
Abstract
Auditory neuropathy (AN) is a unique type of language developmental disorder, with no precise rate of genetic contribution that has been deciphered in a large cohort. In a retrospective cohort of 311 patients with AN, pathogenic and likely pathogenic variants of 23 genes were identified in 98 patients (31.5% in 311 patients), and 14 genes were mutated in two or more patients. Among subgroups of patients with AN, the prevalence of pathogenic and likely pathogenic variants was 54.4% and 56.2% in trios and families, while 22.9% in the cases with proband-only; 45.7% and 25.6% in the infant and non-infant group; and 33.7% and 0% in the bilateral and unilateral AN cases. Most of the OTOF gene (96.6%, 28/29) could only be identified in the infant group, while the AIFM1 gene could only be identified in the non-infant group; other genes such as ATP1A3 and OPA1 were identified in both infant and non-infant groups. In conclusion, genes distribution of AN, with the most common genes being OTOF and AIFM1, is totally different from other sensorineural hearing loss. The subgroups with different onset ages showed different genetic spectrums, so did bilateral and unilateral groups and sporadic and familial or trio groups.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Auditory neuropathy (AN), a unique form of hearing impairment, is characterized by evidence of intact outer hair cell function, accompanied by poor eighth nerve-brainstem responses (Starr A 1996), affecting 1.2–8.4% of cases with hearing loss from different populations (Foerst et al. 2006; Penido and Isaac 2013; Vignesh et al. 2016) and 0.006–0.039% in low-risk newborn population (Korver et al. 2012). The term “auditory neuropathy” was first proposed in 1996, for the patients with normal hair cell activity but abnormal auditory nerve functions (Kaga et al. 1996; Starr A 1996). As a heterogeneous disorder, AN varies in several measures including age of onset (either congenital or acquired), hereditary (either sporadic or familial), affected sites (bilateral or unilateral), presence of peripheral/optic neuropathy (either non-syndromic or syndromic), and clinical manifestation. The site of lesion causing AN may involve the presynaptic site, the synapses, and the postsynaptic site.
The diagnosis of AN mainly relies on audiological and electrophysiological testing. Previous studies of pathophysiological mechanisms of AN have shown the diagnostic criteria for presynaptic disorders (including inner hair cells and synapse disorders) and postsynaptic disorders (Rance and Starr 2015). Confusingly, patients with AN, who have similar clinical manifestations, may have different damaged anatomical site. It is difficult to distinguish dysfunction of the inner hair cells and synapses related AN from auditory nerve disorders. The precision lesion site of AN is difficult to be identified by a single pathophysiological test. It can be caused by damage of inner hair cell, synapse, auditory nerve, or the combination of these sites. Genetic testing may provide a new perspective for the diagnosis and management of AN.
Both genetic factors and environmental risk factors are attributed to AN, such as hyperbilirubinemia, anoxia, and immunological and infectious factors (Starr A 1996; Starr et al. 2000), with about 50% having no defined etiology. Thirteen genes have been reported to be related to AN, showing implications for expected cochlear implantation outcomes (Shearer and Hansen 2019), including genes that affected presynaptic site (OTOF), synaptic site (CACNA1D, CABP2, SLC17A8), postsynaptic site (spiral ganglion, DIAPH3, OPA1, ROR1, ATP1A3), and auditory nerve (spiral ganglion cell bodies and proximal axons, TIMM8A, AIFM1, NARS2, MPZ, PMP22). Genetic diagnosis for patients suffering from AN has critical implications for treatment, prognosis, and development of precision medicine strategies. However, to date, no large AN cohort has been built to decipher the diagnostic yield of high-throughput sequencing.
The molecular diagnostic rate of AN varies greatly, which is supposed to be different from other genetic hearing loss, the most common attributing genes of which are GJB2 and SLC26A4. Recently, genetic studies are playing an increasingly crucial role in disease diagnosis and management. Next-generation sequencing has revolutionized the discovery of rare Mendelian disease-related gene variants (Yang et al. 2014). The yield of genetic testing of AN varies based on the accompanied phenotype (syndromic and non-syndromic AN), onset age (infant and non-infant AN), affected sides, etc. In this study, we recruited 311 cases who were clinically diagnosed as AN and performed genome sequencing to detect and quantify genetic variants and assess existing therapeutic effects (Fig. 1). Our findings have implications for the future development of precision medicine strategies.

Study design and flowchart. Trios, patient with auditory neuropathy plus both parents; AN, auditory neuropathy; P, pathogenic; LP, likely pathogenic; PP, possible pathogenic; HHL, hereditary hearing loss; BAN, bilateral auditory neuropathy; and UAN, unilateral auditory neuropathy
Materials and methods
Study participants (cases recruitment and clinical evaluations)
A total of 311 cases clinically diagnosed with AN were identified by the Institute of Otolaryngology, Chinese PLA General Hospital, between January 2003 and December 31, 2020 (Fig. 1). Personal or family medical reports were identified by a team of experienced physicians and audiologists. Audiometric evaluations included audiogram, auditory brainstem responses (ABR), distortion product otoacoustic emission (DPOAE), electrocochleography (EcochG), and speech discrimination score (SDS). Pure-tone averages (PTA) were defined as average threshold of 500, 1000, 2000, and 4000 Hz frequencies (Thorpe et al. 2021), as well as in all frequencies. Children under six years old were evaluated by auditory steady-state response (ASSR) and behavioral audiometry. The severity of hearing impairment was defined as mild (26–40 dB HL), moderate (41–55 dB HL), moderately severe (56–70 dB HL), severe (71–90 dB HL), or profound (> 90 dB HL) according to middle frequencies PTA. Cochlear microphonic (CM) was conducted including the use of inserting earphones, reverse polarity, and blocking the stimulus tube to eliminate electrical artifact interference. High-resolution computed tomography (HRCT) and/or MRI was also performed on some of the cases to verify whether the family members had complications other than hearing disorders.
Inclusion criteria were cases who were diagnosed as AN by 1) the presence of OAE and/or CM and 2) the abnormal or absence of ABR (Chinese Multi-center Research Collaborative Group on Clinical et al. 2022; Constantina Georga et al. 2019; Deborah Hayes et al. 2008; Judy Gravel et al. 2004; Mona M. Dworsack-Dodge et al. 2012; Rachel Feirn et al. 2013). Exclusion criteria were 1) acoustic neuroma and 2) cases or the guardians who refused genetic testing. For the cases with family history, only the proband was recruited in this study, and the family members were only recruited for segregation analysis, instead of phenotype analysis.
