
Abstract
Nuclear speckles are small, membrane-less organelles that reside within the nucleus. Nuclear speckles serve as a regulatory hub coordinating complex RNA metabolism steps including gene transcription, pre-mRNA splicing, RNA modifications, and mRNA nuclear export. Reflecting the importance of proper nuclear speckle function in regulating normal human development, an increasing number of genetic disorders have been found to result from mutations in the genes encoding nuclear speckle proteins. To denote this growing class of genetic disorders, we propose “nuclear speckleopathies”. Notably, developmental disabilities are commonly seen in individuals with nuclear speckleopathies, suggesting the particular importance of nuclear speckles in ensuring normal neurocognitive development. In this review article, a general overview of nuclear speckle function, and the current knowledge of the mechanisms underlying some nuclear speckleopathies, such as ZTTK syndrome, NKAP-related syndrome, TARP syndrome, and TAR syndrome, are discussed. These nuclear speckleopathies represent valuable models to understand the basic function of nuclear speckles and how its functional defects result in human developmental disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nuclear speckles (NS), previously referred to as interchromatin granule clusters (IGCs), are small, membrane-less organelles that reside within the nucleus (Fig. 1). A large number of proteins are located within NS, and classes of NS proteins include gene transcription regulatory molecules, chromatin remodelers, precursor mRNA (pre-mRNA) splicing, RNA-associated proteins, cleavage and polyadenylation factors, and mRNA export. Based on the known functions of proteins located within the NS, they are considered a hub of complex RNA metabolism steps. In this review article, the current understanding of NS function and genetic disorders associated with NS dysfunction are discussed.

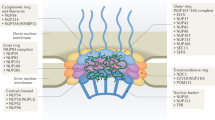
Intranuclear distribution of nuclear speckles and its contents. a Immunofluorescence staining of SRRM2, a core structural protein of nuclear speckles in the HCT 116 colon cancer cell line. Anti-SRRM2 antibody produced in rabbit (Sigma Aldrich: HPA041411-100UL.1:500 dilution) was used for the immunofluorescence staining. Merged image: SRRM2 (red), and DAPI (blue). Bar: 5 µm. b Schema showing the classes of RNA metabolism-regulating proteins located within nuclear speckles
Historical background
NS were originally described by Spanish physician Santiago Ramón y Cajal in (1910). By using acid aniline staining, Dr. Cajal discovered irregular and transparent clumps in the nucleus of a neuron. This structure was later referred to as speckles by Dr. J. Swanson Beck in 1961, in his article describing the nuclear staining patterns of patients with autoimmune disorders (Beck 1961). NS was also called as IGCs that were visualized by electron microscopy (Swift 1959). The first connection between NS/IGCs and RNA processing was established in the early 1980s based on the discovery of small nuclear ribonucleoprotein particles (snRNPs), which catalyze pre-mRNA splicing and colocalize within NS/IGCs (Lerner et al. 1981; Spector et al. 1983). Subsequent studies have revealed the diverse roles of NS beyond mRNA splicing, extending to gene transcriptional regulation, various RNA modifications, and nuclear export.
Mechanism of NS organization
Immunofluorescence staining of NS proteins revealed that NS are irregular, mottled structures found inside the nucleus (Fig. 1). In interphase, the nucleus usually contains 20–50 speckles whose sizes are up to several micrometers (Lamond and Spector 2003). The number of NS and their size possibly vary among different cell types, reflecting the levels of gene expression required for each cell type, because transcriptional suppression or splicing inhibition causes a reduced number of NS accompanied by an enlargement of each NS’s size (Fei et al. 2017; Kim et al. 2019; O'Keefe et al. 1994; Spector et al. 1991).
NS demonstrate a layered structure. SON and SRRM2 locate at the core of NS, and these core proteins are surrounded by MALAT1 (noncoding RNA), U1 and U2 small nuclear RNAs (Fei et al. 2017). SC35 was also considered as a core structural NS protein, but a recent paper raised a possibility that SC35 detected by immunofluorescence was SRRM2 (Ilik et al. 2020). In contrast to organelles, whose borders are demarcated by membranes, NS forms their structures by self-assembly of its protein content. This self-assembly is facilitated by the unique physical properties of NS proteins that demonstrate liquid–liquid phase separation. Phase separation enables the formation of droplet-like structures. These physical properties facilitate the formation of membrane-less NS, whose core proteins (SRRM2 and SON) facilitate NS formation (Ilik et al. 2020; Xu et al. 2022). NS proteins tend to contain arginine repeats, and arginine-enriched mixed-charge domains form condensates that drive speckle assembly (Greig et al. 2020).
Contents of NS
The NS is composed of RNA and various RNA metabolism-regulating proteins. The previous studies have revealed the presence of RNAs, particularly poly (A)-attached RNAs and noncoding RNAs, in NS (Carter et al. 1991; Shopland et al. 2002). On the contrary, NS contain little or no DNA (Carter et al. 1991; Spector and Lamond 2011). More than 50% of the proteins residing within the NS are involved in transcription or splicing regulation (Galganski et al. 2017) (Fig. 1b). Examples of NS proteins include speckle core proteins (SRRM2 and SON), transcriptional regulatory molecules (Pol II subunits, CDK12, and CDK13), RNA-binding proteins (RBM10 and RBM8A), and snRNP (SF3B4 and SF3B2).
For the comprehensive identification of NS proteins, we obtained a list of 380 NS proteins residing within the NS from a public database, the Human Protein Atlas database [https://www.proteinatlas.org/] (Supplemental Table 1) (Uhlen et al. 2015). Using the obtained list, we performed gene ontology (GO) analysis using DAVID functional annotation tool (Dennis et al. 2003). As expected, “nuclear speckles” were the top-ranked GO terms of the cellular component (Supplemental Table 2). Other cellular component terms enriched in NS proteins were “nuclear body,” “nucleoplasm,” “spliceosomal complex,” “chromosome,” “ribonucleoprotein complex,” and “chromatin.” Biological process GO term ranking indicated that “mRNA processing” was the top-ranked term, followed by “RNA splicing,” “gene expression,” and “regulation of transcription.” (Supplemental table 3). Molecular function GO terms enriched were “nucleic acid binding,” “DNA binding,” “RNA binding,” and “RNA polymerase II regulatory region DNA binding.” (Supplemental table 4). We acknowledge the limitations of the use of the Human Protein Atlas database and those include antibody specificities and cell fixation conditions, because this database relied on immunofluorescence staining experiments using fixed cells. Of note, cell fixation conditions are known to affect localization of certain NS proteins (Dopie et al. 2020). As a result, it is possible that the obtained NS protein list may include false positives or negatives. Despite these limitations, similar sets of proteins were identified as NS proteins in studies using mass spectrometry (Dopie et al. 2020; Saitoh et al. 2004). Collectively, these findings support the notion that gene expression or mRNA processing/splicing regulation are the main functions of NS proteins.
Functions of NS
Consistent with the findings of GO analyses of NS proteins, NS is currently regarded as a site of gene expression regulation from transcription to RNA processing. Pol II catalyzes DNA transcription to synthesize pre-mRNA. This active transcription occurs at the periphery of the NS, and gene expression is increased by their association with NS (Kim et al. 2020). During active gene transcription, Pol II undergoes serial protein modifications, including phosphorylation of the C-terminal domain of Pol II. Various cyclin-dependent kinases (CDKs) are involved in the phosphorylation of Pol II, and CDK12/CDK13, NS proteins, are the CDKs involved in Pol II phosphorylation that determines the activity of Pol II (Fan et al. 2020).The repertoire of phosphorylated Pol II-interacting proteins includes a large number of spliceosomal proteins found in NS, suggesting that Pol II is recruited to the NS during active transcriptional elongation (Nojima et al. 2018). Of note, phosphorylation of Pol II facilitates the incorporation of Pol II into splicing condensates (Guo et al. 2019; Lu et al. 2018), while it remains to be determined whether these splicing condensates are NS or not. It is possible that condensate formation of Pol II may be involved in the mechanism linking gene transcription to subsequent RNA-processing steps within NS.
Pre-mRNA is converted to mRNA after several processing steps, including splicing. As many NS proteins are known to regulate these pre-mRNA-processing steps (Dias et al. 2010), NS is thought to be involved in the regulation of these steps. However, it remains to be determined whether these steps occur within NS or not. During pre-mRNA synthesis, the spliceosome, composed of five snRNPs (U1, U2, U4/U6, and U5) and non-snRNP splicing factors, is recruited to the pre-mRNA, where it catalyzes splicing reactions. Splicing is a stepwise process that includes catalytic steps (splice-site recognition, intron lariat formation, and intron excision) and post-catalytic steps (exonic ligation and mRNA release from the spliceosome). Throughout this process, the spliceosome undergoes dynamic component changes wherein many proteins join and leave the complex. For example, U2 snRNP, containing SF3B4 and SF3B2, is recruited during splicing steps (Sun 2020), RBM8A is recruited to the exon junction complex (EJC) (Chuang et al. 2015), and RBM10 is recruited to the pre-mRNA during splicing (Inoue et al. 2014; Wang et al. 2013). These splicing processes are intricately integrated into the Pol II transcription process and Pol II regulation, and the pre-mRNA splicing steps are interdependent. For example, alterations in Pol II activity can affect pre-mRNA splicing (Fong et al. 2014). Conversely, pre-mRNA splicing factors, such as SC35, can influence the Pol II transcription rate (Ji et al. 2013; Lin et al. 2008). A detailed review of pre-mRNA splicing can be found in Lee and Rio (Lee and Rio 2015).
