
Abstract
Neutrophil extracellular traps or NETs are released by highly activated neutrophils in response to infectious agents, sterile inflammation, autoimmune stimuli and cancer. In the cells, the nuclear envelop disintegrates and decondensation of chromatin occurs that depends on peptidylarginine deiminase 4 (PAD4) and neutrophil elastase (NE). Subsequently, proteins from neutrophil granules (e.g., NE, lactoferrin and myeloperoxidase) and the nucleus (histones) bind to decondensed DNA and the whole structure is ejected from the cell. The DNA decorated with potent antimicrobials and proteases can act to contain dissemination of infection and in sterile inflammation NETs were shown to degrade cytokines and chemokines via serine proteases. On the other hand, overproduction of NETs, or their inadequate removal and prolonged presence in vasculature or tissues, can lead to bystander damage or even initiation of diseases. Considering the pros and cons of NET formation, it is of relevance if the stage of neutrophil maturation (immature, mature and senescent cells) affects the capacity to produce NETs as the cells of different age-related phenotypes dominate in given (pathological) conditions. Moreover, the immune system of neonates and elderly individuals is weaker than in adulthood. Is the same pattern followed when it comes to NETs? The overall importance of individual and neutrophil age on the capacity to release NETs is reviewed in detail and the significance of these facts is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neutrophils, polymorphonuclear cells (PMNs), are the first leukocytes to reach the site of inflammation where they perform their effector functions, phagocytose microbes and kill them intracellularly. Alternatively, neutrophils fight pathogens extracellularly in either of two ways: upon discharge of potent antimicrobials and proteases from their granules or on release of neutrophil extracellular traps (NETs; Kolaczkowska and Kubes 2013).
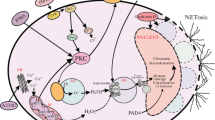
The first report on NETs revealed that neutrophils stimulated by agents such as lipopolysaccharide (LPS), interleukin 8 (IL-8) or phorbol 12-myristate 13-acetate (PMA) form and release structures similar to the network, hence their name (Brinkmann et al. 2004). Detailed studies of NETs by electron scanning and confocal microscopy as well as proteomic analyses showed that NETs are composed of thin chromatin fibers that are decorated with some 30 neutrophil proteins, including neutrophil elastase (NE), bactericidal/permeability-increasing protein (BPI), defensins, cathelicidin (LL-37), proteinase 3 and cathepsin G of granular origin and nuclear histones (Brinkmann et al. 2004; Urban et al. 2009) (Fig. 1). NETs can take different forms, from a band form, by a cloud-like structure, when the NET is fully hydrated, to a network-like shape, exceeding 10–15 times the volume of the releasing cell (Brinkmann et al. 2004; Brinkmann and Zychlinsky 2012). More recent studies, applying atomic force microscopy to reveal their nanoscale properties, reported that NETs are branching filament networks with a substantially organized porous structure and with openings in the size range of small pathogens (Pires et al. 2016). Importantly, proteases attached to NETs secure assembly of the whole structure and its mechanical properties. While such a structure increases the efficiency of catching pathogens, it can also favor collateral damage (Pires et al. 2016). The latter observation directly relates to pros and cons of NET formation.

Basic characteristics of neutrophil extracellular traps (NETs): I) inducing factors, II) involved pathways, III) composition and IV) fate of NET-releasing neutrophils. The image captures NETs formed upon LPS stimulation of murine neutrophils (green arrows extracellular DNA, red arrows citrullinated histone H3), scale bar 50 μm. PAMP pathogen-associated molecular pattern, DAMP damage-associated molecular pattern, PMA phorbol 12-myristate 13-acetate, ROS reactive oxygen species, NO nitric oxide, PAD4 peptidylarginine deiminase 4, MPO myeloperoxidase, BPI bactericidal/permeability-increasing protein, LL-37 cathelicidins cathelicidin
Ying and yang of NETs
There are multiple reports on NETs being able to capture, immobilize and neutralize pathogens. The microbes caught by NET include both Gram-positive (e.g., Staphylococcus aureus) and Gram-negative bacteria (e.g., Salmonella typhimurium and Shigella flexneri; Brinkmann et al. 2004), fungi (e.g., Candida albicans; Urban et al. 2006) and viruses (Saitoh et al. 2012; Jenne et al. 2013). More controversial is their capacity to kill trapped pathogens. As NETs are decorated with antimicrobial proteins and proteases, their killing potential seemed to be unavoidable and in fact it was repeatedly reported to occur (Brinkmann et al. 2004; Urban et al. 2006; Guimarães-Costa et al. 2009). However, some studies ruled it out (Gabriel et al. 2010). A recent paper by Menegazzi et al. (2012) challenged the technical approach applied in the majority of the studies, most of which were performed on isolated neutrophils and revealed that the results depended on the chosen strategy; i.e., incubation with DNase prior or post-addition of bacteria to the NET forming neutrophils. Overall, the study concluded that NETs entrap but do not kill microbes (Menegazzi et al. 2012). This is in line with some in vivo studies showing that, after intravascular application of DNase, colony-forming units (CFUs) of S. aureus do not increase despite strong deposition of NETs in the vasculature of mice with S. aureus sepsis (Kolaczkowska et al. 2015). But even if NETs indeed do not kill pathogens, their role in capturing and immobilizing microbes should not be underestimated as NETs prevent microbial dissemination throughout the body. This was, for example, shown in the course of Escherichia coli sepsis (McDonald et al. 2012). Moreover, one can speculate that NETs can indirectly contribute to pathogen killing, as immobilized microbes are exposed to microenvironmental immune factors present in serum or tissues as well as cytotoxic leukocytes (macrophages and NK cells). In addition, by means of proteases attached to NETs, virulence factors of pathogens can be shed from their surface limiting their virulency, e.g., IpaB on S. flexneri is being removed by NE decorating the traps (Brinkmann et al. 2004). Another important, anti-inflammatory function of NETs comes from studies on sterile inflammation, as during gout, serine proteases attached to NETs were shown to degrade pro-inflammatory cytokines and chemokines contributing to the resolution of the immune response (Schauer et al. 2014).
The importance of NETs is further strengthened by four facts: (1) their evolutionary conservation, (2) release by multiple populations of leukocytes, (3) release of the NET backbone (DNA) from either nucleus or mitochondria and (4) strategies of pathogens developed to escape from NETs. It turns out that DNA decorated with antimicrobials and proteases is preserved in evolution; not only do all vertebrates (only data on amphibians are missing) release extracellular traps (ETs; Brinkmann et al. 2004; Alghamdi and Foster 2005; Palić et al. 2007; Pijanowski et al. 2013; Reichel et al. 2015) but also invertebrate species (Ng et al. 2013; Homa et al. 2016) and even plants (Wen et al. 2009, 2017) and social amoebae (Zhang et al. 2016) do so. Moreover, although not all cells releasing ETs are leukocytes or leukocyte-like, they all seem to perform a kind of defense function, including root border cells of plants (Hawes et al. 2000) and sentinel cells of the multicellular slug stage of the social amoeba functioning as a primitive innate immune system (Chen et al. 2007). Thus, it is not surprising that, in vertebrates, as depicted in detail in mammals, ET formation is universal among innate immune leukocytes and also characterizes monocytes (Granger et al. 2017), macrophages (Chow et al. 2010; Liu et al. 2014), eosinophils (Yousefi et al. 2008), basophils (Morshed et al. 2014) and mast cells (von Köckritz-Blickwede et al. 2008). Furthermore, the source of DNA can vary since neutrophils and eosinophils not only eject DNA of nuclear but also of mitochondrial origin (mNETs; Yousefi et al. 2008, 2009). The studies on neutrophils revealed that DNA of mNETs indeed contains mitochondrial (e.g., Cyb) and not nuclear (e.g., Gapdh) genes (Yousefi et al. 2008). Interestingly, mNETs are released by vital neutrophils and they prolong survival of the releasing cells (Yousefi et al. 2009). Finally, different strategies of pathogens to avoid trapping by NETs, or to escape from the released chromatin fibers, have been described. Streptococcus pneumoniae and S.aureus are good examples of bacteria armed against NETs but fungi (Lee et al. 2015; Rocha et al. 2015; Johnson et al. 2016) and parasites (Guimarães-Costa et al. 2014) have also developed such mechanisms. S. pneumoniae possesses the ability to form polysaccharide capsules protecting them from binding to NETs (Wartha et al. 2007) and their endonucleases degrade the network (Beiter et al. 2006). Moreover, S. pneumoniae can change the electrical charge of their membrane to positive, by incorporation of D-alanine residues into LTAs (lipoteichoic acids). This strategy protects them against positively-charged residues on NET antimicrobials and proteases preventing the trapping (Beiter et al. 2006). S. aureus also releases nucleases but not only to desintegrate NETs (Berends et al. 2010), as they also degrade NET-DNA to intermediate products that are converted to 2′-deoxyadenosine. The latter deoxyribonucleoside induces apoptosis of macrophages that otherwise could phagocytose pathogens immobilized in NETs (Thammavongsa et al. 2013).
Having described the adventages of NET release, one must also acknowledge the side effects of their formation leading to either initiation of bystander damage or even diseases. Numerous studies have reported that uncontrolled and/or excessive release of NETs, as well as their inadequate removal, leads, or at least contributes, to various pathological conditions, including rheumatoid arthritis (RA; Sur Chowdhury et al. 2014; Carmona-Rivera et al. 2017), systemic lupus erythematosus (SLE; Lande et al. 2011; Villanueva et al. 2011), atherosclerosis (Knight et al. 2014; Wang et al. 2017), vasculitis (Kessenbrock et al. 2009; Söderberg and Segelmark 2016), thrombosis (Gould et al. 2014; Martinod and Wagner 2014), sepsis (Kolaczkowska et al. 2015) and cancer (Berger-Achituv et al. 2013; Tohme et al. 2016). SLE and sepsis are representative examples of excessive/inapropiate NET release and inadequate removal, respectively. SLE is manifested by benign skin lesions to life-threatening symptoms resulting from overproduction of autoantibodies and loss of tolerance to their own antigens (Crispín et al. 2010; Dörner et al. 2011). The autoantibodies, anti-neutrophil cytoplasmic antibodies (ANCAs) are directed against PR3, MPO, NE and the anti-nuclear antibodies (ANAs) against DNA and histones, all of which are components of NETs (Fauzi et al. 2004; Yu and Su 2013; Gajic-Veljic et al. 2015). Characteristic for SLE NETs is the presence of LL-37 and human neutrophil peptide (HNP). The DNA/LL-37/HNP complexes activate plasmacytoid dendritic cells (pDCs) resulting in increased production of IFN-α (Lande et al. 2011), which plays a central role in the pathogenesis of SLE by promoting immune system activation that contributes to tissue and organ inflammation and damage (Crow 2014). In addition, NETs of SLE patients are inadequately degraded as they are protected by DNase inhibitors (Hakkim et al. 2010) but also complement C1q bound to NET (Leffler et al. 2012), while LL-37 can protect DNA from degradation (Lande et al. 2011). Of importance, during SLE, numbers of circulating immature neutrophils are elevated (Bennett et al. 2003).
