
Abstract
The human solute carrier (SLC) superfamily of transporters is comprised of over 400 membrane-bound proteins, and plays essential roles in a multitude of physiological and pharmacological processes. In addition, perturbation of SLC transporter function underlies numerous human diseases, which renders SLC transporters attractive drug targets. Common genetic polymorphisms in SLC genes have been associated with inter-individual differences in drug efficacy and toxicity. However, despite their tremendous clinical relevance, epidemiological data of these variants are mostly derived from heterogeneous cohorts of small sample size and the genetic SLC landscape beyond these common variants has not been comprehensively assessed. In this study, we analyzed Next-Generation Sequencing data from 141,456 individuals from seven major human populations to evaluate genetic variability, its functional consequences, and ethnogeographic patterns across the entire SLC superfamily of transporters. Importantly, of the 204,287 exonic single-nucleotide variants (SNVs) which we identified, 99.8% were present in less than 1% of analyzed alleles. Comprehensive computational analyses using 13 partially orthogonal algorithms that predict the functional impact of genetic variations based on sequence information, evolutionary conservation, structural considerations, and functional genomics data revealed that each individual genome harbors 29.7 variants with putative functional effects, of which rare variants account for 18%. Inter-ethnic variability was found to be extensive, and 83% of deleterious SLC variants were only identified in a single population. Interestingly, population-specific carrier frequencies of loss-of-function variants in SLC genes associated with recessive Mendelian disease recapitulated the ethnogeographic variation of the corresponding disorders, including cystinuria in Jewish individuals, type II citrullinemia in East Asians, and lysinuric protein intolerance in Finns, thus providing a powerful resource for clinical geneticists to inform about population-specific prevalence and allelic composition of Mendelian SLC diseases. In summary, we present the most comprehensive data set of SLC variability published to date, which can provide insights into inter-individual differences in SLC transporter function and guide the optimization of population-specific genotyping strategies in the bourgeoning fields of personalized medicine and precision public health.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The solute carrier (SLC) gene superfamily is one of two major human gene families encoding transporters of endogenous and exogenous compounds. SLCs constitute the second-largest family of membrane proteins in the human genome with over 400 proteins classified into 65 subfamilies based on sequence similarity (Fredriksson et al. 2008; Höglund et al. 2011; Schlessinger et al. 2013). Substrate specificity differs substantially across the various subfamilies. While some subfamilies, such as the carbohydrate and long chain fatty acid transporters of the SLC2 and SLC27 subfamilies, transport only few physicochemically homogenous substrates (Anderson and Stahl 2013; Mueckler and Thorens 2013), transporters of the SLC22 family mediate the translocation of various dissimilar ions, including organic cations, anions, and zwitterions (Koepsell 2013). Most SLC transporters are equilibrative, making use of electrochemical and concentration gradients to facilitate the uptake of their substrates into cells. Transport mechanisms can differ within subfamilies, however, as seen in the secondary active symporters and antiporters of the SLC4 bicarbonate transporter family (Romero et al. 2013).
Due to their essential roles in the transport of a plethora of essential organic and inorganic substrates and the high number (> 100) of SLC transporters that have been associated with human genetic disorders, SLC transporters are being increasingly investigated as potential drug targets. One prominent example is the development of blockbuster SGLT2 (encoded by SLC5A2) inhibitors for the treatment of diabetic hyperglycaemia, which was inspired by associations between SLC5A2 mutations and familial renal glucosuria (OMIM identifier 233100). Besides their role as drug targets, SLC transporters play fundamental roles in the disposition of numerous drugs, including various chemotherapeutics, antidiabetics, and diuretics. Given the extensive genomic coverage and the critical role that SLC transporters play in mediating drug pharmacokinetics and -dynamics (PK/PD), the genetic variability of SLC genes is of considerable interest for human genetics, as well as for drug discovery and development programs.
In the last decade, seminal studies have contributed substantially to our understanding of the link between SLC variability and drug response. Prominent examples include the association between variants in SLC22A1, encoding OCT1, and pharmacokinetics and response to metformin (Dujic et al. 2015; Sundelin et al. 2017) and variations in SLC19A1, encoding the reduced folate transporter RFT, with toxicity of antifolate metabolites (Bohanec Grabar et al. 2012; Corrigan et al. 2014; Lima et al. 2014). However, these studies were only powered to detect associations with common variations. Importantly, recent population-scale sequencing projects revealed that rare variations with minor allele frequencies (MAF) < 1% greatly outnumber common variants in genes involved in drug absorption, distribution, metabolism, and excretion (ADME) (Bush et al. 2016; Kozyra et al. 2017; Wright et al. 2018; Zhou and Lauschke 2018). Rare variations are enriched in variants with functional consequences and commonly have increased effect sizes compared to common variants when analyzed in relation to disease (Ingelman-Sundberg et al. 2018; Manolio et al. 2009). While the extent and functional importance of rare variants is becoming increasingly appreciated, SLC transporters are understudied (César-Razquin et al. 2015) and their genetic landscape remains to be systematically analyzed.