NS also contains proteins involved in the regulation of other mRNA maturation processes including 5′ end capping, 3′ end polyadenylation (poly(A)), N6-methyladenosine (m6A) modification, and mRNA nuclear export. 5′ capping involves the addition of 7-methylguanosine triphosphate to the 5′ end of mRNA transcripts (Reviewed in (Ramanathan et al. 2016)). 3′ tail polyadenylation involves the addition of a series of adenine repeats to the end of the transcripts (Reviewed in (Liu et al. 2022)). Interestingly, NS contain Star-PAP, which differentially regulates mRNA 3′ end processing of certain transcripts depending on oxidative/DNA damage pathways (Li et al. 2013; Mellman et al. 2008). Monomethylated adenosine at the N6 position (m6a) is the most well-characterized epitranscriptomic modification. m6A provides a binding site for m6A-binding proteins such as YTH family proteins (Reviewed in (Boulias and Greer 2022)). These mRNA modifications serve as a method to stabilize mRNA and facilitate the transfer of mature mRNA to the ribosome for translation. After the completion of RNA processing, processed transcripts are transported to the cytosol by nuclear export factors such as the TREX complex (Reviewed in (Khan et al. 2022)). Depletion of TREX complex components enhances the association of mRNA with NS, supporting the role of NS in mRNA nuclear export (Dias et al. 2010).
In addition, a role for NS in genome organization has been proposed. Knockdown of SRRM2, a core structural NS protein, resulted in the reduction of intra-chromosomal interactions within active chromatin compartments (Hu et al. 2019). These findings were supported by genomic and cytological studies, as NS resides near the highly transcribed genes of active compartment A, which are mutually exclusive to lamina-associated inactive compartment B (Chen et al. 2018; Su et al. 2020). These observations suggest a role for NS in maintaining chromatin interactions in active compartments.
Although chromatin remodeler proteins, including SETD1A, SETD2, and HDAC4, are located within NS, the role of these proteins in NS function remains unclear. Interestingly, only subsets of chromatin remodelers reside within NS. Based on the known functions of these proteins (Wang et al. 2021b, 2014; Xu et al. 2019), chromatin remodelers located within the NS are expected to influence gene transcription or splicing efficacy.
Although the above-mentioned proteins mainly localize within NS, they also present outside the NS. Therefore, the interpretation of their role in NS requires caution, as their canonical function can be executed outside of NS.
NS target genes and transcripts
To support the notion that NS is involved in gene expression regulation, gene expression levels correlates inversely with distance to nuclear speckles (Zhang et al. 2020). Although a sparse amount of DNA resides within the NS, genomic loci located near the NS have been evaluated using tyramide signal amplification sequencing (TSA-seq) (Chen et al. 2018) and split-pool recognition of interactions by tag extension (SPRITE) (Quinodoz et al. 2018). Considering that NS proteins function as universally required transcriptional and splicing regulatory molecules, it is expected that the majority of Pol II-transcribed genes require RNA processing steps orchestrated by NS. However, NS tend to localize close proximity to genomic regions of certain features, such as genes residing within the active A chromatin compartment, highly expressed genes, housekeeping genes, genes with low transcriptional pausing, and super-enhancers (Chen et al. 2018; Quinodoz et al. 2018). HSPA1 genes (HSPA1A, HSPA1B, and HSPA1L), that encode heat shock 70 kDa proteins, represents examples of well-characterized NS target genes (Kim et al. 2019). There is a significant overlap between the NS target and p53 target genes, suggesting the role of p53 in determining NS target genomic regions (Alexander et al. 2021). Considering that many NS proteins lack DNA-binding domains, transcription factors, including p53, may determine the genomic loci targeted by NS.
Unsurprisingly, NS-associated transcripts demonstrated a high degree of overlap with NS-associated genomic loci. NS-associated coding transcripts are characterized by a high GC content, and their transcript lengths tend to be shorter than those associated with other nuclear components (Barutcu et al. 2022). Speckle-associated transcripts were also enriched for transcripts with retained short introns, suggesting the role of NS in the splicing of shorter introns (Barutcu et al. 2022). To support this notion, SRRM2 deficiency causes exon skipping of short introns surrounded by weak splice sites (Xu et al. 2022). Notably, exon skipping with weak splice sites is influenced by the transcriptional rate (Dujardin et al. 2014). Therefore, it is possible that the observed splicing defects may be the consequence of transcriptional rate changes.
To explain the high gene expression levels of NS target genes, recent cytological investigations have revealed that NS is involved in the gene expression bursting of NS-associated genes (Alexander et al. 2021; Kim et al. 2019). By maintaining the close proximity between the genomic locus and NS, ample amounts of transcriptional and splicing regulatory molecules are furnished to these genomic loci, resulting in gene expression amplification. However, it remains to be determined whether this gene expression bursting is at the transcriptional versus post-transcriptional level.
NS and human genetic disorders
Reflecting the importance of proper nuclear speckle function in regulating normal human development, an increasing number of genetic disorders have been found to result from mutations in the genes encoding NS proteins. Several notable examples include ZTTK syndrome due to SON mutations, TARP syndrome due to RBM10 mutations, and TAR syndrome due to RBM8A mutations.
We investigated the involvement of genes encoding NS proteins in the pathogenesis of human genetic diagnoses using public databases (OMIM: Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD) https://omim.org/ and HPO browsers: (Kohler et al. 2021)). A systematic search of NS genes within the “genemap2.txt” file obtained from the OMIM database was conducted on 11/3/2022. A total of 291 out of 380 genes, whose encoded proteins localize within NS based on Protein Atlas database, were found to be associated with human disease phenotypes. Strikingly, among the top 100 annotated NS proteins reported by Dopie et al. (2020), 95 out of 100 genes were found to be associated with human disease phenotype. Therefore, most NS proteins have been implicated in the pathogenesis of human diseases.
Next, we investigated the clinical features commonly observed in disorders due to mutations in genes encoding NS proteins based on the Protein Atlas data analysis. A table listing the associations between genes and Human Phenotype Ontology (HPO) terms was obtained from the HPO browser (genes_to_phenotype.txt; available at: http://purl.obolibrary.org/obo/hp/hpoa/genes_to_phenotype.txt; data queried on 11/3/22). The HPO terms associated with NS genes were ranked by frequency, and the results are presented in Table 1. The most common HPO term associated with NS genes was “global developmental delay.” Similarly, HPO terms associated with brain developmental defects, such as “intellectual disability” and “seizures” were ranked highly in the table. Similar result was obtained by using NS proteins identified by Dopie et al. (Supplemental table 5). These findings are of particular interest as most of the NS proteins are ubiquitously expressed. Therefore, the enrichment of neurological symptoms indicates the importance of NS function in human brain development. Alternative splicing has been proposed as a method to diversify protein repertoires, particularly in the brain (Zhang et al. 2016); therefore, it is possible that brain tissues may be more susceptible to NS dysfunction, which could trigger splicing alterations, compared to other organ systems.
Nuclear speckleopathies due to mutations in genes encoding NS proteins
To denote genetic disorders caused by mutations in genes encoding NS proteins, we propose a term “nuclear speckleopathies” (Table 2). The current knowledge of various nuclear speckleopathies and the molecular mechanisms underlying these disorders are discussed herein.
ZTTK syndrome due to SON mutations
ZTTK (Zhu-Tokita-Takenouchi-Kim) syndrome (MIM# 617,140) is characterized by developmental delay/intellectual disability, hypotonia, skeletal abnormalities, and facial dysmorphisms (Kim et al. 2016b; Takenouchi et al. 2016; Tokita et al. 2016; Zhu et al. 2015). Recent large cohort studies have revealed that developmental delay/intellectual disability are almost universally observed in individuals with ZTTK syndrome, and the range of intellectual disability ranges from mild to severe (Dingemans et al. 2022; Kushary et al. 2021). Brain structural abnormalities, such as hypoplasia/agenesis of the corpus callosum and ventricular enlargement, are common in individuals with ZTTK syndrome (83–90%). Seizures (50–60%) and hypotonia (60%) are also common in individuals with ZTTK syndrome. This syndrome is also associated with structural birth defects, including congenital heart defects (18–33%, particularly atrial septal defect or ventricular septal defect) and genitourinary malformations (31–47%) (Dingemans et al. 2022; Kushary et al. 2021). Visual and hearing impairments and immunological abnormalities have also been observed in individuals with ZTTK syndrome (Dingemans et al. 2022). Facial dysmorphic features were commonly observed; however, no unifying, recognizable features were identified (Dingemans et al. 2022).
Germline heterozygous loss of function (LoF) mutations in SON cause ZTTK syndrome. Most of the identified mutations are predicted LoF variants, such as frameshift, nonsense, or splice-site variants (Dingemans et al. 2022; Kushary et al. 2021). A small number of missense or in-frame deletion variants have been reported as candidate variants; however, the clinical significance of these variants remains unclear (Dingemans et al. 2022; Kushary et al. 2021).