Correspondingly, during sepsis, NETs contribute to bystander damage of endothelium due to activity of histones (Xu et al. 2009; Saffarzadeh et al. 2012; McDonald et al. 2017) and NE (Kolaczkowska et al. 2015) of NETs that are not timely removed. Also, sepsis is characterized by a rapid recruitment to blood of immature neutrophils (Mare et al. 2015) and not fully mature neutrophils are also present in tumors where they display a pro-tumorgenic phenotype (Sagiv et al. 2015). These data suggest that the age of neutrophils might not only impact the phenotype of neutrophils but also their contribution to disease pathology.
On how NETs are created
Thirteen years into NET research and still we know little about the mechanisms of NET formation, although numerous studies have been published on this topic. Not to underestimate any of the studies, one must keep in mind that, to our estimation, approximately 90% of studies on NETs are performed on isolated neutrophils or tissues collected post-mortem. This does not reflect on a complex in vivo milieu and behavior of neutrophils and other leukocytes in situ, in blood or tissues. However, the main concern is that most of what we know on the mechanisms of NETs come from studies in which PMA was used a sole stimulant. PMA is a syntetic phorbol 12-myristate 13-acetate, a robust activator of two of the three families of protein kinase C (PKC; Liu and Heckman 1998; Neeli and Radic 2013) and, as such, enforces particular signaling pathways. A recent paper re-examing kinetics and signaling pathways of NETs induced by various agents concluded that “PMA stimulation should be regarded as mechanistically distinct from NET formation induced by natural triggers” (van der Linden et al. 2017).
Very early in NET research, dependence on reactive oxygen species (ROS) generated by the NADPH oxidase pathway was reported to be a prerequisite for their formation (Fuchs et al. 2007). The studies were subsequently strongly supported by observation that patients with chronic granulomatous disease (CGD), with impaired NADPH oxidase activity, did not release NETs but that this could be restored by a targeted gene therapy (Bianchi et al. 2009). Subsequently, the Raf-MEK-ERK pathway was identified as being involved in NET formation through activation of NADPH oxidase (Hakkim et al. 2011). But then numerous studies reported ROS-independence of NET formation, which resulted from both in vitro (Gabriel et al. 2010; Byrd et al. 2013; Pijanowski et al. 2013; Mejía et al. 2015) and in vivo studies (Chen et al. 2012; Kolaczkowska et al. 2015; Barth et al. 2016a) utilizing NADPH inhibitors and knockout mice. This discrepancy in the data on ROS involvement in NET release is difficult to explain at this stage. It might be resulting from the experimental milieu or the nature of NET-inducing factors as not all agents activate NADPH oxidase (Farley et al. 2012). The latter study reports on an interesting discrepancy: PMA but not platelet-activating factor (PAF), generated ROS but the NADPH oxidase inhibitor (DPI) reduced NET release by both PMA and PAF. These data indicate that, once again, results from PMA studies should be carefully reviewed unless supported by data from concominant studies applying pathogen- or immune response-related agents to induce NETs. However, most importantly, the study suggests an interesting explanation of ROS involvement in NET formation as DPI also inhibits a range of flavoenzymes including mitochondrial oxidase and nitric oxide synthase (Stuehr et al. 1991; Li and Trush 1998), which could “substitute” for phagosomal ROS. Thus, in some circumstances, NET formation might depend on phagosomal ROS (NADPH-dependent; e.g., Fuchs et al. 2007) but also on mitochondrial ROS (as shown in Lood et al. 2016) or NO (as reported in Patel et al. 2010) or none. It is also of note that the only family of endogenous inhibitors of NETs known to date does not inhibit ROS formation and instead blocks PAD4-dependent citrullination (see “NET formation in neonates”) (Yost et al. 2016).
Another mechanism putatively involved in NET formation is autophagy. This process is critical for the turnover of damaged organelles and proteins during homeostasis but, during infection, plays a role in the killing of phagocytosed pathogens and down-regulation of inflammasome activation (Birmingham et al. 2006; Jabir et al. 2014). The majority of studies showing involvement of autophagy in NET formation applied pharmacological inhibitors of key pathways or molecules involved in this process that however, were also inhibiting ROS (Remijsen et al. 2011; McInturff et al. 2012; Kenno et al. 2016; Ullah et al. 2017). Recently, the involvement of autophagy in NET release was studied in transgenic mice with conditionally deleted atg5 (its product is critical for autophagosome formation) in either neutrophils or eosinophils (Germic et al. 2017). The study ruled out a role of autophagy in NET formation. A similar controversy concerns the involvement of necroptosis (a programmed necrosis-like cell death), which is well illustrated by two contradictory papers published recently head-to-head (Amini et al. 2016; Desai et al. 2016).
However, there are two enzyme-based mechanisms of NET formation that were confirmed to operate independently of the in vitro or in vivo settings and the inducing agents. These include the involvement of NE and peptidylarginine deiminase 4 (PAD4) (Fig. 1). PAD4 belongs to the group of Ca2+-dependent enzymes and is located in the nucleus and granules of neutrophils (Asaga et al. 2001; Nakashima et al. 2002; Kearney et al. 2005). The enzyme is involved in catalyzing the citrullination of histones H2/H3/H4, which is a post-translational modification converting the methylarginine residues to citrulline to form a carbonyl group (Hagiwara et al. 2002; Arita et al. 2006; György et al. 2006). The conversion of positively charged methylarginine to neutral side chains of citrulline affects protein (histone)-DNA stabilization and leads to chromatin decondensation and NET release (Neeli et al. 2008; Wang et al. 2009). Studies on PAD4 knockout mice (PAD4−/−) showed impaired ability to form NETs in comparison to WT animals independently of stimuli, be it LPS or ionomycin (Martinod et al. 2013). Similarly, the PAD4 inhibitor (Cl-amidine) also diminishes NET release both in vitro (Li et al. 2010; Kusunoki et al. 2016) and in vivo (Knight et al. 2013, 2014). However, recently, PMA-induced NET formation was reported not to be connected with histone deamination (no citrullinated H3 histones were detected in PMA-induced NETs), which was explained by the fact that PMA activates the PKCα isoform that inhibits PAD4 while it is the PMA-irresponsive PKCζ that activates deamination (Neeli and Radic 2013). Nevertheless, there are also studies reporting deposition of citrullinated histones in PMA-stimulated NETs, although to a lower degree than upon other inducers (Martinod et al. 2016; van der Linden et al. 2017).
Another enzyme required to form NETs is a serine protease: neutrophil elastase. The proposed mechanism of its action is specific degradation of histones that destabilizes chromatin (Papayannopoulos et al. 2010). In addition, blockade of NET formation was also demonstrated in vivo on NE KO mice infected with Gram-negative bacteria (Papayannopoulos et al. 2010; Farley et al. 2012) or Gram-positive bacteria (Kolaczkowska et al. 2015). Also, the use of NE inhibitor resulted in the inhibition of C. albicans-induced NET formation (Papayannopoulos et al. 2010). However, Martinod et al. (2016) showed that numerous neutrophils derived from NE−/− mice ejected NETs upon in vitro ionomycin stimulation, while 40% of them did not (Martinod et al. 2016). Interstingly, during mouse sterile thrombosis, only 20% fewer NETs were produced by NE KO neutrophils (Martinod et al. 2016). This indicates that both PAD4 and NE are involved in NET formation but might be more or less redundant depending on the disease state and/or stimuli. For example, during S. aureus sepsis, NE−/− neutrophils did not produce NETs while some PAD4−/− PMNs (c. 20%) did (Kolaczkowska et al. 2015), whereas during deep vein thrombosis, 80% of NE−/− neutrophils released NETs (Martinod et al. 2016) but no such structures were cast by the PAD4−/− cells (Martinod et al. 2013). These findings reflect well on the diversity of NET types. The traps seem to vary not only in their appearance, involved molecules and pathways but also in the consequences for the producing cells. The first report on the existence of NETs presented many arguments supporting that the trap-releasing cells remain viable (Brinkmann et al. 2004) but subsequent studies reported on the process being lethal (Fuchs et al. 2007) and eventually a term NETosis was coined (Steinberg and Grinstein 2007). However, Yipp et al. (2012) showed by means of intravital microscopy of S. aureus-inflamed skin that anuclear neutrophils that released NETs remain alive and keep moving and phagocytosing (Yipp et al. 2012). This seems more economical and efficient than the beneficial suicide and was detected in the milieu of the live organism. Successively, viable NET-forming neutrophils were also reported in in vitro settings (Yousefi et al. 2009; Pilsczek et al. 2010). Most probably, the two modes represent another set of parallel mechanisms by which NETs are released, either upon cell rupture (Fuchs et al. 2007) or vesicular transport to the cell surface (Pilsczek et al. 2010).
We still do not know how to understand this variety of involved mechanisms and whether reported NETs are always “NETs”, as adequate, multicomponent detection is a key but not a golden standard. This issue is even becoming a topic of open discussions with “healthy critisism” such as the one of Nauseef and Kubes (2016).