Searching for AN-associated genes in OMIM
We used the clinical synopsis advanced search to find 494 entries for “auditory neuropathy” from Online Mendelian Inheritance in Man (OMIM), which is a database that contains all known Mendelian inheritance diseases, as well as clinical synopses that can be used to find a target gene. The following were the inclusion criteria: 1) phenotypic title includes “auditory neuropathy”, 2) clinical synopsis description includes “auditory neuropathy”, or 3) clinical aspects of reported patients include “hearing loss” or “deafness”. After that, we assembled 138 items including phenotype, gene, and inheritance information. When we combined genes from PubMed, the MGI database and common genes for genetic hearing loss, we got a total of 178 genes (Supplementary Data 1) that could be associated to AN. The genetic pattern of these 178 genes varies: 44 genes are autosomal dominant, 94 genes are autosomal recessive, 23 genes have a complicated genetic pattern that includes autosomal dominant (AD), autosomal recessive (AR), and multifactorial, and 11 genes are linked to the X chromosome.
We examined the interaction among these genes using STRING (https://www.string-db.org/), and the PPI enrichment p-value was less than 1.0e−16, indicating that the majority of these genes were related to one another and could form a network with substantial interactions. Outside of the network were 19 genes (PTRH2, PORCN, RAI1, MCM3AP, TMEM43, FITM2, ADPRHL2, ORC1, ROR1, MORC2, PIGB, KIAA0586, TMEM30B, GPRASP2, SLC52A2, SNX10, TSHZ1, and GGT1). To rule out the possibility of this link being random, we used random sampling to obtain three groups of genes with the same number of genes associated with AN (178). There seemed to be no substantial interaction among any of the three gene sets.
Sequencing and bioinformatics analysis
Whole-genome sequencing (WGS, 233 cases) and clinical targeted panel sequencing (hereditary hearing loss (HHL) Panel, 78 cases) were all options for our study. The HHL Panel approach had previously been described, which focused on genes related to monogenic hearing loss (Wang et al. 2014, 2018) (Supplementary Data 2). The WGS pipeline consists of eliminating low-quality reads and adaptor reads, mapping reads to the reference genome (GRCh37) by BWA and SOAPnuke, detecting single-nucleotide polymorphisms (SNPs) and small insertion or deletion (InDel) via GATK, and finally generating a VCF file for each sample. We used VEP to annotate the detail information for SNP and small InDel containing the gene name, transcript, mutation consequence, and allele frequency in public databases (gnomAD, 1000 Genome Project, ExAC) based on the VCF file. We matched variants to all versions of a gene’s transcript depending on physical location throughout the annotation process and then output two sets of annotation results: the most severe transcript result and the authoritative transcript result. This set was to avoid missing disease-causing variants when just utilizing an authenticated transcript.
We used the following criteria to filter SNPs and small InDels from WGS data: 1) removing variants with population frequencies greater than 0.005 in the public database (gnomAD v2.1.1); 2) reserving variants located on the coding region of 178 AN-associated genes; and 3) reserving variants that may affect the structure and function of protein through consequence type (splice_acceptor_variant, splice_donor_variant, stop_gained, frameshift_variant, stop_lost, start_lost, inframe_insertion, inframe_deletion, missesnse_variant, TFBS_amplification). Following the filtering process, each sample yields fewer variants.
Then, using the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) (Oza et al. 2018; Patel et al. 2021; Richards et al. 2015), we classified the variants identified in WGS and panel sequencing as pathogenic (P), likely pathogenic (LP), variants with uncertain significance (VUS), likely benign (LB), and benign (B). Then, in two steps—true variant verification and co-segregation analysis within pedigrees—we employed Sanger sequencing to verify the variation classified as P, LP, or VUS and further classifed VUS to VUS, VUS|LP, and VUS|P. For patients with these variants, we classified the genetic testing result according to the inheritance pattern, ACMG variant classification, heterozygous or homozygous into pathogenic, likely pathogenic, possible pathogenic, carrier, and unknown (Supplementary Table 1).
Drug gene interaction analysis
Drug-Gene Interaction database (http://dgidb.org/) was used for drug–gene interaction analysis; “search Drug-Gene Interactions” and “search Potential Druggability” were selected, respectively, to search for the known pathogenic genes of AN. All the analyses and visualizations were conducted in R 4.2.1. The following R package was used: ggplot2 package.
Gene function analysis
Enrichment analysis was performed on the candidate genes using the database established by the Gene Ontology Consortium, focusing on biological process (BP), cellular component (CC), and molecular function (MF). All the analyses and visualizations were performed in R 4.2.1. The R package involved was clusterProfiler package (used for enrichment analysis). Sankey + dot plot was plotted by https://www.bioinformatics.com.cn (last accessed on July 10, 2023), an online platform for data analysis and visualization.
Gene expression analysis
The expression sites of the AN new genes were analyzed using single-cell sequencing data published by van der (van der Valk et al. 2023) and Philippe Jean (Jean et al. 2023). All the data mentioned were publicly available and were analyzed online using gGAR: Gene Expression Analysis Resource portal for community-driven, multi-omic data exploration. The gene expression pattern map was created with BioRender.com.
Cochlear implantation/hearing aid management of AN in clinic
Bilateral AN patients in this study were followed up via phone interviews, during which management strategies, aided hearing sensitivity, and speech recognition after cochlear implantation or hearing aids were collected through a unified common questionnaire. Auditory and speech abilities were evaluated by the categories of CAP and SIR (Archbold et al. 1995; Miyamoto et al. 1999; Song et al. 2021).
Statistical analysis
Data were analyzed using the Prism 6.0 statistical analysis program (GraphPad). A two-tailed value for p < 0.05 was considered statistically significant. Continuous data were presented as the mean and standard deviation (SD), and categorical variables were reported as the frequency and percentage. Fisher’s exact test or chi-square test was used to assess the difference in the mutation frequencies between the persons from different subgroups of auditory neuropathy.
Results
Demographic features of patients
A total of 311 AN cases received high-throughput sequencing were ascertained in this study (Table 1), showing no gender preference: 47.9% (149/311) of our samples were female, and 52.1% (162/311) were male. The ethnic background of the probands was primarily Han (n = 285, 91.6%). The cases with newborn risk factors of jaundice or hyperbilirubinemia, infection, ototoxic drugs, and hypoxia or asphyxia were 19, 12, 12, and 9, respectively.
The onset age of AN occurred at all ages, but the typical onset age of AN was in the early childhood and during adolescence. The medium age of disease onset in this cohort was 10.9 ± 7.7 years old, ranging from 0 to 40 years old for bilateral AN, 3.7 ± 3.3 for unilateral AN. Patients were classified based on onset age: infant (0–3 years old, 92 patients, 29.6%) and non-infant (> 3 years old, 219 patients, 70.4%, including 1 case with unclear onset age, and 49 cases ≥ 18 years old); affected sides: bilateral AN (291 patients, 93.6%) and unilateral AN (20 patients, 6.4%); and family history: sporadic AN (295 patients, 94.9%) and familial AN with more than one member were diagnosed as AN in a family (16 patients, 5.1%, Supplementary Fig. 1).