Molecular mechanism
SON was originally identified as a NS protein by comprehensive proteomic analysis (Saitoh et al. 2004). SON functions as a scaffolding protein for RNA-processing factors in NS (Sharma et al. 2010). SON plays various transcriptional regulatory roles, including splicing and epigenetic regulation (Kim et al. 2016a; Lu et al. 2013). A recent study has demonstrated that SON is a core structural component of NS (Ilik et al. 2020).
Genome-wide transcriptome analysis has not been performed to date using samples from patients with ZTTK syndrome; however, misexpression and alternative splicing events of a selected set of 11 genes have been reported in the context of ZTTK syndrome using blood samples obtained from patients (Kim et al. 2016b). A recent report by Ueda et al. investigating the neuronal effects of Son in a mouse model has demonstrated that Son knockdown results in neural migration defects and dendritic spine abnormalities, which are reversed via induction of the human wild-type SON gene, suggesting the importance of SON in regulating brain development (Ueda et al. 2020).
Neurodevelopmental disorder due to SRRM2 mutation
A novel neurodevelopmental disorder due to LoF variants of SRRM2 has been recently reported in a cohort of twenty-two patients by Cuinat et al. (Cuinat et al. 2022). Clinical features include mild developmental delay, variable intellectual disability, predominant speech delay, autism or attention-deficit/hyperactivity disorder, overfriendliness, generalized hypotonia, increased weight, and dysmorphic facial features. Variant analysis of the patient cohort identified twelve frameshift variants, eight nonsense variants, and two microdeletions of 66 and 270 kb (Cuinat et al. 2022).
Molecular mechanism
SRRM2 is a highly conserved core scaffolding protein component of NS (Miyagawa et al. 2012). SRRM2 forms a complex that facilitates pre-mRNA maturation as one of the main catalytic components of the spliceosome and promotes the interaction between pre-mRNA and splicing factors (Blencowe et al. 2000; Sawada et al. 2000). In a study by Hu et al., wherein Hi-C experiments using Srrm2 knockdown models in mouse hepatocytes were performed to characterize the effects of SRMM2 on 3D genome organization and gene expression, it was demonstrated that the loss of SRRM2 resulted in a global decrease in intra-topologically associating domain (TAD) chromosomal interactions in active (type A) compartments and an increase in repressive (type B) compartments (Hu et al. 2019). They also observed that Srrm2 knockdown resulted in changes in the expression of over 1000 genes. Even though these results illustrate the mechanisms by which SRRM2 maintains specific patterns of genomic interactions in NS, these effects were not associated with corresponding changes in gene expression at the TAD level. Together, these results suggest that while loss of SRRM2 causes a reduction in TAD interactions, particularly in A1 sub-compartments, TAD-mediated transcription is preserved, and the observed differences in gene expression may be a result of alternative or downstream components of genome organization.
NKAP-related syndrome due to NKAP mutations
NKAP-related syndrome (MIM# 301039) is characterized by developmental delay, behavioral abnormalities, cardiac abnormalities, tall stature, scoliosis, camptodactyly, and obesity (Fiordaliso et al. 2019). NKAP-related syndrome is caused by hemizygous missense mutations in NKAP. In the human genome, NKAP resides on the X-chromosome. NKAP mutations primarily cause symptoms in males. Females with heterozygous NKAP mutations are unaffected or significantly less affected than their male counterparts, although the full clinical spectrum of females with heterozygous NKAP mutations remains unknown. Notably, the mutations identified in individuals with NKAP-related syndrome showed clear clustering within the C-terminus of the NKAP protein.
Molecular mechanism
NKAP was originally identified as a nuclear protein that promotes NF-κB activation (Chen et al. 2003). Pajerowski et al. demonstrated that NKAP interacts with HDAC3 and functions as a transcriptional regulator of the Notch signaling pathway (Pajerowski et al. 2009), although the role of NKAP-HDAC3 in global transcriptional regulation remains unknown. Burgute et al. demonstrated that NKAP localizes within NS and binds to RNA to regulate mRNA splicing (Burgute et al. 2014).
Previously, a conditional Nkap knockout model was reported. Similar to human NKAP, mouse Nkap also resides on the X-chromosome. Hematopoietic lineage-specific Nkap hemizygous null male mice exhibit embryonic lethality (Pajerowski et al. 2010). However, the role of NKAP in the development and function of other organ systems was not studied. A recent study has documented strong Nkap expression in the mouse brain, indicating a potential role for NKAP in neuronal development and function (Worlitzer and Schwamborn 2014).
Considering that the mutations identified in NKAP-related syndrome are clustered in the C-terminal region of NKAP, it is possible that a particular aspect of NKAP function is disrupted in NKAP-related syndrome. Notably, the C-terminus of NKAP harbors an HDAC3-interacting domain and a post-catalytic spliceosomal P-complex interacting domain (Fica et al. 2019; Pajerowski et al. 2009). Therefore, in a previous study, we hypothesized that NKAP mutations alter transcription. To test this hypothesis, transcriptome analysis was performed using RNA sequencing in lymphoblastoid cell lines obtained from individuals with NKAP mutations and controls. Consistent with the role of NKAP as a post-catalytic P-complex subunit, NKAP mutations had a minimal impact on splicing but did demonstrate a large number of differentially expressed genes in NKAP-related syndrome (Fiordaliso et al. 2019). Thus, we concluded that NKAP mutations cause only quantitative transcriptome changes without qualitative alternative splicing in NKAP-related syndrome. However, the mechanism underlying this transcriptome profile remains unknown.
Congenital heart defects, dysmorphic facial features, and intellectual developmental disorder (CHDFIDD) due to CDK13 mutations
CHDFIDD (MIM #617360) was first reported by Sirfrim et al. in a cohort of seven children harboring heterozygous missense mutations within the CDK13 kinase domain (Sifrim et al. 2016). The patients exhibited the following clinical features to a variable extent: atrial and/or ventricular septal heart defects, pulmonary valve abnormalities, dysmorphic features, global developmental delay, intellectual disability, seizures, microcephaly, structural brain malformations, feeding difficulties, and digital anomalies (Sifrim et al. 2016). In a subsequent report, Hamilton et al. described nine additional pediatric patients presenting with developmental delays, variable intellectual disability with autistic features, common facial gestalt, digital anomalies, and poor feeding. Notably, only two patients in this cohort had structural heart defects, and one had seizures (Hamilton et al. 2018).
Molecular mechanism
CDK13, a cyclin–dependent kinase (CDK), belongs to a class of ATP-dependent serine-threonine protein kinases that mediate extracellular and intracellular signals to regulate cell cycle progression and gene expression. Researchers have sought to define the role of CDK13 in NS. A recent study has supported the role of CDK13 in transcriptional regulation by activating RNA Pol II (Fan et al. 2020). The N-terminus of CDK13 contains arginine and serine-rich (RS) motifs, which generally regulate splicing (Marques et al. 2000). The localization of CDK12 and CDK13 to NS is dependent upon their respective RS motifs (Even et al. 2006; Ko et al. 2001). Even et al. previously demonstrated that CDK13 is highly concentrated in NS and regulates splicing by controlling the phosphorylation status and activity of splicing factors (Even et al. 2006). Liang et al. systematically identified protein-binding partners for CDK12 and CDK13 and found that RNA-processing factors, spliceosomes, and NS components were significantly enriched in purifications for both CDK12 and CDK13 (Liang et al. 2015).
The role of CKD13 in NS has previously been reported in cancer and HIV; however, its role in the pathophysiology of CHDFIDD remains to be established (Berro et al. 2008; Greenleaf 2019). To date, the reported variants associated with CHDFIDD are heterozygous missense mutations isolated from the CDK13 protein kinase domain (Hamilton et al. 2018; Sifrim et al. 2016). Molecular modeling analysis of the variants predicted structural changes resulting in a significant loss of catalytic activity of the enzyme, including deleterious effects on ATP binding, magnesium binding, and cyclin K interactions (Hamilton et al. 2018; Sifrim et al. 2016). However, functional studies have not yet established the predicted effects of these observed variants. Numerous LoF variants in CDK13 have been reported in relatively healthy individuals within the gnomAD and Decipher databases, suggesting that LoF mutations alone may not account for the severe clinical phenotypes observed in patients with CHDFIDD and that dominant negative effects may contribute, wherein the resultant inactive enzymatic complexes could compete with active complexes for binding substrates (Hamilton et al. 2018). Considering that the reported CHDFIDD variants are predicted to retain the ability to bind cyclin K, it is plausible that cyclin K could be sequestered into inactive complexes (Hamilton et al. 2018). Pathogenic variants in CDK13 associated with CHDFIDD have not been observed in the RS domains; thus, it is possible that the localization of CDK13 to NS is preserved in these patients, although the predicted reduction in catalytic activity may result in the loss of phosphorylation of splicing factors within the NS.