NETs and age of neutrophils
Immature neutrophils versus mature neutrophils
Neutrophils arise and mature in the bone marrow. The maturation consists of the mitotic stage (myeloblasts, promyelocytes and myelocytes) and postmitotic stage (metamyelocyte, neutrophil band and mature segmented neutrophils) (Borregaard 2010; Amulic et al. 2012; Lahoz-Beneytez et al. 2016). Neutrophil secretion from the bone marrow into circulation is controlled by circadian oscillations (Casanova-Acebes et al. 2013) and depends on the interactions between the CXCL12 chemokine and its CXCR4 receptor (retention of neutrophils in the bone marrow) and the CXCL1 ligand with the CXCR2 receptor (release of neutrophils into blood) (Martin et al. 2003; Eash et al. 2010). In circulation, neutrophil age and human neutrophil half-life is less than 1 day, about 19 h (Lahoz-Beneytez et al. 2016) and about 12 h in mice (Pillay et al. 2010a). Expression of CXCR4 increases on aging cells and causes neutrophils to return to the bone marrow, where they are removed by macrophages (Furze and Rankin 2008; Casanova-Acebes et al. 2013) but the cells can also be removed in the spleen and the liver (Shi et al. 2001; Suratt et al. 2001). In turn, this leads to secretion from the bone marrow of a correspondingly small number of mature but not immature (Bruegel et al. 2004; Nierhaus et al. 2013), neutrophils to the circulation (Semerad et al. 2002). As shown recently, the process is controlled by gut microbiota (Zhang et al. 2015) and most probably also by exosomes whose numbers and content change during aging (Prattichizzo et al. 2017). If during their life neutrophils are recruited to the site of inflammation, their life-span is prolonged and their death by apoptosis is delayed (Simon 2003; Milot and Filep 2011). During inflammation, especially the systemic one, both mature and immature neutrophils are recruited from the bone marrow (Drifte et al. 2013). Interestingly, a recent study showed that the first neutrophils to arrive at the site of inflammation are aged neutrophils and they are followed by nonaged cells (Uhl et al. 2016). The fact that aged cells disappear from circulation, neatly explains why fresh cells are recruited to the blood from the bone marrow in the course of inflammation.
Immature and mature neutrophils differ in their gene expression, the former having higher expression of genes controlling their differentiation and granular protein synthesis, including NE, MPO and BPI, whereas genes controlling chemotaxis or apoptosis are down-regulated in immature neutrophils (Martinelli et al. 2004). Comparison of human immature (bone marrow) and mature (blood) neutrophils in their capacity to produce NETs upon IFN-α/γ priming and following stimulation with complement factor C5, showed that only the mature neutrophils released the traps (Martinelli et al. 2004). Other studies revealed diminished yet detectable NET release by immature neutrophils. In the study by Taneja et al. (2008), circulating neutrophils consisted of c. 35% of immature cells (vs. 5% in healthy volunteers) during sepsis. And similar results were obtained by Pillay et al. (2010b). The immature neutrophils had a lower ratio of phagocytosis and Ca2+ signaling (Taneja et al. 2008), antimicrobial recognition and killing and ROS generation (Pillay et al. 2010b). Also, in patients with sterile burn injury, immature neutrophils were numerously present in circulation and these patients had higher levels of circulating free DNA (cfDNA) and citH3, clinical markers of systemic formation of NETs (Hampson et al. 2017). This was especially apparent at times when numbers of immature neutrophils dominated in circulation. However, when neutrophils were isolated from blood and ex vivo-stimulated with PMA, the cells (a mixture of mature and immature neutrophils) of patients with thermal injury released fewer NETs (Hampson et al. 2017).
There is also a report on normal production of NETs by human immature neutrophils present in circulation that comes from studies on bone marrow transplantation (Glenn et al. 2016). Important, although not direct, information on NET production by immature neutrophils comes from studies on diseases during which the undeveloped cells are either present in blood or tissues. One such example is SLE, as lupus patients display a varying degree of neutrophil maturation (Denny et al. 2010; Villanueva et al. 2011). In particular, two neutrophil subpopulations, low-density granulocytes (LDGs) and high-density neutrophils, were identified in the course of the disease. The LDGs do not carry any specific markers identified to date but their nuclear morphology (c. 40% cells have lobular, band or myelocyte-like nuclei vs. c. 60% with segmented nuclei) suggests that many of these cells represent the immature phenotype of neutrophils (Denny et al. 2010). The cells have higher expression of azurophilic granule genes, including those encoding NE and MPO and exhibit increased spontaneous NET production and overall release more traps (Villanueva et al. 2011). A similar subset of neutrophils was also described in the course of psoriasis and psoriatic LDGs also tend to form NETs without any stimulation, in contrast to control or psoriasis mature neutrophils (Lin et al. 2011). Low-density neutrophils, consisting of both immature and mature neutrophils, have also been described in cancer (Sagiv et al. 2015). Unlike high-density neutrophils, the low-density cells have a pro-tumor phenotype (i.e., decreased chemotaxis, phagocytosis and ROS production). The two phenotypes of tumor-associated neutrophils (TANs), i.e., high-density and low-density neutrophils, are also termed N1 and N2, respectively (Fridlender et al. 2009). The N2 phenotype dominates in the presence of TGF-β but is diminished by IFN-β (Fridlender et al. 2009; Andzinski et al. 2016). It was shown that blood neutrophils collected from mice with tumors in which N2 phenotype was suggested to dominate (IFN-β KOs), produced fewer NETs, either spontaneously or upon PMA ex vivo stimulation (Andzinski et al. 2016). These, however, were not TANs and the exact phenotype of circulating neutrophils was not examined, nevertheless immature neutrophils present in a course of disease might not always release spontaneously higher amounts of NETs. In addition, the tumor environment is unique and thus we can speculate that NET release increases anti-tumoral response as NET components might damage tumor cells. But NETs could also function as scaffolds of tumor antigens, facilitating their take-up by DCs and macrophages. On the other hand, NETs can trigger metastasis, e.g., high-mobility group box 1 (HMGB1) released from NETs activates the TLR9-dependent pathway in cancer cells promoting their adhesion, proliferation, migration and invasion (Berger-Achituv et al. 2013; Tohme et al. 2016). Similar results came from a study on immature and mature granulocytes present in leukemic patients (Lukášová et al. 2013). In this study, only data on PMA-induced NETs were reported and acute myeloid leukemia (ALM) granulocytes were shown not to produce the traps as opposed to granulocytes isolated from peripheral blood of healthy donors (Lukášová et al. 2013). The immature cells expressed heterochromatin protein 1 γ (HP1γ) and dimethylated histone H3 at lysine 9 (H3K9me2). The two proteins interact to preserve the spreading of heterochromatin and HP1γ is absent in mature granulocytes. Terminally differentiated mature neutrophils are characterized by a tightly condensed chromatin and gene repression, while immature cells do not (Lukášová et al. 2013). Lukášová et al. (2013) hypothesized that it might be necessary for chromatin to be condensed to facilitate PAD4 action and for this NET formation to be weaker in immature cells.
One has to bear in mind that the majority of data on NET formation by immature neutrophils come from ill patients (with sepsis, SLE, psoriasis or cancer). Nevertheless, many of them, although not all, report on spontaneous release of the traps by immature neutrophils (if this aspect was studied/reported) and diminished, or at least not futher increased, production of NETs upon stimulation (mostly with PMA) (Fig. 2). In addition, at least one study reported on concomitantly elevated markers of NETs in circulation. Considering all the above data, one might hypothesize that immature neutrophils present in blood tend to spontaneously release NETs, hence the presence of their markers in circulation and thus, when isolated and ex vivo-stimulated to produce the traps, fail to form them. This is either due to an exhausted phenotype of the cells or the fact that all neutrophils with a potential to release NETs have already done so once in vasculature. Especially, it is only about 25% of neutrophils that release NETs (Nauseef and Kubes 2016).

Neurophil maturation- and age-dependent changes in neutrophil extracellular traps (NETs) formation. To strengthen the graphical visualization, potential to form NETs is marked with – and +, where + < ++ < +++; −/+ indicates that, for immature neutrophils stimulated ex vivo, some studies reported a lack of NET formation (−) whereas others reported some NET release although weak (+). Phenotype of mature versus aged neutrophils is defined by high or low expression of CXCR4 and CD62L. Immature neutrophils were mostly defined by their nucleus morphology. Reference data are included and discussed in the main text
Aged or senescent neutrophils
Not much is known about NET production by senescent neutrophils. Aging neutrophils up-regulate CXCR4 and progressively lose CD62L (L-selectin) expression that facilitates their re-direction to the bone marrow (Zhang et al. 2015). However, they exhibit enhanced adhesion molecules (e.g., Mac-1, ICAM-1) and TLR4 expression (Zhang et al. 2015), which is in line with their rapid recruitment to the site of inflammation, prior to mature but not aged, neutrophils (Uhl et al. 2016). This aging phenotype is regulated by microbiota and is lost in mice treated with broad-range antibiotics or germ-free animals but restored by application of LPS or fecal transplantation (Zhang et al. 2015). The CD62LloCXCR4hi aged neutrophils are significantly numerous in Selp−/− mice (P-selectin KOs) or anti-P-selectin-treated animals (Zhang et al. 2015; Uhl et al. 2016). When NET production was studied in the latter mice, neutrophils stimulated ex vivo with LPS dramatically increased trap formation. This was further confirmed in an endotoxemic model by intravital imaging of NETs in liver vasculature (Zhang et al. 2015). Therefore, in the case of scenescent neutrophils, the ex vivo and in vivo data clearly correlated, indicating their enhanced capacity to release NETs, which is in line with a pro-inflammatory phenotype of these cells (Fig. 2). However, no data on human scenescent neutrophils are available.
NETs and age of individuals
Immune system matures during fetal development and then declines as we age. These facts have important impacts on susceptibility to infection and the chances of surviving it. And, as such, it is also important how NET release changes with age. Especially, the world is undergoing a shift in demographics with low birth rates and aging of populations (Boule and Kovacs 2017). Independently of the age of mothers, not only fewer babies are being born but also many of them are born preterm and therefore they are more likely to become ill or die, as preterm infants are more vulnerable to infection (Urquhart et al. 2017). In line with this, the risk of severe sepsis in neonates increases dramatically with decreasing gestational age (Sperandio et al. 2013). On the other hand, the global population is aging and the number of indivuduals older than 65 years will double by 2050 (Boule and Kovacs 2017). Elderly people are more susceptible to infection due to inflamm-aging or immunosenescence, i.e., the age-related dysfunction of the immune system but they also develop chronic inflammatory states (Boe et al. 2017).
