consensus document. Nephrol Dial Transplant 29 Suppl 4:iv87–94
Beara-Lasic L, Cogal A, Mara K, Enders F, Mehta RA, Haskic Z, Furth SL, Trachtman H, Scheinman SJ, Milliner DS, Goldfarb DS, Harris PC, Lieske JC, investigators of the Rare Kidney Stone Consortium (2020) Prevalence of low molecular weight proteinuria and dent disease 1 CLCN5 mutations in proteinuric cohorts. Pediatr Nephrol 35:633–640
Hall AM, Bass P, Unwin RJ (2014) Drug-induced renal fanconi syndrome. QJM 107:261–269
Greene WB (1996) Genu varum and genu valgum in children: Differential diagnosis and guidelines for evaluation. Compr Ther 22:22–29
Tan JG, Vasanwala RF, Yap F, Lek N, Ho CKM (2019) What are the appropriate reference limits for the diagnosis of hypophosphataemia in paediatric patients? J Clin Pathol 72:569–572
Brodehl J, Krause A, Hoyer PF (1988) Assessment of maximal tubular phosphate reabsorption: Comparison of direct measurement with the nomogram of bijvoet. Pediatr Nephrol 2:183–189
Brodehl J (1994) Assessment and interpretation of the tubular threshold for phosphate in infants and children. Pediatr Nephrol 8:645
Brodehl J, Gellissen K, Weber HP (1982) Postnatal development of tubular phosphate reabsorption. Clin Nephrol 17:163–171
Alon U, Hellerstein S (1994) Assessment and interpretation of the tubular threshold for phosphate in infants and children. Pediatr Nephrol 8:250–251
Walton RJ, Bijvoet OL (1975) Nomogram for derivation of renal threshold phosphate concentration. Lancet 2:309–310
Payne RB (1998) Renal tubular reabsorption of phosphate (TmP/GFR): Indications and interpretation. Ann Clin Biochem 35(Pt 2):201–206
Turan S, Topcu B, Gökçe İ, Güran T, Atay Z, Omar A, Akçay T, Bereket A (2011) Serum alkaline phosphatase levels in healthy children and evaluation of alkaline phosphatase z-scores in different types of rickets. J Clin Res Pediatr Endocrinol 3:7–11
Adeli K, Higgins V, Trajcevski K, White-Al Habeeb N (2017) The canadian laboratory initiative on pediatric reference intervals: A CALIPER white paper. Crit Rev Clin Lab Sci 54:358–413
Fischer DC, Mischek A, Wolf S, Rahn A, Salweski B, Kundt G, Haffner D (2012) Paediatric reference values for the C-terminal fragment of fibroblast-growth factor-23, sclerostin, bone-specific alkaline phosphatase and isoform 5b of tartrate-resistant acid phosphatase. Ann Clin Biochem 49:546–553
Higgins V, Truong D, White-Al Habeeb NMA, Fung AWS, Hoffman B, Adeli K (2018) Pediatric reference intervals for 1,25-dihydroxyvitamin D using the DiaSorin LIAISON XL assay in the healthy CALIPER cohort. Clin Chem Lab Med 56:964–972
Ewert A, Leifheit-Nestler M, Hohenfellner K, Büscher A, Kemper MJ, Oh J, Billing H, Thumfart J, Stangl G, Baur AC, Föller M, Feger M, Weber LT, Acham-Roschitz B, Arbeiter K, Tönshoff B, Zivicnjak M, Haffner D (2020) Bone and mineral metabolism in children with nephropathic cystinosis compared with other CKD entities. J Clin Endocrinol Metab 105:dgaa267. https://doi.org/10.1210/clinem/dgaa267
Fukumoto S (2021) FGF23-related hypophosphatemic rickets/osteomalacia: Diagnosis and new treatment. J Mol Endocrinol 66:R57–R65
Giralt M, Chocron S, Ferrer R, Ariceta G (2021) Plasma intact fibroblast growth factor 23 level is a useful tool for diagnostic approach of renal hypophosphatemia. Pediatr Nephrol 36:1025–1028
Dirks NF, Smith ER, van Schoor NM, Vervloet MG, Ackermans MT, de Jonge R, Heijboer AC (2019) Pre-analytical stability of FGF23 with the contemporary immunoassays. Clin Chim Acta 493:104–106
Carpenter TO, Insogna KL, Zhang JH, Ellis B, Nieman S, Simpson C, Olear E, Gundberg CM (2010) Circulating levels of soluble klotho and FGF23 in X-linked hypophosphatemia: Circadian variance, effects of treatment, and relationship to parathyroid status. J Clin Endocrinol Metab 95:352
Imel EA, DiMeglio LA, Hui SL, Carpenter TO, Econs MJ (2010) Treatment of X-linked hypophosphatemia with calcitriol and phosphate increases circulating fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab 95:1846–1850
Piketty ML, Brabant S, Souberbielle JC, Maruani G, Audrain C, Rothenbuhler A, Prié D, Linglart A (2020) FGF23 measurement in burosumab-treated patients: An emerging treatment may induce a new analytical interference. Clin Chem Lab Med 58:e267–e269
Endo I, Fukumoto S, Ozono K, Namba N, Tanaka H, Inoue D, Minagawa M, Sugimoto T, Yamauchi M, Michigami T, Matsumoto T (2008) Clinical usefulness of measurement of fibroblast growth factor 23 (FGF23) in hypophosphatemic patients: Proposal of diagnostic criteria using FGF23 measurement. Bone 42:1235–1239