TARP syndrome due to RBM10 mutations
TARP syndrome, characterized by talipes equinovarus, atrial septal defect, Robin sequence, and persistent left superior vena cava, is an X-linked disorder with frequently observed male lethality that was originally discovered by Gorlin et al. (Gorlin et al. 1970), and is caused by inactivating mutations in RBM10 (RNA-binding motif 10; MIM#300080) (Johnston et al. 2010; Kaeppler et al. 2018; Niceta et al. 2019). Notably, the four cardinal features are not consistently present, and additional nonclassical findings have been observed (Kaeppler et al. 2018). Other clinical features include feeding difficulties, developmental delay, facial dysmorphisms, other distal-limb and digital anomalies, various renal anomalies, deafness, oral masses, chorio-retinal atrophy, bronchopulmonary pathology, skeletal anomalies, cerebral and cerebellar anomalies, and multiple other cardiac lesions, including hypertrophic cardiomyopathy (Kaeppler et al. 2018; Niceta et al. 2019). Pathogenic mutations reported to date include frameshift and nonsense variants predicted to lead to destabilization of the protein structure as well as induction of nonsense-mediated mRNA decay (Johnston et al. 2014, 2010; Kaeppler et al. 2018; Niceta et al. 2019). Recent studies with expanded clinical follow-up have also identified male patients with nontruncating and truncating variants with milder phenotypes that have survived into adolescence (Hojland et al. 2018; Niceta et al. 2019).
Molecular mechanism
RBM10, also known as S1-1, is a ubiquitously expressed protein possessing two RNA recognition motifs, a nuclear localization signal, two zinc finger motifs and a G patch domain that is frequently observed in splicing regulatory RNA-binding proteins (Niceta et al. 2019). RBM10 has been proposed to coordinate diverse post-translational processes and interact with more than 200 spliceosome proteins (Inoue et al. 2014; Wang et al. 2013). Interestingly, RBM10 exists in a dynamic state between the nucleoplasm and membrane-less nuclear bodies called S1-1 NBs (Inoue 2021; Wang et al. 2021a). S1-1 NBs are often spatially overlap with NS and show a similar change in size in response to transcriptional states of cells, indicating a close functional relationship between NS and S1-1 NBs (Inoue et al. 2008). Wang et al. showed that RBM10 is sequestered in S1-1 NBs when it is not engaged in alternative splicing or when transcription decreases, and can then dissociate from S1-1 NBs into the nucleoplasm when transcription increases; therefore, it may participate in alternative splicing (Wang et al. 2021a). Given the severe, pleiotropic effects of RBM10 variants in patients with TARP syndrome, it is possible that the role of RBM10 within S1-1 NBs has broad effects on gene expression and developmental processes; however, the exact molecular mechanisms by which effects on RNA binding and alternative splicing contribute to disease remain to be elucidated.
TAR syndrome due to RBM8A mutations
Thrombocytopenia-absent radius (TAR) syndrome [MIM #274000] is a well-recognized clinical entity characterized by hypomegakaryocytic thrombocytopenia and bilateral radial aplasia with the presence of both thumbs. Additional associations include congenital heart disease, genitourinary tract abnormalities, structural brain differences, hip dislocation, knee subluxation, cow milk intolerance, and resultant eosinophilia (Klopocki et al. 2007). Klopocki et al. demonstrated that affected individuals commonly harbor de novo or inherited deletions on chromosome 1q21.1; however, the authors hypothesized that an additional genetic modifier must be present to cause the disease, given its complex mode of inheritance and sporadic nature (Klopocki et al. 2007). Albers et al. discovered compound inheritance mechanisms that cause TAR syndrome involving a rare null allele (deletion, frameshift mutation, or premature stop codon) and one of the two low-frequency noncoding single nucleotide polymorphisms (SNPs) in RBM8A (Albers et al. 2012).
Molecular mechanism
RBM8A (also known as Y14) is a component of the EJC, which is required for several mRNA regulatory processes, including splicing, mRNA export, and nonsense-mediated decay (Chuang et al. 2015). Albers et al. showed that two regulatory SNPs observed in patients with TAR syndrome resulted in diminished RBM8A transcription in vitro and reduced expression in patient platelets. The authors concluded that TAR syndrome was a result of EJC dysfunction due to Y14 insufficiency (Albers et al. 2012). Notably, EJC proteins accumulate in NSs alongside most other splicing-related factors, and such interactions with NS occur in both transient and long-term states (Schmidt et al. 2009). Future functional studies are needed to fully characterize the downstream effects of the complex genetic etiology underlying TAR syndrome on EJC functions that are coordinated with NS to regulate mRNA metabolism.
Nager syndrome due to SF3B4 mutations
Nager syndrome is a representative disorder of acrofacial dysostosis (AFDs) [MIM #155440]. Clinical features include midface retrusion, severe micrognathia, down-slanted palpebral fissures, hypoplastic or triphalangeal thumbs, radial hypoplasia or aplasia, radioulnar synostosis, upper-limb phocomelia, and lower-limb defects. Occasionally, patients exhibit short stature, cardiac malformations, costovertebral anomalies, and renal malformations. Notably, unlike most nuclear speckleopathies, patients with Nager syndrome typically have normal intelligence (Bernier et al. 2012; Petit et al. 2014). Most cases of Nager syndrome (~ 60%) are associated with autosomal dominant LoF mutations in SF3B4, including nonsense, frameshift, and splice-site variants (Bernier et al. 2012; Czeschik et al. 2013; Petit et al. 2014). Highly variable phenotypic severity with intra-family variability was noted as well (Petit et al. 2014).
Molecular mechanism
SF3B4 encodes the splicing promoter protein SAP49, an integral component of the SF3B complex that tethers the U2 snRNP branch site (Champion-Arnaud and Reed 1994). The full spectrum of phenotypic effects of SF3B4 haploinsufficiency may also be accounted for via diverse mechanisms in addition to its impact on spliceosome function. SAP49 interaction with the BMP signaling pathway underlying osteochondral cell differentiation has also been proposed as a molecular mechanism leading to the skeletal features of Nager syndrome (Petit et al. 2014). Considering that approximately one-third of patients with Nager syndrome do not possess detectable LoF SF3B4 variants, future work will be needed to clarify potential genetic heterogeneity inside and outside the spliceosome.
Craniofacial microsomia (CFM) due to SF3B2 mutations
CFM is an asymmetrical congenital malformation disorder (historically referred to as hemifacial microsomia, oculo-auricular-vertebral spectrum (OAVS), or Goldenhar syndrome) that can be inherited in an autosomal dominant manner [MIM #164210]. CFM presents with a spectrum of phenotypes and core clinical features, including hemifacial mandibular hypoplasia, microtia, facial and preauricular skin tags, epibulbar dermoids, and lateral oral clefts. At the mild end of the spectrum, patients present with microtia in the absence of other anomalies. Patients on the more severe end of the spectrum can exhibit more extensive multisystem disease, including anomalies of the cardiac, nervous, vertebral, and renal systems (Timberlake et al. 2021). Genetic and environmental etiologies have been proposed to explain this disorder (Barisic et al. 2014; Beleza-Meireles et al. 2014). LoF variants leading to haploinsufficiency of SF3B2 represents the most common genetic cause of CFM (Timberlake et al. 2021). Consistent observations of external ear malformations involving the tragus and mandibular hypoplasia suggest that SF3B2 variants predominantly influence pharyngeal arch I development. Functional studies have demonstrated that Sf3b2 knockdown disrupts cranial neural crest cell formation in Xenopus embryos (Timberlake et al. 2021).
Molecular mechanism
SF3B is a component of the U2 snRNP complex that interacts directly with SF3B4 to anchor U2 snRNP to pre-mRNAs. Interestingly, recent studies have implicated spliceosomal dysfunction in retaining highly conserved “poison exons” that harbor premature termination codons and are typically spliced out from pre-mRNAs in genes, which have substantial roles in developmental processes (Thomas et al. 2020). It is not yet understood how dysfunctional splicing that leads to the retention of such exons may contribute to the pathogenesis of CFM. Future studies focused on understanding the fundamental functions of SF3B4 and its potential interactions with other regulatory components of NS may identify additional “second-hit” mechanisms accounting for the phenotypic variability observed in CFM.
Nager syndrome due to SF3B4 mutations and CFM due to SF3B2 mutations are also referred to as “spliceosomopathies” (Beauchamp et al. 2020). Considering that these conditions are associated with a unique craniofacial phenotype without neurocognitive impairment, the underlying pathogenic mechanisms of these syndromes may differ from those of other NS component dysfunctions.