NET formation in neonates
The immune system plays a very important role during pregnancy, with the purpose of protecting the mother and the developing fetus (Mor et al. 2011). Pregnancy is a period that is characterized by modulation of the immune system associated with both the course and stage of pregnancy, as well as the exposure to pathogens. Moreover, the pregnancy is characterized by a pro-inflammatory phase (first trimester), the anti-inflammatory phase (second trimester) and by the end of the pregnancy returns to the pro-inflammatory phase (Mor and Cardenas 2010). Pregnant women have an increase in the total number of leukocytes, which correlates with the course of pregnancy (the highest level is in the third trimester) of which the most abundant cells are circulating neutrophils (Crocker et al. 2000). These neutrophils display a decreased respiratory burst during the second and third trimesters; however, this activity returns to normal within 7 weeks post-partum (Crocker et al. 2000). With respect to NETs, increased levels of cfDNA (nucleosome/MPO complexes) are observed in pregnant women’s serum, compared to nonpregnant women (Sur Chowdhury et al. 2016). Interestingly, the tendency to form such complexes increasingly relates to the duration of pregnancy. Nevertheless, the highest serum cfDNA level is observed in preeclampsia women, as opposed to women with normal pregnancy and nonpregnant women (Lo et al. 1999; Sur Chowdhury et al. 2016). Moreover, the level of both fetal and maternal circulating plasma DNA from preeclampsia women correlates with the degree of disease severity (Zhong et al. 2001).
The fetus, which is located in the uterus, develops its own immune system (Dauby et al. 2012). After birth, both preterm (<37 weeks) and term (37–42 weeks) neonates are characterized by a tolerogenic immune response due to the reduced number of immune cells, including neutrophils or lymphocytes, which increase in the first weeks of life (Walker et al. 2011; Nguyen et al. 2016). In the developing human fetus, a small number of neutrophils begin to appear in the clavicular marrow after 11–12 weeks post-conception with the majority observed after 13–15 weeks (Slayton et al. 1998a, b). However, neutropoiesis starts prior to this in the fetal liver (around week 5; Slayton et al. 1998a) and yolk sac (around week 3; Sperandio et al. 2013). Neutrophils of a mature individual display a capacity to migrate to the site of inflammation and effectively fight pathogens through phagocytosis or degranulation (Kolaczkowska and Kubes 2013). In term neonates, the phagocytosis and degranulation are equally efficient as in adults but not in preterm neonates (Bektas et al. 1990; Nupponen et al. 2002). However, both preterm and term neonates show impaired migration of neutrophils to the inflammatory focus (McEvoy et al. 1996; Nussbaum et al. 2013). Hence, the young organism is not able to defend itself as efficiently as the adult and therefore neonates are highly susceptible to infections, including sepsis, which directly affect increased morbidity and mortality (Gardner 2009; Lawn et al. 2010). Makoni et al. (2016) suggested that impairment of the neonatal neutrophils may be due to the increased number of developmentally immature neutrophils at birth rather than other abnormalities such as the expression of surface adhesion molecules, which is low at birth but increases over time (Carr et al. 1992; Makoni et al. 2016). Another reason could be keeping down immunity to prevent side effects that might result from its overactivity.
Furthermore, the formation of NETs in preterm or term infants/neonates has been reported to be weaker (Fig. 3). Neutrophils isolated from infants/neonates displayed impaired NET production after stimulation with LPS, PAF and fMLP, in contrast to neutrophils collected from adult individuals (Yost et al. 2009; Lipp et al. 2017). This was despite the presence of functional receptors that recognize these molecules and uncompromised phagocytosis. Nevertheless, when bacteria (E. coli, S. aureus) or PMA were used to induce NETs, neonatal neutrophils did not form NETs (Yost et al. 2009). On the other hand, Lipp et al. (2017) reported that the cells of term infants release some NETs in response to PMA and those of preterm babies release significantly fewer of these structures. Importantly, the defect of NET formation by neutrophils of preterm and term neonates was not rescued by the ROS donor (glucose oxidase) (Yost et al. 2009). Also, the study by Byrd et al. (2016) showed that NETs induced by neonates in response to a combination of fibronectin (Fn) with purified fungal β-glucan or Fn with C. albicans hyphae are ROS-independent, although in this case, NETs were formed normally. Thus, neonate neutrophils seem to be sensitive to fungal stimulation but not necessarily the bacterial components (Byrd et al. 2016). However, in contrast to Lipp et al. (2017), Marcos et al. (2009) showed that neonatal neutrophils can cast NETs upon LPS (as well as other numerous TLR agonists) although at first the signal is weaker (Marcos et al. 2009). Direct comparison of the two studies indicates that neonate neutrophils release NETs but they require a longer time for their maximal formation. In fact, further studies revealed that even the most prematurely born infants gain the capacity to release NET by day 3 post-birth and maximal capacity to cast NETs is achieved between day 3 and 14 of life (Yost et al. 2016). This characteristic seems also to be present in other mammals, as the same phenomenon was observed in pigs (Nguyen et al. 2016). Also, neutrophils of 21-day-old mice produced fewer NETs than the cells of 60-day-old animals (Barth et al. 2016b).

Impact of individual (human) age on neutrophil extracellular trap (NET) release. Graphical representation of neutrophil capacity to produce NETs upon stimulation. In the case of neonates, neutrophil potential to release the traps changes in time. To strengthen the graphical visualization, potential to form NETs is marked with – and + where + < ++ ; −/+ indicates that in the case of newborn infants some studies reported a lack of NET formation (−) whereas others reported some NET release (+). The presence of endogenous NET inhbitors shortly after birth is indicated by circles with a diagonal line. Reference data are included and discussed in the main text
In the search for mechanisms of impaired/delayed NET formation by neonates, a family of endogenous inhibitors of NETs was discovered (Yost et al. 2016). The family, called nNIF-related peptides (NRPs), after the first identified peptide (NET-inhibitory factor, nNIF), also consists of cancer-associated SCM recognition, immune defense suppression and serine protease protection peptide (CRISPP) and a 44–amino acid carboxy terminus cleavage fragment of A1AT (α1-antitrypsin), A1ATm358 (Yost et al. 2016). The levels of inhibitors rapidly decrease in the circulation of the infant after delivery. This might explain why, in some studies, differences in NET formation were reported between preterm and term infants. The inhibitors were detected in different tissues/body fluids - umbilical blood (nNIF), placenta (A1ATm358), plasma (CRISPP-related peptides) - underlinging their importance. They also inhibited NET formation induced by bacteria (S. aureus), damage-associated molecular pattern (DAMP; heme) and PMA (Yost et al. 2016) but did not destroy them. A mechanism of their action is also very intriguing, as NRPs do not affect ROS production nor NE activity (although, after entering the cell, they localize in its close proximity) but inhibit PAD4 and histone citrullination. Importantly from a therapeutical point of view, the injection of either nNIF or CRISPP into adult mice infected with E. coli or LPS prevented formation of NETs and decreased mortality (Yost et al. 2016).
Why would such inhibitors function only in fetuses/neonates? nNIF levels were negligible in healthy adults and undetectable in the plasma of adult individuals with chronic inflammatory disorders (Yost et al. 2016). It is possibly because, during pregnancy, NET-inducing stimuli are present/generated at the maternal fetal interface (Gupta et al. 2005; Marder et al. 2016; Mizugishi and Yamashita 2017) and thus excessive formatiom of the traps could cause inflammatory pathology in the fetomaternal environment. But then, shortly after birth, the inhbitors are degraded or neutralized by unknown means. Intriguingly, the latter correlates with the time when resident microbiota inhabits the human infant and the microbiota was indeed shown to regulate granulocytosis and host resistance to sepsis in the neonate (Deshmukh et al. 2014). The impact of microbiota on the life-span and functioning of neutrophils in adulthood has just been established (see “NETs and age of neutrophils”).
NET release by elderly individuals
An aging organism, like the newborn, is susceptible to a variety of inflammatory pathogenesis, leading to increased morbidity, which is due to impaired immune function (Collerton et al. 2012; Tseng et al. 2012; Boe et al. 2017). Therefore, the term immunosenescence has been introduced. Immunsenescence, or inflamm-aging, is associated with low-grade, chronic, pro-inflammatory status, resulting from an imbalance between pro-inflammatory agents and anti-inflammatory factors (Franceschi et al. 2007; Collerton et al. 2012). It is characterized by elevated levels of pro-inflammatory cytokines, including IL-6 and TNF-α, in physiological conditions (Bruunsgaard et al. 2000; Krabbe et al. 2004; Ferrucci et al. 2005). One hypothesis says it is because of the constant immune challenges over the lifetime leading to a higher basal activation state of cells of the innate immune system (Fulop et al. 2017). In addition, a recent study reports that these age-associated changes depend on the microbiota (Thevaranjan et al. 2017). On the other hand, the elderly have a weaker response to vaccination (Goodwin et al. 2006; Sasaki et al. 2011), which might result from an impaired ability to present antigens to T cells, the latter leading to a dysfunctional immune response (De Martinis et al. 2004; Plowden et al. 2004; Wong and Goldstein 2013).
Neutrophils of elderly individuals are characterized by impaired bactericidal activity (Wenisch et al. 2000), chemotaxis (Fulop et al. 2004), phagocytosis (Butcher et al. 2001; Simell et al. 2011) and decreased ability to perform a respiratory burst (Wenisch et al. 2000). However, some parameters are either preserved (chemokinesis) or up-regulated (degranulation) (Sapey et al. 2014). The changes are believed to reflect on the behavior of the cells in aged individuals. They perform aberrant migration (altered chemotaxis/chemokinesis ratio) so they can spread more efficiently than those from younger individuals and, because they release more protease (as shown for NE; Sapey et al. 2014), possibly to facilitate migration through the ECM, more collateral damage can occur.
Interestingly, high levels of NE, along with eleventated pro-inflammatory cytokine levels, are also characteristic for the low-grade inflammatory state accompanying obesity (Talukdar et al. 2012). In fact, it is recognized now that such an inflammatory state connects aging, metabolic syndrome and cardiovascular disease (Guarner and Rubio-Ruiz 2015).