Among the 291 bilateral AN cases, 4.9% (4/82) of infant AN and 65.1% (136/209) of non-infant AN complained tinnitus; 1.2% (1/82) of infant AN and 5.7% (12/209) of non-infant AN with vertigo or dizziness; 4.9% (4/82) of infant AN and 8.6% (18/209) of non-infant AN with movement disorder and numbness of limbs; 26.8% (22/82) of infant AN and 4.3% (9/209) of non-infant AN showed severe communication disorders; and 4.9% (4/82) of infant AN and 18.2% (17/209) of non-infant AN complained vision loss, among whom five cases were diagnosed of optic nerve atrophy.
Audiological, electrophysiological, and imaging (Table 2 and Supplementary Table 2)
Pure-tone thresholds ranged from normal to total deafness, with the PTA in all frequencies 49.9 dB HL. The PTA in all frequencies of the patients in infant group and non-infant group were 90.4 and 44.0 dB HL, respectively. The audiograms were various, and the most common type was ascending in non-infant group and total deaf in infant group. Most of the bilateral AN cases (81%) had symmetrical audiograms (Supplementary Fig. 2). A total of 298 ears had speech audiometry data in the first consultation, the PTA was 42.13 ± 18.65 dB HL, and the SDS was 40.27 ± 30.07%. There was a significant correlation between speech recognition rate and average hearing threshold (p = 0.001, Supplementary Fig. 3). To compare the correlation between the pure-tone hearing threshold of each frequency or the ASSR threshold of each frequency and the speech recognition rate, the ASSR (especially 1 kHz on the left) may have a better correlation with speech recognition rate, from the perspective of the correlation coefficient for bilateral AN (Supplementary Fig. 3).
The elicitation rate of DPOAE in different frequencies ranged from 57.6 to 81.95%, and the 500 Hz had the lowest elicitation rate. The overall elicitation rate in infant group was lower than non-infant group, and the low frequency (0.5–1.5 kHz) extraction rate was less than half; while in the non-infant group, the extraction rate was relatively low in 500 Hz (Supplementary Fig. 4). A total of 241 ears could record CM waves, and the presence rate in infant and non-infant group was 79.9% and 26.4%, respectively. For ABR testing, most ears (92.4%, 556/602) did not elicit a response at the maximum sound intensity (> 100 dBnHL), and 46 ears (7.6%) could elicit ABR waveforms, but the waveform differentiation was severely abnormal, with three ears (6.5%) only showing I wave, four ears (8.7%) eliciting I.III.V wave, but the amplitudes were abnormal, and the remaining 39 ears (84.8%) only had V wave. A total of 244 ears were conducted electrocochleogram, 17 ears showed no response, and 1 ear only recorded action potential (AP), 28 ears only recorded summing potential (SP), the SP/AP ratios were available in 198 ears, with 20 ears < 0.6, 67 ears ranged from 0.6 to 1, and 111 ears > 1, respectively. Audiological and electrophysiological features of the affected ears of unilateral AN were shown in Supplementary Data 3.
Fifty-five probands received temporal CT, and most of them showed no obvious abnormalities. Ninety-five cases, including 12 unilateral auditory neuropathy (UAN) cases and 83 bilateral auditory neuropathy (BAN) cases, were performed MRI scanning, among which 25 cases (26.3%, 25/95) showed unilateral or bilateral auditory nerve abnormalities, including slender, unclear, or missing auditory nerves. Among the 25 cases with abnormal MRI, six cases were unilateral AN, and 19 cases were BAN.
Following-up
For patients who have visited our hospital more than two times (139 cases in total), the audiological testing of the latest and the first time was compared. The average follow-up period was 42.84 ± 4.54 months, ranging from 1 to 301 months. The middle-frequency PTA was used to evaluate the progression of the affected ears, and PTA increase more than 10 dB, change ≤ 10 dB, and decrease more than 10 dB were determined as deterioration, no change (or fluctuation), and improvement. The deterioration, no change, and improvement ears were 57 (33.7%), 80 (47.3%), and 32 (19.0%), respectively. The progression showed no difference between infant and non-infant groups. Thirty-two cases (63 ears) with the following SDS were analyzed, with a follow-up period of 47.03 ± 12.87 months, ranging from 1 to 301 months. There was no significant difference between the first and last SDS, the average scores of which were 28.16 ± 2.953%, and 32.86 ± 3.237%, respectively.
Genetic spectrum in this AN cohort
For the WGS data from 233 cases, we had a total of 541 candidate disease-related SNPs and InDel located on 138 genes after the selection process, including variants classified as VUS, LP, and P. A total of 69 variants were shared by two or more samples, and 472 variants appeared only once. A total of 200 AN patients had these variants.
If more than two patients have the same genotype, the evidence for a link between genotype and phenotype is higher. Further, according to ACMG variant classification, inheritance pattern, and heterozygous or homozygous type, we classified the variants as carrier, unknown, possible pathogenic, likely pathogenic, and pathogenic variants from the perspective of subjects instead of variants only (Supplementary Table 1). A total of 90 cases (38.6%, 90/233) were identified with possible pathogenic variants, and 73 cases (31.3%, 73/233) were identified with pathogenic or likely pathogenic results. For the cases with pathogenic or likely pathogenic variants, the top gene was AIFM1, which had likely pathogenic or pathogenic variations in 23 samples, followed by OTOF, which had compound heterozygous variations in 17 of the samples. MYO7A (nine samples), TWNK (seven samples), WFS1 (six samples), and ATP1A3 (five samples) were the next four genes. More than five samples shared each of these six genes. Totally 142 cases were carriers, with 233 variants of 68 recessive genes, the top three genes were SLC52A2, SLC52A3, and SLC26A4, with 18, 15, and 14 carriers, respectively.
Eleven genes were identified by both of the methods, including AIFM1, ATP1A3, FDXR, GJB2, MYO7A, NARS2, OPA1, OTOF, SLC17A8, SLC26A4, TIMM8A, TSPEAR, and WFS1. Among these genes, likely pathogenic and pathogenic variants were identified in AIFM1 (5 samples), ATP1A3 (2 samples), OPA1 (4 samples), OTOF (12 samples), TIMM8A (1 sample) from the panel sequencing.
Cross WGS and panel sequencing methods, 73 cases (31.3%, 73/233) by WGS methods and 25 cases (32.0%, 25/78) from panel sequencing with likely pathogenic or pathogenic variants were identified. In summary, the eight most common genes in our cohort were OTOF (29 samples), AIFM1 (28 samples), MYO7A (9 samples), ATP1A3 (7 samples), WFS1 (6 samples), OPA1 (5 samples), NOTCH3 (4 samples), and TWNK (4 samples) (Table 3). For the most common gene in hearing loss population, one case had GJB2 compound heterozygous mutation (1,007,133), with infant onset bilateral profound hearing loss and presence of otoacoustic emission (OAE), and two cases found GJB2 heterozygous mutation (1,507,400 and 1,006,855).