Summary
Here, we review the current knowledge of NS and the genetic diagnoses associated with NS dysfunction. Recent studies have revealed a critical role of NS in diverse aspects of RNA metabolism regulation, including transcriptional and splicing reactions, RNA modifications, and mRNA export. As expected from the pleiotropic functions of NS, an increasing number of genetic disorders are linked to NS dysfunction. However, as NS regulates multiple aspects of RNA metabolism, it has been challenging to identify the molecular step(s) whose perturbation is directly related to the clinical phenotype of nuclear speckleopathies. Since NS orchestrate complex gene expression regulation, mutations within genes encoding NS protein could affect a part of NS function. These mutations could also affect the encoded proteins’ function not only within NS, but also outside of NS. With the development of cutting-edge next-generation sequencing applications enabling transcriptome-wide RNA metabolism investigations, such as nascent RNA sequencing, and cross-linking immunoprecipitation sequencing (CLIP-seq), it has become technically feasible to carefully characterize the type of RNA metabolism defects associated with nuclear speckleopathies. In addition to proteins regulating RNA metabolism, NS contain chromatin remodelers such as SETD1A, SETD2, and HDAC4. Mutations in genes encoding chromatin proteins, collectively called chromatinopathies, cause developmental disabilities similar to nuclear specleopathies. As recent findings indicate the role of NS in chromatin organizational regulation (Hu et al. 2019), there may exist a common molecular perturbation between chromatinopathies and nuclear specleopathies. Elucidating the mechanisms underlying nuclear speckleopathies will contribute to the basic knowledge of NS and how its functional defects result in human developmental disorders.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, Jolley JD, Cvejic A, Kostadima M, Bertone P, Breuning MH, Debili N, Deloukas P, Favier R, Fiedler J, Hobbs CM, Huang N, Hurles ME, Kiddle G, Krapels I, Nurden P, Ruivenkamp CA, Sambrook JG, Smith K, Stemple DL, Strauss G, Thys C, van Geet C, Newbury-Ecob R, Ouwehand WH, Ghevaert C (2012) Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 44(435–9):S1-2. https://doi.org/10.1038/ng.1083
Alexander KA, Cote A, Nguyen SC, Zhang L, Gholamalamdari O, Agudelo-Garcia P, Lin-Shiao E, Tanim KMA, Lim J, Biddle N, Dunagin MC, Good CR, Mendoza MR, Little SC, Belmont A, Joyce EF, Raj A, Berger SL (2021) p53 mediates target gene association with nuclear speckles for amplified RNA expression. Mol Cell 81:1666-1681 e6. https://doi.org/10.1016/j.molcel.2021.03.006
Barisic I, Odak L, Loane M, Garne E, Wellesley D, Calzolari E, Dolk H, Addor MC, Arriola L, Bergman J, Bianca S, Doray B, Khoshnood B, Klungsoyr K, McDonnell B, Pierini A, Rankin J, Rissmann A, Rounding C, Queisser-Luft A, Scarano G, Tucker D (2014) Prevalence, prenatal diagnosis and clinical features of oculo-auriculo-vertebral spectrum: a registry-based study in Europe. Eur J Hum Genet 22:1026–1033. https://doi.org/10.1038/ejhg.2013.287
Barutcu AR, Wu M, Braunschweig U, Dyakov BJA, Luo Z, Turner KM, Durbic T, Lin ZY, Weatheritt RJ, Maass PG, Gingras AC, Blencowe BJ (2022) Systematic mapping of nuclear domain-associated transcripts reveals speckles and lamina as hubs of functionally distinct retained introns. Mol Cell 82:1035-1052 e9. https://doi.org/10.1016/j.molcel.2021.12.010
Beauchamp MC, Alam SS, Kumar S, Jerome-Majewska LA (2020) Spliceosomopathies and neurocristopathies: two sides of the same coin? Dev Dyn 249:924–945. https://doi.org/10.1002/dvdy.183
Beck JS (1961) Variations in the morphological patterns of “autoimmune” nuclear fluorescence. Lancet 1:1203–1205. https://doi.org/10.1016/s0140-6736(61)91944-4
Beleza-Meireles A, Clayton-Smith J, Saraiva JM, Tassabehji M (2014) Oculo-auriculo-vertebral spectrum: a review of the literature and genetic update. J Med Genet 51:635–645. https://doi.org/10.1136/jmedgenet-2014-102476
Bernier FP, Caluseriu O, Ng S, Schwartzentruber J, Buckingham KJ, Innes AM, Jabs EW, Innis JW, Schuette JL, Gorski JL, Byers PH, Andelfinger G, Siu V, Lauzon J, Fernandez BA, McMillin M, Scott RH, Racher H, Majewski J, Nickerson DA, Shendure J, Bamshad MJ, Parboosingh JS, Consortium FC (2012) Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am J Hum Genet 90:925–933. https://doi.org/10.1016/j.ajhg.2012.04.004
Berro R, Pedati C, Kehn-Hall K, Wu W, Klase Z, Even Y, Geneviere AM, Ammosova T, Nekhai S, Kashanchi F (2008) CDK13, a new potential human immunodeficiency virus type 1 inhibitory factor regulating viral mRNA splicing. J Virol 82:7155–7166. https://doi.org/10.1128/JVI.02543-07
Blencowe BJ, Bauren G, Eldridge AG, Issner R, Nickerson JA, Rosonina E, Sharp PA (2000) The SRm160/300 splicing coactivator subunits. RNA 6:111–120. https://doi.org/10.1017/s1355838200991982
Boulias K, Greer EL (2022) Biological roles of adenine methylation in RNA. Nat Rev Genet. https://doi.org/10.1038/s41576-022-00534-0
Burgute BD, Peche VS, Steckelberg AL, Glockner G, Gassen B, Gehring NH, Noegel AA (2014) NKAP is a novel RS-related protein that interacts with RNA and RNA binding proteins. Nucleic Acids Res 42:3177–3193. https://doi.org/10.1093/nar/gkt1311
Cajal S (1910) El nucleo de las celulas piramidales del cerebro humano y de algunos mamiferos. Trab Lab Invest Biol 8:27–62
Carter KC, Taneja KL, Lawrence JB (1991) Discrete nuclear domains of poly(A) RNA and their relationship to the functional organization of the nucleus. J Cell Biol 115:1191–1202. https://doi.org/10.1083/jcb.115.5.1191
Champion-Arnaud P, Reed R (1994) The prespliceosome components SAP 49 and SAP 145 interact in a complex implicated in tethering U2 snRNP to the branch site. Genes Dev 8:1974–1983. https://doi.org/10.1101/gad.8.16.1974
Chen D, Li Z, Yang Q, Zhang J, Zhai Z, Shu HB (2003) Identification of a nuclear protein that promotes NF-kappaB activation. Biochem Biophys Res Commun 310:720–724
Chen Y, Zhang Y, Wang Y, Zhang L, Brinkman EK, Adam SA, Goldman R, van Steensel B, Ma J, Belmont AS (2018) Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J Cell Biol 217:4025–4048. https://doi.org/10.1083/jcb.201807108
Chuang TW, Lee KM, Tarn WY (2015) Function and pathological implications of exon junction complex factor Y14. Biomolecules 5:343–355. https://doi.org/10.3390/biom5020343
Cuinat S, Nizon M, Isidor B, Stegmann A, van Jaarsveld RH, van Gassen KL, van der Smagt JJ, Volker-Touw CML, Holwerda SJB, Terhal PA, Schuhmann S, Vasileiou G, Khalifa M, Nugud AA, Yasaei H, Ousager LB, Brasch-Andersen C, Deb W, Besnard T, Simon MEH, Amsterdam KH, Verbeek NE, Matalon D, Dykzeul N, White S, Spiteri E, Devriendt K, Boogaerts A, Willemsen M, Brunner HG, Sinnema M, De Vries BBA, Gerkes EH, Pfundt R, Izumi K, Krantz ID, Xu ZL, Murrell JR, Valenzuela I, Cusco I, Rovira-Moreno E, Yang Y, Bizaoui V, Patat O, Faivre L, Tran-Mau-Them F, Vitobello A, Denomme-Pichon AS, Philippe C, Bezieau S, Cogne B (2022) Loss-of-function variants in SRRM2 cause a neurodevelopmental disorder. Genet Med 24:1774–1780. https://doi.org/10.1016/j.gim.2022.04.011
Czeschik JC, Voigt C, Alanay Y, Albrecht B, Avci S, Fitzpatrick D, Goudie DR, Hehr U, Hoogeboom AJ, Kayserili H, Simsek-Kiper PO, Klein-Hitpass L, Kuechler A, Lopez-Gonzalez V, Martin M, Rahmann S, Schweiger B, Splitt M, Wollnik B, Ludecke HJ, Zeschnigk M, Wieczorek D (2013) Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Hum Genet 132:885–898. https://doi.org/10.1007/s00439-013-1295-2
Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA (2003) DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 4:P3