Endo I, Fukumoto S, Ozono K, Namba N, Inoue D, Okazaki R, Yamauchi M, Sugimoto T, Minagawa M, Michigami T, Nagai M, Matsumoto T (2015) Nationwide survey of fibroblast growth factor 23 (FGF23)-related hypophosphatemic diseases in japan: Prevalence, biochemical data and treatment. Endocr J 62:811–816
Thacher TD, Pettifor JM, Tebben PJ, Creo AL, Skrinar A, Mao M, Chen CY, Chang T, San Martin J, Carpenter TO (2019) Rickets severity predicts clinical outcomes in children with X-linked hypophosphatemia: Utility of the radiographic rickets severity score. Bone 122:76–81
Thacher TD, Fischer PR, Pettifor JM, Lawson JO, Manaster BJ, Reading JC (2000) Radiographic scoring method for the assessment of the severity of nutritional rickets. J Trop Pediatr 46:132–139
Holm IA, Nelson AE, Robinson BG, Mason RS, Marsh DJ, Cowell CT, Carpenter TO (2001) Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab 86:3889–3899
De Souza VC, Rabilloud M, Cochat P, Selistre L, Hadj-Aissa A, Kassai B, Ranchin B, Berg U, Herthelius M, Dubourg L (2012) Schwartz formula: Is one k-coefficient adequate for all children? PLoS One 7:e53439
KDOQI Work Group (2009) KDOQI clinical practice guideline for nutrition in children with CKD: 2008 update. executive summary. Am J Kidney Dis 53:11
Lam V, Dhaliwal SS, Mamo JC (2013) Adjustment of ionized calcium concentration for serum pH is not a valid marker of calcium homeostasis: Implications for identifying individuals at risk of calcium metabolic disorders. Ann Clin Biochem 50:224–229
Soldin SJ, Brugnara C, Wong EC (2003) Pediatric reference ranges, 4th, edition. AACC-Press, Washington, p 150
Greenberg BG, Winters RW, Graham JB (1960) The normal range of serum inorganic phosphorus and its utility as a discriminant in the diagnosis of congenital hypophosphatemia. J Clin Endocrinol Metab 20:364–379
Stark H, Eisenstein B, Tieder M, Rachmel A, Alpert G (1986) Direct measurement of TP/GFR: A simple and reliable parameter of renal phosphate handling. Nephron 44:125–128
Schumann G, Klauke R, Canalias F, Bossert-Reuther S, Franck PF, Gella FJ, Jørgensen PJ, Kang D, Lessinger JM, Panteghini M, Ceriotti F (2011) IFCC primary reference procedures for the measurement of catalytic activity concentrations of enzymes at 37 °C. part 9: Reference procedure for the measurement of catalytic concentration of alkaline phosphatase international federation of clinical chemistry and laboratory medicine (IFCC) scientific division, committee on reference systems of enzymes (C-RSE) (1)). Clin Chem Lab Med 49:1439–1446
Matos V, van Melle G, Boulat O, Markert M, Bachmann C, Guignard JP (1997) Urinary phosphate/creatinine, calcium/creatinine, and magnesium/creatinine ratios in a healthy pediatric population. J Pediatr 131:252–257
Pak CY, Oata M, Lawrence EC, Snyder W (1974) The hypercalciurias. causes, parathyroid functions, and diagnostic criteria. J Clin Invest 54:387–400
Spanaus K, von Eckardstein A (2017) Evaluation of two fully automated immunoassay based tests for the measurement of 1α,25-dihydroxyvitamin D in human serum and comparison with LC-MS/MS. Clin Chem Lab Med 55:1305–1314
Kruse K, Kracht U, Göpfert G (1982) Renal threshold phosphate concentration (TmPO4/GFR). Arch Dis Child 57:217–223
Blind E, Schmidt-Gayk H, Scharla S, Flentje D, Fischer S, Göhring U, Hitzler W (1988) Two-site assay of intact parathyroid hormone in the investigation of primary hyperparathyroidism and other disorders of calcium metabolism compared with a midregion assay. J Clin Endocrinol Metab 67:353–360
Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, Murad MH, Weaver CM; Endocrine Society (2011) Evaluation, treatment, and prevention of vitamin D deficiency: An endocrine society clinical practice guideline. J Clin Endocrinol Metab 96:1911–1930
Schnabel D, Haffner D (2005) Rickets. diagnosis and therapy. Orthopade 34:703–706
Haffner D, Linglart A (2021) Renal hypophosphatemia. In: Emma F, Bagga A, Bates C, Shroff R (eds) Pediatric Nephrology. Springer, Berlin, Heidelberg, pp 1–29
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
D.H. and D.S. received speaker fees, consultation fees, and research grants from Kyowa Kirin. All other authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Answers
1. c; 2. d; 3. e; 4. b; 5. a
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Haffner, D., Leifheit-Nestler, M., Grund, A. et al. Rickets guidance: part I—diagnostic workup. Pediatr Nephrol 37, 2013–2036 (2022). https://doi.org/10.1007/s00467-021-05328-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-021-05328-w