Collectively, 584 candidate variants in 139 genes of 243 cases were identified to be P, LP, or VUS, accounting for 11.6% (68/584), 15.9% (93/584), and 72.4% (423/584), respectively. The distribution of the top 50 genes can be seen in Fig. 2A, based on the number of variants and whether they were novel or reported. Previously reported variants were those identified to be damaging according to ClinVar or published studies.

Overview of the genetic spectrum in the AN cohort. A Distribution of the top 50 genes of the number of sites, including both novel and reported variants. B The diagnostic rates for infant and non-infant groups. C Genes contribution of the 98 patients with P/LP variants. D The mutation type of P/LP variants in cases of infant and non-infant groups. E The prevalence of P/LP variants in cases of infant and non-infant groups. P/LP, pathogenic or likely pathogenic
A total of 154 variants spanning 23 genes were identified and classified as pathogenic or likely pathogenic in 98 patients (31.5%) (Fig. 2C and Supplementary Data 4). Among them, 73/154 variants (47.4%) were previously reported as pathogenic, and 81/154 variants (52.6%) were novel. Among the 154 variants, 113 were missense variants, 13 were frameshift variants, 10 were stop_gained variants, 9 were splicing variants, 7 were inframe variants, and 1 was start_lost variant (Fig. 2D). Further categorization of the genotypic and phenotypic data of 98 patients with confirmed pathogenic genes was conducted (Supplementary Data 5).
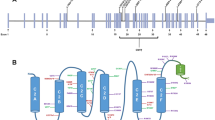
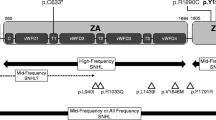
Among the 23 genes with pathogenic or likely pathogenic variants, 7 genes were known AN causative genes, and 16 genes were novel candidate genes (Fig. 3A). The P/LP/VUS ratio of each gene and the number of gene sites in infant/non-infant groups were shown in Fig. 3B and C. Furthermore, the Drug-Gene Interaction database (DGIdb) was used to search for drug–gene interactions and potential druggability (Supplementary Data 6, Supplementary Table 3). The results of potential druggability were visualized in an upset plot (Fig. 3D).

Overview of genes with P/LP variants. A The genes into known causative genes and novel candidate genes. B The P/LP/VUS ratio of each gene. C The number of gene sites in infant/non-infant groups. D The upset plot of potential druggability of each gene. P, pathogenic; LP, likely pathogenic; and VUS, variants with uncertain significance
The mode of inheritance was autosomal recessive for 30 cases, autosomal dominant for 29 cases, X-linked for 26 cases, and 13 cases with more than two diagnoses (Table 4).
Genetic spectrum in different subgroups
Testing of probands concurrently with parents (trio; n = 68) significantly improved the molecular diagnostic yield from 22.9% (52/227, proband-only) to 54.4% (37/68) (chi-square test, p < 0.05). Among trios with positive diagnostic results (n = 37), 64.9% (24/37) were inherited in an autosomal recessive manner, 25.0% (9/36) were inherited in an autosomal dominant manner, including six autosomal dominant de novo variants, and 11.1% (4/36) were X-linked. Testing of families with more than one patient also improved the molecular diagnostic yield, 56.2% (9/16) families were identified as likely pathogenic or pathogenic variants, including five X-linked (AIFM1 gene), two autosomal dominant (OPA1 gene), and two autosomal recessive (OTOF gene) inheritance.
The subgroups with different onset ages of hearing loss showed different variants spectrum. The diagnostic rate in the infant group was 45.7% (42/92), which was significantly higher than non-infant patients who had disease onset more than 3 years old (25.6%, 56/219) (Fig. 2B). The diagnostic rates of the 23 genes with P or LP variants between infant and non-infant groups were plotted (Fig. 2E). In total, we identified 50 likely pathogenic and pathogenic variants in OTOF, only one patient with AN onset within the non-infant age, and the distribution showed a significant difference. We identified 13 variants in AIFM1 gene, no (0%) patients with AN in the infant period, and the distribution showed a significant difference (p < 0.0001).
No pathogenic or likely pathogenic variants were identified in the cases with unilateral auditory neuropathy.
Gene function and expression analysis
Gene function clustering analysis was performed for biological process, cell component, and molecular function on the 139 genes of 584 candidate variants (Fig. 4 and Supplementary Data 7). The top 5 biological processes were sensory perception of sound, sensory perception of mechanical stimulus, myelination, ensheathment of neurons, and axon ensheathment. The top 5 cell components were presynapse, peroxisomal membrane, microbody membrane, peroxisome, and microbody. The top 5 molecular functions were NADH dehydrogenase activity, oxidoreduction-driven active transmembrane transporter activity, NADH dehydrogenase (ubiquinone) activity, NADH dehydrogenase (quinone) activity, and NAD(P)H dehydrogenase (quinone) activity.

Genes function clustering analysis of the 139 genes. A The top 5 biological processes of the 139 genes. B The top 5 cell components of the 139 genes. C The top 5 molecular functions of the 139 genes
In this study, a total of 34 genes with pathogenic, likely pathogenic, or possible pathogenic variants were defined as novel AN genes (Supplementary Table 4). The expression analysis of these novel AN genes was performed using the single-cell sequencing data of the human cochlea published by van der (van der Valk et al. 2023). A comparative heatmap of the expression between 9.2-week-old infants (cochlea not fully differentiated) and adult human cochlea at the single-cell level was presented in Supplementary Fig. 5, to explore the expression patterns of the new genes during different stages of cochlear development. Further analysis was made on the expression of AN new genes in different cells of the cochlea for the data of adult human cochlea, and the results were shown in Supplementary Fig. 5B.
Since the data analyzed in van der et al.’s article were classified according to the cochlea at the developmental stage, the cells such as inner and outer hair cells of cochlea could not be distinguished. Further, using the single-cell sequencing results of mouse cochlea published by Philippe Jean (Jean et al. 2023), detailed cell type expression analysis of the expression location of the novel AN gene was performed. The expression of different cell types in P8 and P20 mice was analyzed, respectively, and the results were shown in Supplementary Fig. 6A and B and were further drawn into the gene expression pattern map, as shown in Supplementary Fig. 6C and D.