Dias AP, Dufu K, Lei H, Reed R (2010) A role for TREX components in the release of spliced mRNA from nuclear speckle domains. Nat Commun 1:97. https://doi.org/10.1038/ncomms1103
Dingemans AJM, Truijen KMG, Kim JH, Alacam Z, Faivre L, Collins KM, Gerkes EH, van Haelst M, van de Laar I, Lindstrom K, Nizon M, Pauling J, Heropolitanska-Pliszka E, Plomp AS, Racine C, Sachdev R, Sinnema M, Skranes J, Veenstra-Knol HE, Verberne EA, Vulto-van Silfhout AT, Wilsterman MEF, Ahn EE, de Vries BBA, Vissers L (2022) Establishing the phenotypic spectrum of ZTTK syndrome by analysis of 52 individuals with variants in SON. Eur J Hum Genet 30:271–281. https://doi.org/10.1038/s41431-021-00960-4
Dopie J, Sweredoski MJ, Moradian A, Belmont AS (2020) Tyramide signal amplification mass spectrometry (TSA-MS) ratio identifies nuclear speckle proteins. J Cell Biol. https://doi.org/10.1083/jcb.201910207
Dujardin G, Lafaille C, de la Mata M, Marasco LE, Munoz MJ, Le Jossic-Corcos C, Corcos L, Kornblihtt AR (2014) How slow RNA polymerase II elongation favors alternative exon skipping. Mol Cell 54:683–690. https://doi.org/10.1016/j.molcel.2014.03.044
Even Y, Durieux S, Escande ML, Lozano JC, Peaucellier G, Weil D, Geneviere AM (2006) CDC2L5, a Cdk-like kinase with RS domain, interacts with the ASF/SF2-associated protein p32 and affects splicing in vivo. J Cell Biochem 99:890–904. https://doi.org/10.1002/jcb.20986
Fan Z, Devlin JR, Hogg SJ, Doyle MA, Harrison PF, Todorovski I, Cluse LA, Knight DA, Sandow JJ, Gregory G, Fox A, Beilharz TH, Kwiatkowski N, Scott NE, Vidakovic AT, Kelly GP, Svejstrup JQ, Geyer M, Gray NS, Vervoort SJ, Johnstone RW (2020) CDK13 cooperates with CDK12 to control global RNA polymerase II processivity. Sci Adv. https://doi.org/10.1126/sciadv.aaz5041
Fei J, Jadaliha M, Harmon TS, Li ITS, Hua B, Hao Q, Holehouse AS, Reyer M, Sun Q, Freier SM, Pappu RV, Prasanth KV, Ha T (2017) Quantitative analysis of multilayer organization of proteins and RNA in nuclear speckles at super resolution. J Cell Sci 130:4180–4192. https://doi.org/10.1242/jcs.206854
Fica SM, Oubridge C, Wilkinson ME, Newman AJ, Nagai K (2019) A human postcatalytic spliceosome structure reveals essential roles of metazoan factors for exon ligation. Science 363:710–714. https://doi.org/10.1126/science.aaw5569
Fiordaliso SK, Iwata-Otsubo A, Ritter AL, Quesnel-Vallieres M, Fujiki K, Nishi E, Hancarova M, Miyake N, Morton JEV, Lee S, Hackmann K, Bando M, Masuda K, Nakato R, Arakawa M, Bhoj E, Li D, Hakonarson H, Takeda R, Harr M, Keena B, Zackai EH, Okamoto N, Mizuno S, Ko JM, Valachova A, Prchalova D, Vlckova M, Pippucci T, Seiler C, Choi M, Matsumoto N, Di Donato N, Barash Y, Sedlacek Z, Shirahige K, Izumi K (2019) Missense mutations in NKAP cause a disorder of transcriptional regulation characterized by marfanoid habitus and cognitive impairment. Am J Hum Genet 105:987–995. https://doi.org/10.1016/j.ajhg.2019.09.009
Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T, Diener K, Jones K, Fu XD, Bentley DL (2014) Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev 28:2663–2676. https://doi.org/10.1101/gad.252106.114
Galganski L, Urbanek MO, Krzyzosiak WJ (2017) Nuclear speckles: molecular organization, biological function and role in disease. Nucleic Acids Res 45:10350–10368. https://doi.org/10.1093/nar/gkx759
Gorlin RJ, Cervenka J, Anderson RC, Sauk JJ, Bevis WD (1970) Robin’s syndrome. A probably X-linked recessive subvariety exhibiting persistence of left superior vena cava and atrial septal defect. Am J Dis Child 119:176–178
Greenleaf AL (2019) Human CDK12 and CDK13, multi-tasking CTD kinases for the new millenium. Transcription 10:91–110. https://doi.org/10.1080/21541264.2018.1535211
Greig JA, Nguyen TA, Lee M, Holehouse AS, Posey AE, Pappu RV, Jedd G (2020) Arginine-enriched mixed-charge domains provide cohesion for nuclear speckle condensation. Mol Cell 77:1237-1250 e4. https://doi.org/10.1016/j.molcel.2020.01.025
Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall’Agnese A, Hannett NM, Spille JH, Afeyan LK, Zamudio AV, Shrinivas K, Abraham BJ, Boija A, Decker TM, Rimel JK, Fant CB, Lee TI, Cisse II, Sharp PA, Taatjes DJ, Young RA (2019) Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572:543–548. https://doi.org/10.1038/s41586-019-1464-0
Hamilton MJ, Caswell RC, Canham N, Cole T, Firth HV, Foulds N, Heimdal K, Hobson E, Houge G, Joss S, Kumar D, Lampe AK, Maystadt I, McKay V, Metcalfe K, Newbury-Ecob R, Park SM, Robert L, Rustad CF, Wakeling E, Wilkie AOM, Study T, Twigg SRF, Suri M (2018) Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. J Med Genet 55:28–38. https://doi.org/10.1136/jmedgenet-2017-104620
Hojland AT, Lolas I, Okkels H, Lautrup CK, Diness BR, Petersen MB, Nielsen IK (2018) First reported adult patient with TARP syndrome: a case report. Am J Med Genet A 176:2915–2918. https://doi.org/10.1002/ajmg.a.40638
Hu S, Lv P, Yan Z, Wen B (2019) Disruption of nuclear speckles reduces chromatin interactions in active compartments. Epigenetics Chromatin 12:43. https://doi.org/10.1186/s13072-019-0289-2
Ilik IA, Malszycki M, Lubke AK, Schade C, Meierhofer D, Aktas T (2020) SON and SRRM2 are essential for nuclear speckle formation. Elife 9. https://doi.org/10.7554/eLife.60579
Inoue A (2021) RBM10: Structure, functions, and associated diseases. Gene 783:145463. https://doi.org/10.1016/j.gene.2021.145463
Inoue A, Tsugawa K, Tokunaga K, Takahashi KP, Uni S, Kimura M, Nishio K, Yamamoto N, Honda K, Watanabe T, Yamane H, Tani T (2008) S1–1 nuclear domains: characterization and dynamics as a function of transcriptional activity. Biol Cell 100:523–535. https://doi.org/10.1042/BC20070142
Inoue A, Yamamoto N, Kimura M, Nishio K, Yamane H, Nakajima K (2014) RBM10 regulates alternative splicing. FEBS Lett 588:942–947. https://doi.org/10.1016/j.febslet.2014.01.052
Ji X, Zhou Y, Pandit S, Huang J, Li H, Lin CY, Xiao R, Burge CB, Fu XD (2013) SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell 153:855–868. https://doi.org/10.1016/j.cell.2013.04.028
Johnston JJ, Teer JK, Cherukuri PF, Hansen NF, Loftus SK, Center NIHIS, Chong K, Mullikin JC, Biesecker LG (2010) Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am J Hum Genet 86:743–748. https://doi.org/10.1016/j.ajhg.2010.04.007
Johnston JJ, Sapp JC, Curry C, Horton M, Leon E, Cusmano-Ozog K, Dobyns WB, Hudgins L, Zackai E, Biesecker LG (2014) Expansion of the TARP syndrome phenotype associated with de novo mutations and mosaicism. Am J Med Genet A 164A:120–128. https://doi.org/10.1002/ajmg.a.36212
Kaeppler KE, Stetson RC, Lanpher BC, Collura CA (2018) Infant male with TARP syndrome: review of clinical features, prognosis, and commonalities with previously reported patients. Am J Med Genet A 176:2911–2914. https://doi.org/10.1002/ajmg.a.40645
Khan M, Hou S, Chen M, Lei H (2022) Mechanisms of RNA export and nuclear retention. Wiley Interdiscip Rev RNA. https://doi.org/10.1002/wrna.1755
Kim JH, Baddoo MC, Park EY, Stone JK, Park H, Butler TW, Huang G, Yan X, Pauli-Behn F, Myers RM, Tan M, Flemington EK, Lim ST, Ahn EY (2016a) SON and its alternatively spliced isoforms control MLL complex-mediated H3K4me3 and transcription of leukemia-associated genes. Mol Cell 61:859–873. https://doi.org/10.1016/j.molcel.2016.02.024
Kim JH, Shinde DN, Reijnders MRF, Hauser NS, Belmonte RL, Wilson GR, Bosch DGM, Bubulya PA, Shashi V, Petrovski S, Stone JK, Park EY, Veltman JA, Sinnema M, Stumpel C, Draaisma JM, Nicolai J, University of Washington Center for Mendelian G, Yntema HG, Lindstrom K, de Vries BBA, Jewett T, Santoro SL, Vogt J, Deciphering Developmental Disorders S, Bachman KK, Seeley AH, Krokosky A, Turner C, Rohena L, Hempel M, Kortum F, Lessel D, Neu A, Strom TM, Wieczorek D, Bramswig N, Laccone FA, Behunova J, Rehder H, Gordon CT, Rio M, Romana S, Tang S, El-Khechen D, Cho MT, McWalter K, Douglas G, Baskin B, Begtrup A, Funari T, Schoch K, Stegmann APA, Stevens SJC, Zhang DE, Traver D, Yao X, MacArthur DG, Brunner HG, Mancini GM, Myers RM, Owen LB, Lim ST, Stachura DL, Vissers L, Ahn EYE (2016b) De novo mutations in SON disrupt RNA splicing of genes essential for brain development and metabolism, causing an intellectual-disability syndrome. Am J Hum Genet 99:711–719. https://doi.org/10.1016/j.ajhg.2016.06.029