In physiological conditions, numbers of neutrophils in the bone marrow are similar between old and young mice. However, during inflammatory conditions, such as sepsis induced by cecal ligation and puncture (CLP), fewer neutrophils are observed in the peritoneal lavage in old versus young mice (Xu et al. 2017). Thus, while in healthy aged organisms, the pro-inflammatory state is apparent, in the course of inflammation, the immune response seems to be dimmed. Although not many studies have been undertaken on NET formation by elderly individuals, they all consistently reported weaker production of the traps, in line with the data on other neutrophil activities (Fig. 3). It was observed when the cells were first primed with TNF-α and then activated to form NETs with LPS or IL-8 (Hazeldine et al. 2014), stimulated with Pam3CSK4, a TLR2 ligand (Xu et al. 2017), S. aureus (Tseng et al. 2012) or mitochondrial DNA, a DAMP (Itagaki et al. 2015). Notably, expression of nucleases by S. aureus (vs. the nuclease null strains) led to increased bacterial dissemination in young but not old mice, suggesting that defective NET formation in elderly mice permitted both nuclease and non-nuclease expressing S. aureus to disseminate, altogether leading to more invasive S. aureus infection (Tseng et al. 2012). Interestingly, neutrophils isolated from elderly periodontitis patients also released fewer NETs than the young ones but this was not observed in the case of healthy age-matched controls (Hazeldine et al. 2014). In the studies applying TLR2 and TLR4 ligands, neutrophils collected from elderly people had normal expression of respective receptors required for the cell activation but dimished ROS production (Hazeldine et al. 2014; Xu et al. 2017). And thus the latter was proposed as a mechanism of the lower NET release. However, Hazeldine et al. (2014) as well as Tortorella et al. (2004) showed that there is no impairment in p38 mitogen-activated protein kinase (p38 MAPK) activity, the signaling cascade activated by ROS, in TNF-α-primed neutrophils in both the elderly and younger individuals. Furthermore, PMA, a strong chemical inducer of ROS, induced similar quantities of NETs in both age groups (Tortorella et al. 2004; Hazeldine et al. 2014).
Another possible mechanism leading to dimished NET formation in aged individuals is impaired autophagy. Although involvement of autophagy in NET formation is controversial (see “On how NETs are created”), its impairment was suggested to be co-responsible, along with ROS, for a weak NET release by neutrophils of elderly individuals (Xu et al. 2017). In particular, a defect of Atg5, involved in autphagosome formation, was pointed out to contribute to dimmed NET release. And instead of forming NETs, neutrophils were undergoing apoptosis (Xu et al. 2017) .
There is one report on an increased capacity of neutrophils from aged individuals to produce NETs. This observation comes from studies on aortic lesions in atherosclerotic mice and is strengthened by data from isolated neutrophils activated to produce NETs with 7-ketocholesterol, an athero-relevant stimulus, the most abundant oxysterol in human (Wang et al. 2017). Such an effect resulted from increased mitochondrial oxidative stress, thus mitochondrial (mitoOS) and not cytosolic, ROS generation. The former being indeed associated with atherosclerosis during aging (Vendrov et al. 2015). Considering that numbers of inflammatory neutrophils were the same in aged and young mice, the young animals had smaller lesions and their NET formation was mitoOS-dependent, indicating intrinsic changes in neutrophils of aged subjects. This experimental setting differs from the other studies on NET formation by neutrophils of elderly individuals in clear requirement of mitochondrial ROS and not NADPH oxidase-dependent.
As for what we know to date, neutrophils of elderly subjects in general cast fewer NETs (Fig. 3). No data indicate so far that this is because of active inhibiton of their formation as in neonates but rather it results from dysregulated activity of neutrophils. It is tempting to speculate that one of the mechanisms involved might be connected to the increased release of NE via degranulation, as this enzyme is critical for NET formation. For these NETs that require NADPH oxidase-dependent ROS, a diminished respiratory burst by neutrophils of elderly subjects can provide an additional explanation. However, the observation that increased mitochondrial ROS can in fact increase NET formation by neutrophils of aged individuals suggests that the cells do not lose the capacity to release NETs per se and that this is rather due to upstream dysfunctional pathways.
Conclusions
The phenotype of any given cell reflects either its maturation state or the impact of extrinsic factors and manifests itself by changes in cell morphology, expression pattern of intracellular and extracellular molecules but, foremost, its (altered) functioning. This is also true for neutrophils and their capacity to induce NETs. As, nowadays, NETs are the focus of biomedical research, mostly due to the side effects of their formation, a search for their inhibitors or removing agents dominates the field. Owing to studies on neonate neutrophils is the discovery of endogenous NET inhibitors. This is especially promising in the light of finding that immature neutrophils, which are more abundant in numerous diseases in which NETs play a pivotal role, release the traps spontaneously. Obviously, the cells do not behave uniformly in all conditions and studies on NETs are also technically challenging as they mostly rely on either detection of singular NET components in body fluids or ex vivo stimulation of isolated neutrophils. Although, in the case of mice studies, these limitations can be overcome with intravital microscopy, detecting the traps directly in blood vessels or tissues of live animals, this technique cannot be applied to human studies. And NET inhibition can also be detrimental. For instance, at early stages of sepsis, the structures help to contain dissemination of infection and it is at later time points that their persistent presence causes collateral damage. Thus, NET inhibition or removal should also be timely adjusted, which, however, is difficult to control. Now, a new factor has to be taken into consideration when it comes to the control of NET formation and its consequences, namely the presence of neutrophils of certain ages (immature–mature–senescent) as well as the age of the individuals.
References
Alghamdi AS, Foster DN (2005) Seminal DNase frees spermatozoa entangled in neutrophil extracellular traps. Biol Reprod 73:1174–1181
Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WWL, Simon HU, Yousefi S (2016) NET formation can occur independently of RIPK3 and MLKL signaling. Eur J Immunol 46:178–184
Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A (2012) Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30:459–489
Andzinski L, Kasnitz N, Stahnke S, Wu CF, Gereke M, von Köckritz-Blickwede M, Schilling B, Brandau S, Weiss S, Jablonska J (2016) Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer 138:1982–1993
Arita K, Shimizu T, Hashimoto H, Hidaka Y, Yamada M, Sato M (2006) Structural basis for histone N-terminal recognition by human peptidylarginine deiminase 4. Proc Natl Acad Sci U S A 103:5291–5296
Asaga H, Nakashima K, Senshu T, Ishigami A, Yamada M (2001) Immunocytochemical localization of peptidylarginine deiminase in human eosinophils and neutrophils. J Leukoc Biol 70:46–51
Barth CR, Funchal GA, Luft C, de Oliveira JR, Porto BN, Donadio MVF (2016a) Carrageenan-induced inflammation promotes ROS generation and neutrophil extracellular trap formation in a mouse model of peritonitis. Eur J Immunol 46:964–970
Barth CR, Luft C, Funchal GA, de Oliveira JR, Porto BN, Donadio MVF (2016b) LPS-induced neonatal stress in mice affects the response profile to an inflammatory stimulus in an age and sex-dependent manner. Dev Psychobiol 58:600–613
Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B (2006) An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol 16:401–407
Bektas S, Goetze B, Speer CP (1990) Decreased adherence, chemotaxis and phagocytic activities of neutrophils from preterm neonates. Acta Paediatr Scand 79:1031–1038
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V (2003) Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197:711–723
Berends ETM, Horswill AR, Haste NM, Monestier M, Nizet V, von Köckritz-Blickwede M (2010) Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun 2:576–586
Berger-Achituv S, Brinkmann V, Abed UA, Kühn LI, Ben-Ezra J, Elhasid R, Zychlinsky A (2013) A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol 4:48. https://doi.org/10.3389/fimmu.2013.00048
Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, Reichenbach J (2009) Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 114:2619–2622
Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH (2006) Autophagy controls salmonella infection in response to damage to the salmonella-containing vacuole. J Biol Chem 281:11374–11383
Boe DM, Boule LA, Kovacs EJ (2017) Innate immune responses in the ageing lung. Clin Exp Immunol 187:16–25
Borregaard N (2010) Neutrophils, from marrow to microbes. Immunity 33:657–670
Boule LA, Kovacs EJ (2017) Alcohol, aging, and innate immunity. J Leukoc Biol 102:41–55
Brinkmann V, Zychlinsky A (2012) Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol 198:773–783
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535
Bruegel M, Fiedler GM, Matthes G, Thiery J (2004) Reference values for immature granulocytes in healthy blood donors generated on the Sysmex XE-2100 automated Hematology analyser. Sysmex J Int 14:5–7
Bruunsgaard H, Skinhøj P, Pedersen AN, Schroll M, Pedersen BK (2000) Ageing, tumour necrosis factor-alpha (TNF- alpha) and atherosclerosis. Clin Exp Immunol 121:255–260
Butcher SK, Chahal H, Nayak L, Sinclair A, Henriquez NV, Sapey E, O'Mahony D, Lord JM (2001) Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukoc Biol 70:881–886
Byrd AS, O’Brien XM, Johnson CM, Lavigne LM, Reichner JS (2013) An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J Immunol 190:4136–4148
Byrd AS, O’Brien XM, Laforce-Nesbitt SS, Parisi VE, Hirakawa MP, Bliss JM, Reichner JS (2016) NETosis in neonates: evidence of a reactive oxygen species-independent pathway in response to fungal challenge. J Infect Dis 213:634–639
Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, Liu Y, Bicker KL, Wahamaa H, Hoffmann V, Catrina AI, Thompson P, Buckner JH, Robinson WH, Fox DA, Kaplan MJ (2017) Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol 2:eaag3358. https://doi.org/10.1126/sciimmunol.aag3358
Carr R, Pumford D, Davies JM (1992) Neutrophil chemotaxis and adhesion in preterm babies. Arch Dis Child 67:813–817
Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, A-González N, Kunisaki Y, Zhang D, van Rooijen N, Silberstein LE, Weber C, Nagasawa T, Frenette PS, Castrillo A, Hidalgo A (2013) Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 153:1025–1035
Chen G, Zhuchenko O, Kuspa A (2007) Immune-like phagocyte activity in the social amoeba. Science 317:678–681
Chen K, Nishi H, Travers R, Tsuboi N, Martinod K, Wagner DD, Stan R, Croce K, Mayadas TN (2012) Endocytosis of soluble immune complexes leads to their clearance by FcγRIIIB but induces neutrophil extracellular traps via FcγRIIA in vivo. Blood 120:4421–4431
Chow OA, von Köckritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, Gallo RL, Monestier M, Wang Y, Glass CK, Nizet V (2010) Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe 8:445–454
Collerton J, Martin-Ruiz C, Davies K, Hilkens CM, Isaacs J, Kolenda C, Parker C, Dunn M, Catt M, Jagger C, von Zglinicki T, Kirkwood TBL (2012) Frailty and the role of inflammation, immunosenescence and cellular ageing in the very old: cross-sectional findings from the Newcastle 85+ study. Mech Ageing Dev 133:456–466
Crispín JC, Kyttaris VC, Terhorst C, Tsokos GC (2010) T cells as therapeutic targets in SLE. Nat Rev Rheumatol 6:317–325
Crocker IP, Baker PN, Fletcher J (2000) Neutrophil function in pregnancy and rheumatoid arthritis. Ann Rheum Dis 59:555–564
Crow MK (2014) Advances in understanding the role of type I interferons in SLE. Curr Opin Rheumatol 26:467–474
Dauby N, Goetghebuer T, Kollmann TR, Levy J, Marchant A (2012) Uninfected but not unaffected: chronic maternal infections during pregnancy, fetal immunity, and susceptibility to postnatal infections. Lancet Infect Dis 12:330–340
De Martinis M, Modesti M, Ginaldi L (2004) Phenotypic and functional changes of circulating monocytes and polymorphonuclear leucocytes from elderly persons. Immunol Cell Biol 82:415–420
Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, McCune WJ, Kaplan MJ (2010) A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol 184:3284–3297
Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, Herrmann M, Bilyy R, Müller S, Popper B, Nakazawa D, Weidenbusch M, Thomasova D, Krautwald S, Linkermann A, Anders HJ (2016) PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol 46:223–229
Deshmukh H, Liu Y, Menkiti OR, Mei J, Dai N, O'Leary CE, Oliver PM, Kolls JK, Weiser JN, Worthen GS (2014) The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med 20:524–530
Dörner T, Jacobi AM, Lee J, Lipsky PE (2011) Abnormalities of B cell subsets in patients with systemic lupus erythematosus. J Immunol Methods 363:187–197
Drifte G, Dunn-Siegrist I, Tissières P, Pugin J (2013) Innate immune functions of immature neutrophils in patients with sepsis and severe systemic inflammatory response syndrome. Crit Care Med 41:820–832
Eash KJ, Greenbaum AM, Gopalan PK, Link DC (2010) CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest 120:2423–2431
Farley K, Stolley JM, Zhao P, Cooley J, Remold-O'Donnell E (2012) A SerpinB1 regulatory mechanism is essential for restricting neutrophil extracellular traps generation. J Immunol 189:4574–4581
Fauzi AR, Kong NCT, Chua MK, Jeyabalan V, Idris MN, Azizah R (2004) Antibodies in systemic lupus antineutrophil cytoplasmic erythematosus: prevalence, disease activity correlations and organ system associations. Med J Malaysia 59:372–377
Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub DD, Guralnik JM, Longo DL (2005) The origins of age-related proinflammatory state. Blood 105:2294–2299
Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S (2007) Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev 128:92–105
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM (2009) Polarization of tumor-associated neutrophil (TAN) phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 16:183–194
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176:231–241
Fulop T, Larbi A, Douziech N, Fortin C, Guérard KP, Lesur O, Khalil A, Dupuis G (2004) Signal transduction and functional changes in neutrophils with aging. Aging Cell 3:217–226
Fulop T, Witkowski JM, Le Page A, Fortin C, Pawelec G, Larbi A (2017) Intracellular signalling pathways: targets to reverse immunosenescence. Clin Exp Immunol 187:35–43
Furze RC, Rankin SM (2008) The role of the bone marrow in neutrophil clearance under homeostatic conditions in the mouse. FASEB J 22:3111–3119
Gabriel C, McMaster WR, Girard D, Descoteaux A (2010) Leishmania donovani promastigotes evade the antimicrobial activity of neutrophil extracellular traps. J Immunol 185:4319–4327
Gajic-Veljic M, Bonaci-Nikolic B, Lekic B, Skiljevic D, Ciric J, Zoric S, Stojimirovic B, Nikolic M (2015) Importance of low serum DNase I activity and polyspecific anti-neutrophil cytoplasmic antibodies in propylthiouracil-induced lupus-like syndrome. Rheumatology (Oxford) 54:2061–2070
Gardner SL (2009) Sepsis in the neonate. Crit Care Nurs Clin North Am 21:121–141
Germic N, Stojkov D, Oberson K, Yousefi S, Simon HU (2017) Neither eosinophils nor neutrophils require ATG5-dependent autophagy for extracellular DNA trap formation. Immunology 152:517–525
Glenn JW, Cody MJ, McManus MP, Pulsipher MA, Schiffman JD, Yost CC (2016) Deficient neutrophil extracellular trap formation in patients undergoing bone marrow transplantation. Front Immunol 7:250. https://doi.org/10.3389/fimmu.2016.00250
Goodwin K, Viboud C, Simonsen L (2006) Antibody response to influenza vaccination in the elderly: a quantitative review. Vaccine 24:1159–1169
Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SHC, Weitz JI, Liaw PC (2014) Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol 34:1977–1984
Granger V, Faille D, Marani V, Noël B, Gallais Y, Szely N, Flament H, Pallardy M, Chollet-Martin S, de Chaisemartin L (2017) Human blood monocytes are able to form extracellular traps. J Leukoc Biol 102:775–781
Guarner V, Rubio-Ruiz ME (2015) Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip Top Gerontol 40:99–106
Guimarães-Costa AB, Nascimento MTC, Froment GS, Soares RPP, Morgado FN, Conceição-Silva F, Saraiva EM (2009) Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci U S A 106:6748–6753
Guimarães-Costa AB, DeSouza-Vieira TS, Paletta-Silva R, Freitas-Mesquita AL, Meyer-Fernandes JR, Saraiva EM (2014) 3′-nucleotidase/nuclease activity allows Leishmania parasites to escape killing by neutrophil extracellular traps. Infect Immun 82:1732–1740
Gupta AK, Hasler P, Holzgreve W, Gebhardt S, Hahn S (2005) Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum Immunol 66:1146–1154
György B, Tóth E, Tarcsa E, Falus A, Buzás EI (2006) Citrullination: a posttranslational modification in health and disease. Int J Biochem Cell Biol 38:1662–1677
Hagiwara T, Nakashima K, Hirano H, Senshu T, Yamada M (2002) Deimination of arginine residues in nucleophosmin/B23 and histones in HL-60 granulocytes. Biochem Biophys Res Commun 290:979–983
Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A (2010) Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A 107:9813–9818
Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H (2011) Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol 7:75–77
Hampson P, Dinsdale RJ, Wearn CM, Bamford AL, Bishop JRB, Hazeldine J, Moiemen NS, Harrison P, Lord JM (2017) Neutrophil dysfunction, immature granulocytes, and cell-free DNA are early biomarkers of sepsis in burn-injured patients: a prospective observational cohort study. Ann Surg 265:1241–1249
Hawes MC, Gunawardena U, Miyasaka S, Zhao X (2000) The role of root border cells in plant defense. Trends Plant Sci 5:128–133
Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A, Sapey E, Lord JM (2014) Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell 13:690–698
Homa J, Ortmann W, Kolaczkowska E (2016) Conservative mechanisms of extracellular trap formation by Annelida Eisenia andrei: serine protease activity requirement. PLoS ONE 11:e0159031. https://doi.org/10.1371/journal.pone.0159031
Itagaki K, Kaczmarek E, Lee YT, Tang IT, Isal B, Adibnia Y, Sandler N, Grimm MJ, Segal BH, Otterbein LE, Hauser CJ (2015) Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PLoS ONE 10:e0120549. https://doi.org/10.1371/journal.pone.0120549
Jabir MS, Ritchie ND, Li D, Bayes HK, Tourlomousis P, Puleston D, Lupton A, Hopkins L, Simon AK, Bryant C, Evans TJ (2014) Caspase-1 cleavage of the TLR adaptor TRIF inhibits autophagy and β-interferon production during Pseudomonas aeruginosa infection. Cell Host Microbe 15:214–227
Jenne CN, Wong CHY, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, McFadden G, Kubes P (2013) Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 13:169–180
Johnson CJ, Cabezas-Olcoz J, Kernien JF, Wang SX, Beebe DJ, Huttenlocher A, Ansari H, Nett JE (2016) The extracellular matrix of Candida albicans biofilms impairs formation of neutrophil extracellular traps. PLoS Pathog 12:e1005884. https://doi.org/10.1371/journal.ppat.1005884
Kearney PL, Bhatia M, Jones NG, Yuan L, Glascock MC, Catchings KL, Yamada M, Thompson PR (2005) Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 44:10570–10582
Kenno S, Perito S, Mosci P, Vecchiarelli A, Monari C (2016) Autophagy and reactive oxygen species are involved in neutrophil extracellular traps release induced by C. albicans morphotypes. Front Microbiol 7:879. https://doi.org/10.3389/fmicb.2016.00879
Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, Gröne HJ, Brinkmann V, Jenne DE (2009) Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med 15:623–625
Knight JS, Zhao W, Luo W, Subramanian V, O'Dell AA, Yalavarthi S, Hodgin JB, Eitzman DT, Thompson PR, Kaplan MJ (2013) Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest 123:2981–2993
Knight JS, Luo W, O’Dell AA, Yalavarthi S, Zhao W, Subramanian V, Guo C, Grenn RC, Thompson PR, Eitzman DT, Kaplan MJ (2014) Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res 114:947–956
Kolaczkowska E, Kubes P (2013) Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13:159–175
Kolaczkowska E, Jenne CN, Surewaard BGJ, Thanabalasuriar A, Lee WY, Sanz MJ, Mowen K, Opdenakker G, Kubes P (2015) Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun 6:6673. https://doi.org/10.1038/ncomms7673
Krabbe KS, Pedersen M, Bruunsgaard H (2004) Inflammatory mediators in the elderly. Exp Gerontol 39:687–699