Cochlear implantation interventions and effectiveness
A total of 136 bilateral AN cases were followed up successfully via phone review (a unified common questionnaire, Supplementary Table 5), among which 41 cases (30.15%, 41/136) in this study received cochlear implantation (CI), the surgery age of who ranged from 10 months to 342 (336) months. Of the 41 cochlear implant recipients, 4 cases (9.76%, 4/41) had bilateral implants, 19 cases (46.34%, 19/41) had monaural implants (6 left, 13 right), and 18 cases (43.90%, 18/41) had a hearing aid with a cochlear implant (bimodal hearing). The follow up of intervention was conducted in 41 patients, and the intervention duration was 5.98 ± 3.62 years. Eighty-five patients had no intervention of hearing aid or CI, and 10 patients received hearing aids only. The medium points of categories of auditory performance (CAP) scores in the CI group, CI + hearing aid (HA) group, HA group, and no intervention group were 6 (1–7), 6 (5–6), 5 (4–7), and 5 (0–7), respectively (Supplementary Fig. 7). The medium points of speech intelligibility rating (SIR) score in the CI group, CI + HA group, HA group, and no intervention group were 4 (0–5), 4 (2–5), 3 (2–5), and 5 (1–5), respectively (Supplementary Fig. 7). There was no difference in CAP and SIR scores among different intervention models. Cochlea implant outcome in cases with genetic diagnosis showed in Supplementary Table 6.
Discussion
To the best of our knowledge, this is the largest study interrogating the genetic spectrum in cases with AN, and this cohort study first demonstrates the genetic spectrum in different subgroups of AN, especially the different onset ages. Previous genetic studies of AN focused on cases with one gene based on families, case series, or single case report. In our cohort, AN can be unilateral or bilateral, congenital infant or acquired non-infant, and producing all degrees of hearing loss. Genes distribution of AN is totally different from sensorineural hearing loss, with different molecular diagnostic yields by different targeted genes analysis and in different phenotypic subgroups.
Phenotypic characteristics of the cohort
Most cases (93.6%) showed bilateral auditory neuropathy, and fewer cases (6.4%) showed unilateral AN, with another side presenting normal hearing or sensorineural hearing loss. Among the 20 unilateral AN cases recruited in this study, twelve cases received MRI scanning, and six cases showed cochlear nerve deficiency (CND) (50.0%, 6/12), and relatively rare genetic causes were identified.
Hearing abilities between infant and non-infant AN groups were different, infant AN showing much more severe hearing loss. Hearing threshold showed great individual differences in patients with AN, and the hearing of some cases may be improved, may remain stable for a long time, and may further deteriorate during follow-up period. Averagely, the pure-tone average threshold was stable, but some cases showed progressive worsening hearing ability involving loss of OAEs over time, as well as progressive decreased in speech recognition scores. The PTA and the SDS in quiet varied so great, and neither of them can provide significant information for the evaluation of AN. Only 7.6% of ears in our cohort could elicit at least one wave, lower than the previous study of 25% (Berlin et al. 2010).
The EcochG potential was composed of three components, including CM, SP, and AP. Most ears (81.1%) in our group showed both SP and AP, and the SP/AP ratios were > 1 for more than half ears. In our cohort, EcochG of 244 ears was recorded, which is hard to classify to the three modes being described in the previous study to locate the affected lesion side (Santarelli et al. 2008). Studies have shown that EcochG, which measures cochlear and early neural responses as acoustic signals are transmitted from cochlear to the brain, may have the role of locating the lesions of auditory neuropathy. CM waves were recorded in 241 ears (77.5%); in addition to proving the existence of outer hair cell function, CM may also be used to speculate the location of AN lesions (Shi et al. 2012).
A total of 26.3% of AN cases showed auditory nerve abnormalities in MRI, including 6 unilateral and 19 bilateral cases. In the cases with cochlear nerve deficiency, about 10% showed possible AN with the presence of DPOAE (Matsuura et al. 2021). The previous study showed that most UAN cases were the result of auditory nerve hypoplasia (Buchman et al. 2006); in our cohort, 50% (6/12) of UAN cases showed CND, while another 50% showed normal cochlear nerve, showing no difference with our previous data (38.89%, 7/18) (Song et al. 2021).
Genotype analysis of AN
High-throughput sequencing in AN patients identified pathogenic and likely pathogenic variants with a prevalence of 31.5%. Most AN subjects were sporadic cases with no family history (94.9%, 295/311), and the inheritance pattern include de novo, autosomal dominant, AR, and X-linked. For patients with family history and trio cases concurrently with parents, the diagnostic rate of likely pathogenic and pathogenic variants were 56.2% and 54.4%, higher than the proband-only testing results. To improve the molecular diagnostic rate, we could perform follow-up of the proband-only patients. More than one hundred cases were carriers in our cohort, who can acquire information about whether they have an increased risk of conceiving a child affected with an autosomal recessive or X-linked condition (Sparks 2020; Van Steijvoort et al. 2020). In addition, the analysis of copy number variants for the cases carries one pathogenic and likely pathogenic variants in future may provide much more molecular information for AN, and the underlying mechanism of digenic inheritance of some cases in this cohort.
There were 9 genes that were mutated in single cases and 14 genes that had pathogenic and likely pathogenic variants in two or more patients. The identification of genes mutated in two or more cases provided strong evidence for their role in AN. When genetic counseling was provided, the second pathogenic variant should be taken into consideration. OTOF and AIFM1 are the top two common genes in our cohort, the sites of which are presynaptic and postsynaptic lesions, affecting infant and non-infant AN cases, respectively. OTOF is the most common gene in our infant subgroup, previous studies showed that biallelic OTOF variants cause congenital or early onset (n = 114) hearing impairment mainly, and only few variants have been identified with progressive hearing loss (n = 3) (Vona et al. 2020). Gene therapy has achieved improvement in AN mouse models, and research studies have rescued partial hearing of Otof−/− mouse by single-AAV or dual-AAV mediated system (Akil et al. 2019; Al-Moyed et al. 2019; Rankovic et al. 2020), and further leading to long-lasting hearing reversal by intein-mediated protein recombination (Tang et al. 2022). AIFM1 is the most common gene in our non-infant subgroup, and pathogenic variants in AIFM1 have also been identified in cases with combined oxidative phosphorylation deficiency 6, Cowchock syndrome, and spondyloepimetaphyseal dysplasia. Patients with AIFM1 mutations are supposed to have poor effects from cochlear implantation, and the functional studies of its role in caspase-independent cell death and mitochondrial metabolism may provide potential therapy targets for AIFM1-related AN. AIFM1 is the most common gene in the non-infant subgroup. As far as we are concerned, the disruption of AIFM1 may have a cumulative effect on impairing hearing function and pathogenic auditory pathways. When the auditory pathways are exposed to external sounds, noise, or other forms of negative stress, there is a gradual impairment in normal function, which is manifested as delayed progressive AN.
Exome sequencing, especially HHL-related gene panel, is currently used as a first-tier diagnostic test for individuals with hearing loss. The panels cover most of the genes of HHL, including some of the AN-related genes. The major genetic causes of integral hearing loss are GJB2 and SLC26A4 (Patel et al. 2021), while specific AN is totally different. GJB2 is the most common gene in non-syndromic hearing loss population, and the c.235delC mutation in GJB2 is the most frequently known mutation in some East Asian groups, with a carrier frequency of approximately 1% (Wang et al. 2021a). However, in our study group with AN, only one pair of compound heterozygous mutation and two heterozygous mutations were identified, with no c.235delC. Previous studies showed some patients with GJB2 mutations were audiological confirmed AN (Cheng et al. 2005; Santarelli et al. 2008). It is still confusing and unclear how GJB2 mutation selectively affects the afferent compartment of the cochlea but leaving unchanged out hair cell function (normal OAE and/or CM); further research is needed to clarify the GJB2 genotype and AN phenotype correlation. For the SLC26A4, 5.8% (18/311) cases in our group were carriers, with no definite molecular diagnosis.