Kim J, Han KY, Khanna N, Ha T, Belmont AS (2019) Nuclear speckle fusion via long-range directional motion regulates speckle morphology after transcriptional inhibition. J Cell Sci. https://doi.org/10.1242/jcs.226563
Kim J, Venkata NC, Hernandez Gonzalez GA, Khanna N, Belmont AS (2020) Gene expression amplification by nuclear speckle association. J Cell Biol. https://doi.org/10.1083/jcb.201904046
Klopocki E, Schulze H, Strauss G, Ott CE, Hall J, Trotier F, Fleischhauer S, Greenhalgh L, Newbury-Ecob RA, Neumann LM, Habenicht R, Konig R, Seemanova E, Megarbane A, Ropers HH, Ullmann R, Horn D, Mundlos S (2007) Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am J Hum Genet 80:232–240. https://doi.org/10.1086/510919
Ko TK, Kelly E, Pines J (2001) CrkRS: a novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. J Cell Sci 114:2591–2603. https://doi.org/10.1242/jcs.114.14.2591
Kohler S, Gargano M, Matentzoglu N, Carmody LC, Lewis-Smith D, Vasilevsky NA, Danis D, Balagura G, Baynam G, Brower AM, Callahan TJ, Chute CG, Est JL, Galer PD, Ganesan S, Griese M, Haimel M, Pazmandi J, Hanauer M, Harris NL, Hartnett MJ, Hastreiter M, Hauck F, He Y, Jeske T, Kearney H, Kindle G, Klein C, Knoflach K, Krause R, Lagorce D, McMurry JA, Miller JA, Munoz-Torres MC, Peters RL, Rapp CK, Rath AM, Rind SA, Rosenberg AZ, Segal MM, Seidel MG, Smedley D, Talmy T, Thomas Y, Wiafe SA, Xian J, Yuksel Z, Helbig I, Mungall CJ, Haendel MA, Robinson PN (2021) The human phenotype ontology in 2021. Nucleic Acids Res 49:D1207–D1217. https://doi.org/10.1093/nar/gkaa1043
Kushary ST, Revah-Politi A, Barua S, Ganapathi M, Accogli A, Aggarwal V, Brunetti-Pierri N, Cappuccio G, Capra V, Fagerberg CR, Gazdagh G, Guzman E, Hadonou M, Harrison V, Havelund K, Iancu D, Kraus A, Lippa NC, Mansukhani M, McBrian D, McEntagart M, Pacio-Miguez M, Palomares-Bralo M, Pottinger C, Ruivenkamp CAL, Sacco O, Santen GWE, Santos-Simarro F, Scala M, Short J, Sorensen KP, Woods CG, DDD Study Consortium T, Anyane Yeboa K (2021) ZTTK syndrome: clinical and molecular findings of 15 cases and a review of the literature. Am J Med Genet A 185:3740–3753. https://doi.org/10.1002/ajmg.a.62445
Lamond AI, Spector DL (2003) Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol 4:605–612. https://doi.org/10.1038/nrm1172
Lee Y, Rio DC (2015) Mechanisms and regulation of alternative pre-mRNA splicing. Annu Rev Biochem 84:291–323. https://doi.org/10.1146/annurev-biochem-060614-034316
Lerner EA, Lerner MR, Janeway CA Jr, Steitz JA (1981) Monoclonal antibodies to nucleic acid-containing cellular constituents: probes for molecular biology and autoimmune disease. Proc Natl Acad Sci U S A 78:2737–2741. https://doi.org/10.1073/pnas.78.5.2737
Li W, Laishram RS, Anderson RA (2013) The novel poly(A) polymerase Star-PAP is a signal-regulated switch at the 3’-end of mRNAs. Adv Biol Regul 53:64–76. https://doi.org/10.1016/j.jbior.2012.10.004
Liang K, Gao X, Gilmore JM, Florens L, Washburn MP, Smith E, Shilatifard A (2015) Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol Cell Biol 35:928–938. https://doi.org/10.1128/MCB.01426-14
Lin S, Coutinho-Mansfield G, Wang D, Pandit S, Fu XD (2008) The splicing factor SC35 has an active role in transcriptional elongation. Nat Struct Mol Biol 15:819–826. https://doi.org/10.1038/nsmb.1461
Liu J, Lu X, Zhang S, Yuan L, Sun Y (2022) Molecular Insights into mRNA Polyadenylation and Deadenylation. Int J Mol Sci. https://doi.org/10.3390/ijms231910985
Lu X, Goke J, Sachs F, Jacques PE, Liang H, Feng B, Bourque G, Bubulya PA, Ng HH (2013) SON connects the splicing-regulatory network with pluripotency in human embryonic stem cells. Nat Cell Biol 15:1141–1152. https://doi.org/10.1038/ncb2839
Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, Darzacq X, Zhou Q (2018) Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558:318–323. https://doi.org/10.1038/s41586-018-0174-3
Marques F, Moreau JL, Peaucellier G, Lozano JC, Schatt P, Picard A, Callebaut I, Perret E, Geneviere AM (2000) A new subfamily of high molecular mass CDC2-related kinases with PITAI/VRE motifs. Biochem Biophys Res Commun 279:832–837. https://doi.org/10.1006/bbrc.2000.4042
Mellman DL, Gonzales ML, Song C, Barlow CA, Wang P, Kendziorski C, Anderson RA (2008) A PtdIns4,5P2-regulated nuclear poly(A) polymerase controls expression of select mRNAs. Nature 451:1013–1017. https://doi.org/10.1038/nature06666
Miyagawa R, Tano K, Mizuno R, Nakamura Y, Ijiri K, Rakwal R, Shibato J, Masuo Y, Mayeda A, Hirose T, Akimitsu N (2012) Identification of cis- and trans-acting factors involved in the localization of MALAT-1 noncoding RNA to nuclear speckles. RNA 18:738–751. https://doi.org/10.1261/rna.028639.111
Niceta M, Barresi S, Pantaleoni F, Capolino R, Dentici ML, Ciolfi A, Pizzi S, Bartuli A, Dallapiccola B, Tartaglia M, Digilio MC (2019) TARP syndrome: Long-term survival, anatomic patterns of congenital heart defects, differential diagnosis and pathogenetic considerations. Eur J Med Genet 62:103534. https://doi.org/10.1016/j.ejmg.2018.09.001
Nojima T, Rebelo K, Gomes T, Grosso AR, Proudfoot NJ, Carmo-Fonseca M (2018) RNA polymerase II phosphorylated on CTD serine 5 interacts with the spliceosome during Co-transcriptional splicing. Mol Cell 72:369-379 e4. https://doi.org/10.1016/j.molcel.2018.09.004
O’Keefe RT, Mayeda A, Sadowski CL, Krainer AR, Spector DL (1994) Disruption of pre-mRNA splicing in vivo results in reorganization of splicing factors. J Cell Biol 124:249–260. https://doi.org/10.1083/jcb.124.3.249
Pajerowski AG, Nguyen C, Aghajanian H, Shapiro MJ, Shapiro VS (2009) NKAP is a transcriptional repressor of notch signaling and is required for T cell development. Immunity 30:696–707. https://doi.org/10.1016/j.immuni.2009.02.011
Pajerowski AG, Shapiro MJ, Gwin K, Sundsbak R, Nelson-Holte M, Medina K, Shapiro VS (2010) Adult hematopoietic stem cells require NKAP for maintenance and survival. Blood 116:2684–2693. https://doi.org/10.1182/blood-2010-02-268391
Petit F, Escande F, Jourdain AS, Porchet N, Amiel J, Doray B, Delrue MA, Flori E, Kim CA, Marlin S, Robertson SP, Manouvrier-Hanu S, Holder-Espinasse M (2014) Nager syndrome: confirmation of SF3B4 haploinsufficiency as the major cause. Clin Genet 86:246–251. https://doi.org/10.1111/cge.12259
Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, Trinh V, Aznauryan E, Russell P, Cheng C, Jovanovic M, Chow A, Cai L, McDonel P, Garber M, Guttman M (2018) Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174:744–75724. https://doi.org/10.1016/j.cell.2018.05.024
Ramanathan A, Robb GB, Chan SH (2016) mRNA capping: biological functions and applications. Nucleic Acids Res 44:7511–7526. https://doi.org/10.1093/nar/gkw551
Saitoh N, Spahr CS, Patterson SD, Bubulya P, Neuwald AF, Spector DL (2004) Proteomic analysis of interchromatin granule clusters. Mol Biol Cell 15:3876–3890. https://doi.org/10.1091/mbc.e04-03-0253
Sawada Y, Miura Y, Umeki K, Tamaoki T, Fujinaga K, Ohtaki S (2000) Cloning and characterization of a novel RNA-binding protein SRL300 with RS domains. Biochim Biophys Acta 1492:191–195. https://doi.org/10.1016/s0167-4781(00)00065-8
Schmidt U, Im KB, Benzing C, Janjetovic S, Rippe K, Lichter P, Wachsmuth M (2009) Assembly and mobility of exon-exon junction complexes in living cells. RNA 15:862–876. https://doi.org/10.1261/rna.1387009