Kusunoki Y, Nakazawa D, Shida H, Hattanda F, Miyoshi A, Masuda S, Nishio S, Tomaru U, Atsumi T, Ishizu A (2016) Peptidylarginine deiminase inhibitor suppresses neutrophil extracellular trap formation and MPO-ANCA production. Front Immunol 7:227. https://doi.org/10.3389/fimmu.2016.00227
Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, Niederalt C, Asquith B, Macallan D (2016) Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood 127:3431–3438
Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu YJ, Gilliet M (2011) Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Sci Transl Med 3:73ra19. https://doi.org/10.1126/scitranslmed.3001180
Lawn JE, Kerber K, Enweronu-Laryea C, Cousens S (2010) 3.6 million neonatal deaths–what is progressing and what is not? Semin Perinatol 34:371–386
Lee MJ, Liu H, Barker BM, Snarr BD, Gravelat FN, Al Abdallah Q, Gavino C, Baistrocchi SR, Ostapska H, Xiao T, Ralph B, Solis NV, Lehoux M, Baptista SD, Thammahong A, Cerone RP, Kaminskyj SGW, Guiot MC, Latgé JP, Fontaine T, Vinh DC, Filler SG, Sheppard DC (2015) The fungal exopolysaccharide galactosaminogalactan mediates virulence by enhancing resistance to neutrophil extracellular traps. PLoS Pathog 11:e1005187. https://doi.org/10.1371/journal.ppat.1005187
Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, Bengtsson AA, Blom AM (2012) Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol 188:3522–3531
Li Y, Trush MA (1998) Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun 253:295–299
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207:1853–1862
Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, Bruce AT (2011) Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol 187:490–500
Lipp P, Ruhnau J, Lange A, Vogelgesang A, Dressel A, Heckmann M (2017) Less neutrophil extracellular trap formation in term newborns than in adults. Neonatology 111:182–188
Liu WS, Heckman CA (1998) The sevenfold way of PKC regulation. Cell Signal 10:529–542
Liu P, Wu X, Liao C, Liu X, Du J, Shi H, Wang X, Bai X, Peng P, Yu L, Wang F, Zhao Y, Liu M (2014) Escherichia coli and Candida albicans induced macrophage extracellular trap-like structures with limited microbicidal activity. PLoS ONE 9:e90042. https://doi.org/10.1371/journal.pone.0090042
Lo YMD, Leung TN, Tein MSC, Sargent IL, Zhang J, Lau TK, Haines CJ, Redman CWG (1999) Quantitative abnormalities of fetal DNA in maternal serum in preeclampsia. Clin Chem 45:184–188
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ (2016) Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22:146–153
Lukášová E, Kořistek Z, Klabusay M, Ondřej V, Grigoryev S, Bačíková A, Řezáčová M, Falk M, Vávrová J, Kohútová V, Kozubek S (2013) Granulocyte maturation determines ability to release chromatin NETs and loss of DNA damage response; these properties are absent in immature AML granulocytes. Biochim Biophys Acta 1833:767–779
Makoni M, Eckert J, Anne Pereira H, Nizet V, Lawrence SM (2016) Alterations in neonatal neutrophil function attributable to increased immature forms. Early Hum Dev 103:1–7
Marcos V, Nussbaum C, Vitkov L, Hector A, Wiedenbauer EM, Roos D, Kuijpers T, Krautgartner WD, Genzel-Boroviczény O, Sperandio M, Hartl D (2009) Delayed but functional neutrophil extracellular trap formation in neonates. Blood 114:4908–4911
Marder W, Knight JS, Kaplan MJ, Somers EC, Zhang X, O'Dell AA, Padmanabhan V, Lieberman RW (2016) Placental histology and neutrophil extracellular traps in lupus and pre-eclampsia pregnancies. Lupus Sci Med 3:e000134. https://doi.org/10.1136/lupus-2015-000134
Mare TA, Treacher DF, Shankar-Hari M, Beale R, Lewis SM, Chambers DJ, Brown KA (2015) The diagnostic and prognostic significance of monitoring blood levels of immature neutrophils in patients with systemic inflammation. Crit Care 19:57. https://doi.org/10.1186/s13054-015-0778-z
Martin C, Burdon PCE, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM (2003) Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 19:583–593
Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, Dummer R, Simon HU, Yousefi S (2004) Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem 279:44123–44132
Martinod K, Wagner DD (2014) Thrombosis: tangled up in NETs. Blood 123:2768–2776
Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, Hu J, Wang Y, Wagner DD (2013) Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A 110:8674–8679
Martinod K, Witsch T, Farley K, Gallant M, Remold-O'Donnell E, Wagner DD (2016) Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thromb Haemost 14:551–558
McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P (2012) Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 12:324–333
McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, Jenne CN (2017) Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 129:1357–1367
McEvoy LT, Zakem-Cloud H, Tosi MF (1996) Total cell content of CR3 (CD11b/CD18) and LFA-1 (CD11a/CD18) in neonatal neutrophils: relationship to gestational age. Blood 87:3929–3933
McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, Yost CC (2012) Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood 120:3118–3125
Mejía SP, Cano LE, López JA, Hernandez O, González Á (2015) Human neutrophils produce extracellular traps against Paracoccidioides brasiliensis. Microbiology 161:1008–1017
Menegazzi R, Decleva E, Dri P (2012) Killing by neutrophil extracellular traps: fact or folklore? Blood 119:1214–1216
Milot E, Filep JG (2011) Regulation of neutrophil survival/apoptosis by Mcl-1. ScientificWorldJournal 11:1948–1962
Mizugishi K, Yamashita K (2017) Neutrophil extracellular traps are critical for pregnancy loss in sphingosine kinase–deficient mice on 129Sv/C57BL/6 background. FASEB J. https://doi.org/10.1096/fj.201700399RR
Mor G, Cardenas I (2010) The immune system in pregnancy: a unique complexity. Am J Reprod Immunol 63:425–433
Mor G, Cardenas I, Abrahams V, Guller S (2011) Inflammation and pregnancy: the role of the immune system at the implatation site. Ann N Y Acad Sci 1221:80–87
Morshed M, Hlushchuk R, Simon D, Walls AF, Obata-Ninomiya K, Karasuyama H, Djonov V, Eggel A, Kaufmann T, Simon HU, Yousefi S (2014) NADPH oxidase-independent formation of extracellular DNA traps by basophils. J Immunol 192:5314–5323
Nakashima K, Hagiwara T, Yamada M (2002) Nuclear localization of peptidylarginine deiminase V and histone deimination in granulocytes. J Biol Chem 277:49562–49568
Nauseef WM, Kubes P (2016) Pondering neutrophil extracellular traps with healthy skepticism. Cell Microbiol 18:1349–1357
Neeli I, Radic M (2013) Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol 4:38. https://doi.org/10.3389/fimmu.2013.00038
Neeli I, Khan SN, Radic M (2008) Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol 180:1895–1902
Ng TH, Chang SH, Wu MH, Wang HC (2013) Shrimp hemocytes release extracellular traps that kill bacteria. Dev Comp Immunol 41:644–651
Nguyen DN, Jiang P, Frøkiær H, Heegaard PMH, Thymann T, Sangild PT (2016) Delayed development of systemic immunity in preterm pigs as a model for preterm infants. Sci Rep 6:36816. https://doi.org/10.1038/srep36816
Nierhaus A, Klatte S, Linssen J, Eismann NM, Wichmann D, Hedke J, Braune SA, Kluge S (2013) Revisiting the white blood cell count: immature granulocytes count as a diagnostic marker to discriminate between SIRS and sepsis–a prospective, observational study. BMC Immunol 14:8. https://doi.org/10.1186/1471-2172-14-8
Nupponen I, Turunen R, Nevalainen T, Peuravuori H, Pohjavuori M, Repo H, Andersson S (2002) Extracellular release of bactericidal/permeability-increasing protein in newborn infants. Pediatr Res 51:670–674
Nussbaum C, Gloning A, Pruenster M, Frommhold D, Bierschenk S, Genzel-Boroviczény O, von Andrian UH, Quackenbush E, Sperandio M (2013) Neutrophil and endothelial adhesive function during human fetal ontogeny. J Leukoc Biol 93:175–184
Palić D, Andreasen CB, Ostojić J, Tell RM, Roth JA (2007) Zebrafish (Danio rerio) whole kidney assays to measure neutrophil extracellular trap release and degranulation of primary granules. J Immunol Methods 319:87–97
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A (2010) Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 191:677–691
Patel S, Kumar S, Jyoti A, Srinag BS, Keshari RS, Saluja R, Verma A, Mitra K, Barthwal MK, Krishnamurthy H, Bajpai VK, Dikshit M (2010) Nitric oxide donors release extracellular traps from human neutrophils by augmenting free radical generation. Nitric Oxide 22:226–234
Pijanowski L, Golbach L, Kolaczkowska E, Scheer M, Verburg-van Kemenade BML, Chadzinska M (2013) Carp neutrophilic granulocytes form extracellular traps via ROS-dependent and independent pathways. Fish Shellfish Immunol 34:1244–1252
Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JAM, Tesselaar K, Koenderman L (2010a) In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 116:625–627
Pillay J, Ramakers BP, Kamp VM, Loi ALT, Lam SW, Hietbrink F, Leenen LP, Tool AT, Pickkers P, Koenderman L (2010b) Functional heterogeneity and differential priming of circulating neutrophils in human experimental endotoxemia. J Leukoc Biol 88:211–220
Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FHY, Surette MG, Sugai M, Bowden MG, Hussain M, Zhang K, Kubes P (2010) A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol 185:7413–7425
Pires RH, Felix SB, Delcea M (2016) The architecture of neutrophil extracellular traps investigated by atomic force microscopy. Nano 8:14193–14202
Plowden J, Renshaw-Hoelscher M, Gangappa S, Engleman C, Katz JM, Sambhara S (2004) Impaired antigen-induced CD8+ T cell clonal expansion in aging is due to defects in antigen presenting cell function. Cell Immunol 229:86–92
Prattichizzo F, Micolucci L, Cricca M, De Carolis S, Mensà E, Ceriello A, Procopio AD, Bonafè M, Olivieri F (2017) Exosome-based immunomodulation during aging: a nano-perspective on inflamm-aging. Mech Ageing Dev. https://doi.org/10.1016/j.mad.2017.02.008
Reichel M, Muñoz-Caro T, Sanchez Contreras G, Rubio García A, Magdowski G, Gärtner U, Taubert A, Hermosilla C (2015) Harbour seal (Phoca vitulina) PMN and monocytes release extracellular traps to capture the apicomplexan parasite Toxoplasma gondii. Dev Comp Immunol 50:106–115
Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M, Willems J, Vandenabeele P (2011) Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 21:290–304
Rocha JDB, Nascimento MTC, Decote-Ricardo D, Côrte-Real S, Morrot A, Heise N, Nunes MP, Previato JO, Mendonça-Previato L, DosReis GA, Saraiva EM, Freire-de-Lima CG (2015) Capsular polysaccharides from Cryptococcus neoformans modulate production of neutrophil extracellular traps (NETs) by human neutrophils. Sci Rep 5:8008. https://doi.org/10.1038/srep08008
Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT (2012) Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE 7:e32366. https://doi.org/10.1371/journal.pone.0032366
Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, Damti P, Lumbroso D, Polyansky L, Sionov RV, Ariel A, Hovav AH, Henke E, Fridlender ZG, Granot Z (2015) Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep 10:562–573
Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, Uehata T, Iwasaki H, Omori H, Yamaoka S, Yamamoto N, Akira S (2012) Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 12:109–116
Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, Insall RH, Stockley RA, Lord JM (2014) Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood 123:239–248
Sasaki S, Sullivan M, Narvaez CF, Holmes TH, Furman D, Zheng NY, Nishtala M, Wrammert J, Smith K, James JA, Dekker CL, Davis MM, Wilson PC, Greenberg HB, He XS (2011) Limited efficacy of inactivated influenza vaccine in elderly individuals is associated with decreased production of vaccine-specific antibodies. J Clin Invest 121:3109–3119
Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, Lell M, Manger B, Rech J, Naschberger E, Holmdahl R, Krenn V, Harrer T, Jeremic I, Bilyy R, Schett G, Hoffmann M, Herrmann M (2014) Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med 20:511–517
Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC (2002) G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity 17:413–423
Shi J, Gilbert GE, Kokubo Y, Ohashi T (2001) Role of the liver in regulating numbers of circulating neutrophils. Blood 98:1226–1230
Simell B, Vuorela A, Ekström N, Palmu A, Reunanen A, Meri S, Käyhty H, Väkeväinen M (2011) Aging reduces the functionality of anti-pneumococcal antibodies and the killing of Streptococcus pneumoniae by neutrophil phagocytosis. Vaccine 29:1929–1934
Simon HU (2003) Neutrophil apoptosis pathways and their modifications in inflammation. Immunol Rev 193:101–110
Slayton WB, Juul SE, Calhoun DA, Li Y, Braylan RC, Christensen RD (1998a) Hematopoiesis in the liver and marrow of human fetuses at 5 to 16 weeks postconception: quantitative assessment of macrophage and neutrophil populations. Pediatr Res 43:774–782
Slayton WB, Li Y, Calhoun DA, Juul SE, Iturraspe J, Braylan RC, Christensen RD (1998b) The first-appearance of neutrophils in the human fetal bone marrow cavity. Early Hum Dev 53:129–144
Söderberg D, Segelmark M (2016) Neutrophil extracellular traps in ANCA-associated vasculitis. Front Immunol 7:256. https://doi.org/10.3389/fimmu.2016.00256
Sperandio M, Quackenbush EJ, Sushkova N, Altstätter J, Nussbaum C, Schmid S, Pruenster M, Kurz A, Margraf A, Steppner A, Schweiger N, Borsig L, Boros I, Krajewski N, Genzel-Boroviczeny O, Jeschke U, Frommhold D, von Andrian UH (2013) Ontogenetic regulation of leukocyte recruitment in mouse yolk sac vessels. Blood 121:e118–e128
Steinberg BE, Grinstein S (2007) Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE 2007:pe11. https://doi.org/10.1126/stke.3792007pe11
Stuehr DJ, Fasehun OA, Kwon NS, Gross SS, Gonzalez JA, Levi R, Nathan CF (1991) Inhibition of macrophage and endothelial cell nitric oxide synthase by diphenyleneiodonium and its analogs. FASEB J 5:98–103
Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P (2014) Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther 16:R122. https://doi.org/10.1186/ar4579
Sur Chowdhury C, Hahn S, Hasler P, Hoesli I, Lapaire O, Giaglis S (2016) Elevated levels of total cell-free DNA in maternal serum samples arise from the generation of neutrophil extracellular traps. Fetal Diagn Ther 40:263–267
Suratt BT, Young SK, Lieber J, Nick JA, Henson PM, Worthen GS (2001) Neutrophil maturation and activation determine anatomic site of clearance from circulation. Am J Physiol Lung Cell Mol Physiol 281:L913–L921
Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, Ofrecio J, Lin M, Brenner MB, Olefsky JM (2012) Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18:1407–1412
Taneja R, Sharma AP, Hallett MB, Findlay GP, Morris MR (2008) Immature circulating neutrophils in sepsis have impaired phagocytosis and calcium signaling. Shock 30:618–622
Thammavongsa V, Missiakas DM, Schneewind O (2013) Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 342:863–866
Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, Schertzer JD, Larché MJ, Davidson DJ, Verdú EF, Surette MG, Bowdish DME (2017) Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 21:455–466.e4
Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, Wang Y, Simmons RL, Huang H, Tsung A (2016) Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res 76:1367–1380
Tortorella C, Stella I, Piazzolla G, Simone O, Cappiello V, Antonaci S (2004) Role of defective ERK phosphorylation in the impaired GM-CSF-induced oxidative response of neutrophils in elderly humans. Mech Ageing Dev 125:539–546
Tseng CW, Kyme PA, Arruda A, Ramanujan VK, Tawackoli W, Liu GY (2012) Innate immune dysfunctions in aged mice facilitate the systemic dissemination of methicillin-resistant S. aureus. PLoS ONE 7:e41454. https://doi.org/10.1371/journal.pone.0041454
Uhl B, Vadlau Y, Zuchtriegel G, Nekolla K, Sharaf K, Gaertner F, Massberg S, Krombach F, Reichel CA (2016) Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood 128:2327–2337
Ullah I, Ritchie ND, Evans TJ (2017) The interrelationship between phagocytosis, autophagy and formation of neutrophil extracellular traps following infection of human neutrophils by Streptococcus pneumoniae. Innate Immun 23:413–423
Urban CF, Reichard U, Brinkmann V, Zychlinsky A (2006) Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol 8:668–676
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A (2009) Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 5:e1000639. https://doi.org/10.1371/journal.ppat.1000639
Urquhart C, Currell R, Harlow F, Callow L (2017) Home uterine monitoring for detecting preterm labour. Cochrane Database Syst Rev 2:CD006172. https://doi.org/10.1002/14651858.CD006172.pub4
van der Linden M, Westerlaken GHA, van der Vlist M, van Montfrans J, Meyaard L (2017) Differential signalling and kinetics of neutrophil extracellular trap release revealed by quantitative live imaging. Sci Rep 7:6529. https://doi.org/10.1038/s41598-017-06901-w
Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, Runge MS, Madamanchi NR (2015) NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid Redox Signal 23:1389–1409
Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, Kaplan MJ (2011) Netting neutrophils induce endothelial damage, infiltrate tissues and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 187:538–552
von Köckritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, Medina E (2008) Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 111:3070–3080
Walker JC, Smolders MAJC, Gemen EFA, Antonius TAJ, Leuvenink J, de Vries E (2011) Development of lymphocyte subpopulations in preterm infants. Scand J Immunol 73:53–58
Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA (2009) Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 184:205–213
Wang Y, Wang W, Wang N, Tall AR, Tabas I (2017) Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arterioscler Thromb Vasc Biol 37:e99–e107
Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, Henriques-Normark B (2007) Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol 9:1162–1171
Wen F, White GJ, VanEtten HD, Xiong Z, Hawes MC (2009) Extracellular DNA is required for root tip resistance to fungal infection. Plant Physiol 151:820–829
Wen F, Curlango-Rivera G, Huskey DA, Xiong Z, Hawes MC (2017) Visualization of extracellular DNA released during border cell separation from the root cap. Am J Bot 104:970–978
Wenisch C, Patruta S, Daxböck F, Krause R, Hörl W (2000) Effect of age on human neutrophil function. J Leukoc Biol 67:40–45
Wong C, Goldstein DR (2013) Impact of aging on antigen presentation cell function of dendritic cells. Curr Opin Immunol 25:535–541
Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT (2009) Extracellular histones are major mediators of death in sepsis. Nat Med 15:1318–1321
Xu F, Zhang C, Zou Z, Fan EKY, Chen L, Li Y, Billiar TR, Wilson MA, Shi X, Fan J (2017) Aging-related Atg5 defect impairs neutrophil extracellular traps formation. Immunology 151:417–432
Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, Malawista SE, de Boisfleury CA, Zhang K, Conly J, Kubes P (2012) Infection-induced NETosis is a dynamic process involving neutrophil multitaskng in vivo. Nat Med 18:1386–1393
Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML, Chandler NB, Rodesch CK, Albertine KH, Petti CA, Weyrich AS, Zimmerman GA (2009) Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood 113:6419–6427
Yost CC, Schwertz H, Cody MJ, Wallace JA, Campbell RA, Vieira-de-Abreu A, Araujo CV, Schubert S, Harris ES, Rowley JW, Rondina MT, Fulcher JM, Koening CL, Weyrich AS, Zimmerman GA (2016) Neonatal NET-inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J Clin Invest 126:3783–3798
Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich GJ, Simon HU (2008) Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 14:949–953
Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU (2009) Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 16:1438–1444
Yu Y, Su K (2013) Neutrophil extracellular traps and systemic lupus erythematosus. J Clin Cell Immunol 4:139. https://doi.org/10.4172/2155-9899.1000139
Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, Burk RD, Kunisaki Y, Jang JE, Scheiermann C, Merad M, Frenette PS (2015) Neutrophil ageing is regulated by the microbiome. Nature 525:528–532
Zhang X, Zhuchenko O, Kuspa A, Soldati T (2016) Social amoebae trap and kill bacteria by casting DNA nets. Nat Commun 7:10938. https://doi.org/10.1038/ncomms10938
Zhong XY, Laivuori H, Livingston JC, Ylikorkala O, Sibai BM, Holzgreve W, Hahn S (2001) Elevation of both maternal and fetal extracellular circulating deoxyribonucleic acid concentrations in the plasma of pregnant women with preeclampsia. Am J Obstet Gynecol 184:414–419
Funding
This study was funded by the National Science Centre of Poland (grant number 2014/15/B/NZ6/02519).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ortmann, W., Kolaczkowska, E. Age is the work of art? Impact of neutrophil and organism age on neutrophil extracellular trap formation. Cell Tissue Res 371, 473–488 (2018). https://doi.org/10.1007/s00441-017-2751-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-017-2751-4