Through functional analysis, we found that the biological process of the candidate pathogenic genes in AN was mainly concentrated on sensory perception of sound and sensory perception of mechanical stimulus. Cell components were mainly concentrated in presynapse and peroxisomal membrane. Molecular function was mainly concentrated in NADH dehydrogenase activity and oxidoreduction-driven active transmembrane transporter activity. These results indicate that the pathogenic mechanism of AN is mainly concentrated in the upstream and downstream of neuronal synapses, providing evidence for the molecular biological function and selection of therapeutic targets for AN. Then, the potential drug targets and possible effective medicine were found in transporter, druggable genome, and clinically actionable categories through the analysis of the drug associations of AN pathogenic genes, providing evidence for further drug treatment of AN.
By analyzing the expression sites of AN new genes in the cochlea, it was found that the genes are expressed in different developmental stages of the cochlea, and the expression cells are more diverse in non-infants. The expression sites are mainly concentrated in type I neurons and type II neurons. Our findings provide new evidence for the classification of gene expression sites in AN.
Management and consultation of AN
The identification of pathogenic and likely pathogenic variants in AN cases may result in genomics-informed changes in clinical care for some patients. Genetic diagnosis may provide a better understanding of affected lesions; positive results were identified in OTOF, CACNA1D, OPA1, ATP1A3, TIMM8A, PMP22, and NARS2 in this study. Among these genes, OTOF and CACNA1D are synapse-related genes, OPA1 and ATP1A3 are postsynaptic spiral ganglion-related genes, and patients with pathogenic mutations of these genes are supposed to have significant effects from CI. The effectiveness of CI in patients with OTOF gene mutations has been reported in more than 60 cases (Zheng and Liu 2020). While TIMM8A, PMP22, and NARS2 are postsynaptic auditory nerve-related genes, patients with these gene mutations may receive few benefits from CI. In addition, some patients had variants in genes previously associated with syndromic hearing loss, and AN may be a biomarker for neurodegenerative disease, as the first symptom of patients with neurodegenerative conditions (Rance and Starr 2015; Starr A 1996), such as Friedreich ataxia and Charcot-Marie-Tooth disease, CAPOS (Wang et al. 2021b), MTS (Wang et al. 2019), et al.
The CI results are an area of controversy for AN, which is unpredictable. Since the first report on the implantation of CI in AN patients in 1999 (Miyamoto et al. 1999), many studies have shown effective CI results in AN patients (Daneshi et al. 2018; Shearer and Hansen 2019). In general, CI improves the pure-tone threshold and the speech recognition score (Lin et al. 2022), but there is general agreement that outcomes are poor in patients with an absent cochlear nerve or severely narrowed internal auditory canal (Papsin 2005). The exact sites of the lesion of AN can determine the CI performance. Lesions located in the presynaptic and dendrite are associated with good CI performance, while the lesions in the ganglion cells and proximal axon are not. For example, patients with OTOF homozygous or compound heterozygous mutations are supposed to have good CI outcomes, while patients with AIFM1 or TIMM8A mutations are supposed to have poor CI results. Since patients with AIFM1 or TIMM8A mutation in this study showed no severe or profound hearing impairment, CIs were not chosen by the patients or the guardians.
There are some limitations in this study, indicating the future study directions of AN genotype and phenotype correlation. First, different instead of consistent sequencing strategies were performed on the patients; however, all variants were analyzed and evaluated for pathogenicity using standards guidelines. In this study, we focused on the AN-related genes for analyzing WGS data and HHL-related genes for panel data and further compared the different positive rates of these two methods. Second, some cases in this cohort have not available parental samples to assess for variant inheritance, resulting in a conservative result since some do novo variants may be missed. Third, we did not discuss the subgroups of non-syndromic or syndromic AN in this study, since the presence of peripheral/optic neuropathy and the presence of auditory neuropathy were not present at the same time, and the phenotypes of peripheral/optic neuropathy were not complete in this study. We will perform a follow-up study to decipher the constitution of non-syndromic and syndromic AN. Fourth, we conducted a retrospective analysis on the effect of CI in different genetic AN patients, and some of the cases who have received cochlear implantation could not be followed. We will perform a forward-looking study based on the genetic results in this study, to find out the CI effect of patients with different genes, which are located in different sites, providing a precise treatment strategy to the patients.
In summary, we present the first large study of comprehensive high-throughput sequencing and genotype–phenotype correlation analysis in a Chinese AN cohort, identifying the genetic spectrum of AN cases and deciphering that the subgroups with different onset ages showed a diverse genetic spectrum of AN cases. Further, the molecular diagnostic rate of targeted AN genes and targeted HHL genes was 69.9% and 32.1%, respectively. Molecular advances in understanding the various forms of AN will guide us toward more precise management strategies and abilities to predict outcomes. Further research is needed to understand the clinical implications of these findings.
Data availability
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Information files or from the corresponding author upon reasonable request.