Sharma A, Takata H, Shibahara K, Bubulya A, Bubulya PA (2010) Son is essential for nuclear speckle organization and cell cycle progression. Mol Biol Cell 21:650–663. https://doi.org/10.1091/mbc.E09-02-0126
Shopland LS, Johnson CV, Lawrence JB (2002) Evidence that all SC-35 domains contain mRNAs and that transcripts can be structurally constrained within these domains. J Struct Biol 140:131–139. https://doi.org/10.1016/s1047-8477(02)00507-5
Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Turki SH, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ, Prigmore E, Rajan D, Abdul-Khaliq H, Banka S, Bauer UM, Bentham J, Berger F, Bhattacharya S, Bu’Lock F, Canham N, Colgiu IG, Cosgrove C, Cox H, Daehnert I, Daly A, Danesh J, Fryer A, Gewillig M, Hobson E, Hoff K, Homfray T, Study I, Kahlert AK, Ketley A, Kramer HH, Lachlan K, Lampe AK, Louw JJ, Manickara AK, Manase D, McCarthy KP, Metcalfe K, Moore C, Newbury-Ecob R, Omer SO, Ouwehand WH, Park SM, Parker MJ, Pickardt T, Pollard MO, Robert L, Roberts DJ, Sambrook J, Setchfield K, Stiller B, Thornborough C, Toka O, Watkins H, Williams D, Wright M, Mital S, Daubeney PE, Keavney B, Goodship J, Consortium UK, Abu-Sulaiman RM, Klaassen S, Wright CF, Firth HV, Barrett JC, Devriendt K, FitzPatrick DR, Brook JD, Deciphering Developmental Disorders S, Hurles ME (2016) Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet 48:1060–1065. https://doi.org/10.1038/ng.3627
Spector DL, Lamond AI (2011) Nuclear speckles. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a000646
Spector DL, Schrier WH, Busch H (1983) Immunoelectron microscopic localization of snRNPs. Biol Cell 49:1–10. https://doi.org/10.1111/j.1768-322x.1984.tb00215.x
Spector DL, Fu XD, Maniatis T (1991) Associations between distinct pre-mRNA splicing components and the cell nucleus. EMBO J 10:3467–3481. https://doi.org/10.1002/j.1460-2075.1991.tb04911.x
Su JH, Zheng P, Kinrot SS, Bintu B, Zhuang X (2020) Genome-scale imaging of the 3D organization and transcriptional activity of chromatin. Cell 182:1641-1659 e26. https://doi.org/10.1016/j.cell.2020.07.032
Sun C (2020) The SF3b complex: splicing and beyond. Cell Mol Life Sci 77:3583–3595. https://doi.org/10.1007/s00018-020-03493-z
Swift H (1959) Studies on nuclear fine structure. Brookhaven Symp Biol 12:134–152
Takenouchi T, Miura K, Uehara T, Mizuno S, Kosaki K (2016) Establishing SON in 21q22.11 as a cause a new syndromic form of intellectual disability: possible contribution to Braddock-Carey syndrome phenotype. Am J Med Genet A 170:2587–2590. https://doi.org/10.1002/ajmg.a.37761
Thomas JD, Polaski JT, Feng Q, De Neef EJ, Hoppe ER, McSharry MV, Pangallo J, Gabel AM, Belleville AE, Watson J, Nkinsi NT, Berger AH, Bradley RK (2020) RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat Genet 52:84–94. https://doi.org/10.1038/s41588-019-0555-z
Timberlake AT, Griffin C, Heike CL, Hing AV, Cunningham ML, Chitayat D, Davis MR, Doust SJ, Drake AF, Duenas-Roque MM, Goldblatt J, Gustafson JA, Hurtado-Villa P, Johns A, Karp N, Laing NG, Magee L, University of Washington Center for Mendelian G, Mullegama SV, Pachajoa H, Porras-Hurtado GL, Schnur RE, Slee J, Singer SL, Staffenberg DA, Timms AE, Wise CA, Zarante I, Saint-Jeannet JP, Luquetti DV (2021) Haploinsufficiency of SF3B2 causes craniofacial microsomia. Nat Commun 12:4680. https://doi.org/10.1038/s41467-021-24852-9
Tokita MJ, Braxton AA, Shao Y, Lewis AM, Vincent M, Kury S, Besnard T, Isidor B, Latypova X, Bezieau S, Liu P, Motter CS, Melver CW, Robin NH, Infante EM, McGuire M, El-Gharbawy A, Littlejohn RO, McLean SD, Bi W, Bacino CA, Lalani SR, Scott DA, Eng CM, Yang Y, Schaaf CP, Walkiewicz MA (2016) De Novo truncating variants in SON cause intellectual disability, congenital malformations, and failure to thrive. Am J Hum Genet 99:720–727. https://doi.org/10.1016/j.ajhg.2016.06.035
Ueda M, Matsuki T, Fukada M, Eda S, Toya A, Iio A, Tabata H, Nakayama A (2020) Knockdown of son, a mouse homologue of the ZTTK syndrome gene, causes neuronal migration defects and dendritic spine abnormalities. Mol Brain 13:80. https://doi.org/10.1186/s13041-020-00622-4
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F (2015) Proteomics. Tissue-based map of the human proteome. Science 347:1260419. https://doi.org/10.1126/science.1260419
Wang Y, Gogol-Doring A, Hu H, Frohler S, Ma Y, Jens M, Maaskola J, Murakawa Y, Quedenau C, Landthaler M, Kalscheuer V, Wieczorek D, Wang Y, Hu Y, Chen W (2013) Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol Med 5:1431–1442. https://doi.org/10.1002/emmm.201302663
Wang Z, Qin G, Zhao TC (2014) HDAC4: mechanism of regulation and biological functions. Epigenomics 6:139–150. https://doi.org/10.2217/epi.13.73
Wang LY, Xiao SJ, Kunimoto H, Tokunaga K, Kojima H, Kimura M, Yamamoto T, Yamamoto N, Zhao H, Nishio K, Tani T, Nakajima K, Sunami K, Inoue A (2021a) Sequestration of RBM10 in nuclear bodies: targeting sequences and biological significance. Int J Mol Sci. https://doi.org/10.3390/ijms221910526
Wang S, Bleeck A, Nadif Kasri N, Kleefstra T, van Rhijn JR, Schubert D (2021b) SETD1A mediated H3K4 methylation and its role in neurodevelopmental and neuropsychiatric disorders. Front Mol Neurosci 14:772000. https://doi.org/10.3389/fnmol.2021.772000
Worlitzer MM, Schwamborn JC (2014) The Notch co-repressor protein NKAP is highly expressed in adult mouse subventricular zone neural progenitor cells. Neuroscience 266:138–149. https://doi.org/10.1016/j.neuroscience.2014.02.019
Xu Q, Xiang Y, Wang Q, Wang L, Brind’Amour J, Bogutz AB, Zhang Y, Zhang B, Yu G, Xia W, Du Z, Huang C, Ma J, Zheng H, Li Y, Liu C, Walker CL, Jonasch E, Lefebvre L, Wu M, Lorincz MC, Li W, Li L, Xie W (2019) SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet 51:844–856. https://doi.org/10.1038/s41588-019-0398-7
Xu S, Lai SK, Sim DY, Ang WSL, Li HY, Roca X (2022) SRRM2 organizes splicing condensates to regulate alternative splicing. Nucleic Acids Res 50:8599–8614. https://doi.org/10.1093/nar/gkac669
Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, Ozawa M, Ma J, Yoshida N, Reiter JF, Black DL, Kharchenko PV, Sharp PA, Walsh CA (2016) Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell 166:1147–116215. https://doi.org/10.1016/j.cell.2016.07.025
Zhang L, Zhang Y, Chen Y, Gholamalamdari O, Wang Y, Ma J, Belmont AS (2020) TSA-seq reveals a largely conserved genome organization relative to nuclear speckles with small position changes tightly correlated with gene expression changes. Genome Res 31:251–264. https://doi.org/10.1101/gr.266239.120
Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, Ben-Zeev B, Nissenkorn A, Anikster Y, Oz-Levi D, Dhindsa RS, Hitomi Y, Schoch K, Spillmann RC, Heimer G, Marek-Yagel D, Tzadok M, Han Y, Worley G, Goldstein J, Jiang YH, Lancet D, Pras E, Shashi V, McHale D, Need AC, Goldstein DB (2015) Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med 17:774–781. https://doi.org/10.1038/gim.2014.191
Funding
This work was supported by Children’s Hospital of Philadelphia and Japan Society for the Promotion of Science Grant-in-Aid for Transformative Research Areas (A) 20H05940 and Grant-in-Aid for Scientific Research (C) 21K06833. The authors have no relevant financial or nonfinancial interests to disclose.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
439_2023_2540_MOESM5_ESM.docx
Supplementary Supplemental table 5: Top 20 frequent HPO terms associated with NS proteins identified by Dopie et al. file5 (DOCX 18 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Regan-Fendt, K.E., Izumi, K. Nuclear speckleopathies: developmental disorders caused by variants in genes encoding nuclear speckle proteins. Hum. Genet. 143, 529–544 (2024). https://doi.org/10.1007/s00439-023-02540-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-023-02540-6