References
Akil O, Dyka F, Calvet C, Emptoz A, Lahlou G, Nouaille S, Boutet de Monvel J, Hardelin JP, Hauswirth WW, Avan P, Petit C, Safieddine S, Lustig LR (2019) Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc Natl Acad Sci U S A 116:4496–4501. https://doi.org/10.1073/pnas.1817537116
Al-Moyed H, Cepeda AP, Jung S, Moser T, Kugler S, Reisinger E (2019) A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol Med. https://doi.org/10.15252/emmm.201809396
Archbold S, Lutman ME, Marshall DH (1995) Categories of auditory performance. Ann OTol Rhinol Laryngol Suppl 166:312–314
Berlin CI, Hood LJ, Morlet T, Wilensky D, Li L, Mattingly KR, Taylor-Jeanfreau J, Keats BJ, John PS, Montgomery E, Shallop JK, Russell BA, Frisch SA (2010) Multi-site diagnosis and management of 260 patients with auditory neuropathy/dys-synchrony (auditory neuropathy spectrum disorder). Int J Audiol 49:30–43. https://doi.org/10.3109/14992020903160892
Buchman CA, Roush PA, Teagle HF, Brown CJ, Zdanski CJ, Grose JH (2006) Auditory neuropathy characteristics in children with cochlear nerve deficiency. Ear Hear 27:399–408. https://doi.org/10.1097/01.aud.0000224100.30525.ab
Cheng X, Li L, Brashears S, Morlet T, Ng SS, Berlin C, Hood L, Keats B (2005) Connexin 26 variants and auditory neuropathy/dys-synchrony among children in schools for the deaf. Am J Med Genet A 139:13–18. https://doi.org/10.1002/ajmg.a.30929
Chinese Multi-center Research Collaborative Group on Clinical D, Intervention of Auditory N, Editorial Board of Chinese Journal of Otorhinolaryngology H, Neck S, Society of Otorhinolaryngology H, Neck Surgery CMACD, International Association of Physicians in Audiology SoA, Vestibular M, China International E, Promotive Association for M, Health C (2022) Chinese clinical practice guideline of auditory neuropathy (version 2022). Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 57:241–262. https://doi.org/10.3760/cma.j.cn115330-20220117-00031
Daneshi A, Mirsalehi M, Hashemi SB, Ajalloueyan M, Rajati M, Ghasemi MM, Emamdjomeh H, Asghari A, Mohammadi S, Mohseni M, Mohebbi S, Farhadi M (2018) Cochlear implantation in children with auditory neuropathy spectrum disorder: a multicenter study on auditory performance and speech production outcomes. Int J Pediatr Otorhinolaryngol 108:12–16. https://doi.org/10.1016/j.ijporl.2018.02.004
Dworsack-Dodge MM, Gravel J, Grimes AM, Hunter L, Johnson K, Neault M, Roush J, Sininger Y, Tharpe AM, Yellin W (2012) Audiologic guidelines for the assessment of hearing in infants and young children. The American Academy of Audiology, USA
Feirn R, Sutton G, Parker G, Sirimanna T, Lightfoot G, Wood S (2013) Guidelines for the assessment and management of auditory neuropathy spectrum disorder in young infants. Newborn Hearing screening programme (NHSP) Clinical Group, UK
Foerst A, Beutner D, Lang-Roth R, Huttenbrink KB, von Wedel H, Walger M (2006) Prevalence of auditory neuropathy/synaptopathy in a population of children with profound hearing loss. Int J Pediatr Otorhinolaryngol 70:1415–1422. https://doi.org/10.1016/j.ijporl.2006.02.010
Georga , Lightfoot DG, Hospital RB, Ltd ETC (2019) Recommended procedure: assessment and management of auditory neuropathy spectrum disorder (ANSD) in young infants. British Society of Audiology (BSA), UK
Gravel J, Hood L, Lightfoot G, Mason S, Sirimanna T, Stevens J, Wood S (2004) Assessment and management of auditory neuropathy/auditory dys-synchrony. A recommended protocol. Newborn Hearing Screening Programme (NHSP) Clinical Group, UK
Hayes D, Yvonne SS, Northern J, et al. (2008) Guidelines for identification and management of infants and young children with auditory neuropathy spectrum disorder. Como-Italy
Jean P, Wong Jun Tai F, Singh-Estivalet A, Lelli A, Scandola C, Megharba S, Schmutz S, Roux S, Mechaussier S, Sudres M, Mouly E, Heritier AV, Bonnet C, Mallet A, Novault S, Libri V, Petit C, Michalski N (2023) Single-cell transcriptomic profiling of the mouse cochlea: an atlas for targeted therapies. Proc Natl Acad Sci U S A 120:e2221744120. https://doi.org/10.1073/pnas.2221744120
Kaga K, Nakamura M, Shinogami M, Tsuzuku T, Yamada K, Shindo M (1996) Auditory nerve disease of both ears revealed by auditory brainstem responses, electrocochleography and otoacoustic emissions. Scand Audiol 25:233–238. https://doi.org/10.3109/01050399609074960
Korver AM, van Zanten GA, Meuwese-Jongejeugd A, van Straaten HL, Oudesluys-Murphy AM (2012) Auditory neuropathy in a low-risk population: a review of the literature. Int J Pediatr Otorhinolaryngol 76:1708–1711. https://doi.org/10.1016/j.ijporl.2012.08.009
Lin PH, Wu HP, Wu CM, Chiang YT, Hsu JS, Tsai CY, Wang H, Tseng LH, Chen PY, Yang TH, Hsu CJ, Chen PL, Wu CC, Liu TC (2022) Cochlear implantation outcomes in patients with auditory neuropathy spectrum disorder of genetic and non-genetic etiologies: a multicenter study. Biomedicines. https://doi.org/10.3390/biomedicines10071523
Matsuura K, Yoshimura H, Shinagawa J, Kurozumi M, Takumi Y (2021) Audiological features in 63 patients with cochlear nerve deficiency. Otol Neurotol. https://doi.org/10.1097/MAO.0000000000003365
Miyamoto RT, Kirk KI, Renshaw J, Hussain D (1999) Cochlear implantation in auditory neuropathy. Laryngoscope 109:181–185. https://doi.org/10.1097/00005537-199902000-00002
Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, Shen J, Chapin A, Boczek NJ, Schimmenti LA, Murry JB, Hasadsri L, Nara K, Kenna M, Booth KT, Azaiez H, Griffith A, Avraham KB, Kremer H, Rehm HL, Amr SS, Abou Tayoun AN, ClinGen Hearing Loss Clinical Domain Working G (2018) Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat 39:1593–1613. https://doi.org/10.1002/humu.23630
Papsin BC (2005) Cochlear implantation in children with anomalous cochleovestibular anatomy. Laryngoscope 115:1–26. https://doi.org/10.1097/00005537-200501001-00001
Patel MJ, DiStefano MT, Oza AM, Hughes MY, Wilcox EH, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, Shen J, Chapin A, Boczek NJ, Schimmenti LA, Nara K, Kenna M, Azaiez H, Booth KT, Avraham KB, Kremer H, Griffith AJ, Rehm HL, Amr SS, Tayoun ANA, ClinGen Hearing Loss Clinical Domain Working G (2021) Disease-specific ACMG/AMP guidelines improve sequence variant interpretation for hearing loss. Genet Med 23:2208–2212. https://doi.org/10.1038/s41436-021-01254-2
Penido RC, Isaac ML (2013) Prevalence of auditory neuropathy spectrum disorder in an auditory health care service. Braz J Otorhinolaryngol 79:429–433. https://doi.org/10.5935/1808-8694.20130077
Rance G, Starr A (2015) Pathophysiological mechanisms and functional hearing consequences of auditory neuropathy. Brain 138:3141–3158. https://doi.org/10.1093/brain/awv270
Rankovic V, Vogl C, Dorje NM, Bahader I, Duque-Afonso CJ, Thirumalai A, Weber T, Kusch K, Strenzke N, Moser T (2020) Overloaded adeno-associated virus as a novel gene therapeutic tool for otoferlin-related deafness. Front Mol Neurosci 13:600051. https://doi.org/10.3389/fnmol.2020.600051
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Santarelli R, Cama E, Scimemi P, Dal Monte E, Genovese E, Arslan E (2008) Audiological and electrocochleography findings in hearing-impaired children with connexin 26 mutations and otoacoustic emissions. Eur Arch Otorhinolaryngol 265:43–51. https://doi.org/10.1007/s00405-007-0412-z
Shearer AE, Hansen MR (2019) Auditory synaptopathy, auditory neuropathy, and cochlear implantation. Laryngosc Investig Otolaryngol 4:429–440. https://doi.org/10.1002/lio2.288
Shi W, Ji F, Lan L, Liang SC, Ding HN, Wang H, Li N, Li Q, Li XQ, Wang QJ (2012) Characteristics of cochlear microphonics in infants and young children with auditory neuropathy. Acta Otolaryngol 132:188–196. https://doi.org/10.3109/00016489.2011.630016
Song M, Li J, Lan L, Xie L, Xiong F, Yu L, Shi W, Wang D, Guan J, Wang H, Wang Q (2021) Clinical characteristics of patients with unilateral auditory neuropathy. Am J Otolaryngol 42:103143. https://doi.org/10.1016/j.amjoto.2021.103143
Sparks TN (2020) Expanded carrier screening: counseling and considerations. Hum Genet 139:1131–1139. https://doi.org/10.1007/s00439-019-02080-y
Starr APT, Sininger Y, Hood LJ, Berlin CI (1996) auditory neuropathy. Brain 119:741–753
Starr A, Sininger YS, Pratt H (2000) The varieties of auditory neuropathy. J Basic Clin Physiol Pharmacol 11:215–230. https://doi.org/10.1515/jbcpp.2000.11.3.215
Tang H, Wang H, Wang S, Hu SW, Lv J, Xun M, Gao K, Wang F, Chen Y, Wang D, Wang W, Li H, Shu Y (2022) Hearing of Otof-deficient mice restored by trans-splicing of N- and C-terminal otoferlin. Hum Genet. https://doi.org/10.1007/s00439-022-02504-2
Thorpe RK, Azaiez H, Wu P, Wang Q, Xu L, Dai P, Yang T, Schaefer GB, Peters BR, Chan KH, Schatz KS, Bodurtha J, Robin NH, Hirsch Y, Rahbeeni ZA, Yuan H, Smith RJH (2021) The natural history of OTOF-related auditory neuropathy spectrum disorders: a multicenter study. Hum Genet. https://doi.org/10.1007/s00439-021-02340-w
van der Valk WH, van Beelen ESA, Steinhart MR, Nist-Lund C, Osorio D, de Groot J, Sun L, van Benthem PPG, Koehler KR, Locher H (2023) A single-cell level comparison of human inner ear organoids with the human cochlea and vestibular organs. Cell Rep 42:112623. https://doi.org/10.1016/j.celrep.2023.112623
Van Steijvoort E, Chokoshvili D, Jeffrey WC, Peeters H, Peeraer K, Matthijs G, Borry P (2020) Interest in expanded carrier screening among individuals and couples in the general population: systematic review of the literature. Hum Reprod Update 26:335–355. https://doi.org/10.1093/humupd/dmaa001
Vignesh SS, Jaya V, Muraleedharan A (2016) Prevalence and audiological characteristics of auditory neuropathy spectrum disorder in pediatric population: a retrospective study. Indian J Otolaryngol Head Neck Surg 68:196–201. https://doi.org/10.1007/s12070-014-0759-6
Vona B, Rad A, Reisinger E (2020) The many faces of DFNB9: relating OTOF variants to hearing impairment. Genes (basel). https://doi.org/10.3390/genes11121411
Wang H, Zhao Y, Yi Y, Gao Y, Liu Q, Wang D, Li Q, Lan L, Li N, Guan J, Yin Z, Han B, Zhao F, Zong L, Xiong W, Yu L, Song L, Yi X, Yang L, Petit C, Wang Q (2014) Targeted high-throughput sequencing identifies pathogenic mutations in KCNQ4 in two large Chinese families with autosomal dominant hearing loss. PLoS ONE 9:e103133. https://doi.org/10.1371/journal.pone.0103133
Wang H, Wu K, Guan J, Yang J, Xie L, Xiong F, Lan L, Wang D, Wang Q (2018) Identification of four TMC1 variations in different Chinese families with hereditary hearing loss. Mol Genet Genomic Med 6:504–513. https://doi.org/10.1002/mgg3.394
Wang H, Wang L, Yang J, Yin L, Lan L, Li J, Zhang Q, Wang D, Guan J, Wang Q (2019) Phenotype prediction of Mohr-Tranebjaerg syndrome (MTS) by genetic analysis and initial auditory neuropathy. BMC Med Genet 20:11. https://doi.org/10.1186/s12881-018-0741-3
Wang H, Gao Y, Guan J, Lan L, Yang J, Xiong W, Zhao C, Xie L, Yu L, Wang D, Wang Q (2021a) Phenotypic heterogeneity of post-lingual and/or milder hearing loss for the patients with the GJB2 c.235delC homozygous mutation. Front Cell Dev Biol 9:647240. https://doi.org/10.3389/fcell.2021.647240
Wang W, Li J, Lan L, Xie L, Xiong F, Guan J, Wang H, Wang Q (2021b) Auditory neuropathy as the initial phenotype for patients with ATP1A3 c.2452 G > A: genotype-phenotype study and CI management. Front Cell Dev Biol 9:749484. https://doi.org/10.3389/fcell.2021.749484
Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, Patel A, Boerwinkle E, Beaudet AL, Lupski JR, Plon SE, Gibbs RA, Eng CM (2014) Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312:1870–1879. https://doi.org/10.1001/jama.2014.14601
Zheng D, Liu X (2020) Cochlear implantation outcomes in patients with OTOF mutations. Front Neurosci 14:447. https://doi.org/10.3389/fnins.2020.00447
Acknowledgements
The authors have no other funding, financial relationships, or conflicts of interest to disclose. We would like to express our gratitude to the patients and their families who participated in this study, as well as the authors who provided the publicly available data and the online analysis tool used.
Funding
This study was funded by the grants of the National Natural Science Foundation of China, nos. 82222016, 82271189, 82271171, 82171130. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
QW, HW, and LG conceived and designed the experiments. HW, LG, XW, JG, JL, NL, and YG performed the experiments. HW, LG, XW, KW, YG, DB, JZ, and TS analyzed the data. LL, DL, WW, LX, FX, and WS contributed reagents/materials/analysis tools. HW, LG, and XW wrote the paper. LZ, DW, YY, and QW involved in critical reading and discussion of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
The study was approved by the Committee of Medical Ethics of the Chinese People’s Liberation Army (PLA) General Hospital (No. S2020-228-01), conforming to the Helsinki Declaration of Human and Animal Rights. Written informed consents from all the participants or their guardians were obtained.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H., Guan, L., Wu, X. et al. Clinical and genetic architecture of a large cohort with auditory neuropathy. Hum. Genet. 143, 293–309 (2024). https://doi.org/10.1007/s00439-024-02652-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-024-02652-7