Here, we systematically mapped the genetic variability of the human SLC transporter superfamily by analyzing consolidated whole-exome and whole-genome sequencing data (WES and WGS, respectively) from 141,456 individuals across seven major populations. We profiled the SLC genetic variability, its functional consequences, and ethnogeographic distribution using 13 partly orthogonal computational predictors, as well as structural mapping approaches using experimental high-resolution crystal structures. The obtained data set constitutes the most comprehensive analysis of genetic SLC variability published to date and provides valuable insights into inter-individual and inter-ethnic differences in transporter function with important implications for drug disposition, efficacy, and toxicity, as well as population-specific prevalence of Mendelian SLC diseases.
Materials and methods
Data collection and annotation
Genetic variability data of 401 genes comprising the human SLC superfamily were collected from the Genome Aggregation Database (gnomAD) version 2.1 (Lek et al. 2016). The use of these data did not require separate ethical approval, as the data are released under the Fort Lauderdale Agreement. In total, we analyzed sequencing data of 141,456 unrelated individuals spanning seven worldwide populations (64,603 Non-Finnish Europeans, 12,562 Finns, 12,487 Africans, 9977 East Asians, 15,308 South Asians, 17,720 Latinos, 5185 Ashkenazi Jews, and 3614 from other populations). Variants with low confidence calls were removed. Rare and common genetic SNVs were defined as variants with MAF < 1% and MAF ≥ 1%, respectively. Copy-number variants’ (CNVs) data from 59,451 individuals were obtained from the Exome Aggregation Consortium and analyzed as previously described (Santos et al. 2018). Linkage analysis was performed using LDLink (Machiela and Chanock 2015). Disease associations for the relevant SLC genes were obtained from the Online Mendelian Inheritance in Man (OMIM) database. Deleterious variants in disease-associated genes were filtered for benign variants using ClinVar Miner (Henrie et al. 2018).
Computational functionality predictions
Missense variants were analyzed using an array of partly orthogonal algorithms that predict the functional impact of genetic variations based on sequence information, evolutionary conservation, structural considerations, and functional genomics data. Specifically, we used SIFT (Ng and Henikoff 2001), Polyphen-2 (Adzhubei et al. 2010), Likelihood Ratio Tests (Chun and Fay 2009), MutationAssessor (Reva et al. 2011), FATHMM (Shihab et al. 2012), PROVEAN (Choi et al. 2012), VEST3 (Carter et al. 2013), CADD (Kircher et al. 2014), DANN (Quang et al. 2015), FATHMM-mkl (Shihab et al. 2015), MetaSVM (Dong et al. 2015), MetaLR (Dong et al. 2015), and GERP++ (Davydov et al. 2010). We considered all variants that resulted in the gain of a stop codon, the loss of the start codon, that caused frameshifts or that disrupted canonical splice sites as loss-of-function variants.
Structural modeling
The secondary structures of GLUT1 (SLC2A1) and ENT1 (SLC29A1) were obtained from UniProt (UniProt IDs 4PYP and 6OB6, respectively). The structure of OCT1 (SLC22A1) was predicted using Phyre2 (Kelley et al. 2015) as no high-resolution crystal structure for this transporter was available. Confidence and coverage scores were ≥ 100% and 80%, respectively. Structures were modeled using PyMOL version 2.3.
Results
Overview of the genetic variability of the human SLC superfamily
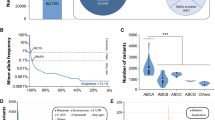
Across 141,456 unrelated individuals, we identified a total of 204,287 exonic single-nucleotide variants (SNVs) and indels (Fig. 1a). In addition, the data set contained 118,597 intronic variations; however, as these were not systematically covered, they were excluded in our further analyses. The majority of exonic variants resulted in amino acid exchanges of the encoded polypeptide (n = 116,300; 57% of all exonic variants). The remaining SNVs included synonymous variants (n = 56,685; 28%) and variants in the untranslated regions (n = 9507; 5% in the 5’ UTRs and n = 7400; 4% in the 3’ UTRs). Furthermore, we identified a multitude of variants that result in putative loss-of-function of the gene product, such as frameshifts (n = 5086), stop-gain variants (n = 3384), and variations in canonical splice sites (n = 3050). In addition to SNVs, we found 3688 copy-number variations (CNVs), comprised of 2532 duplications and 1156 deletions (Fig. 1a). Strikingly, of the 204,287 total exonic variants, 203,968 (99.8%) were identified as rare, with MAFs < 1% (Fig. 1b).

The landscape of genetic variability within the human SLC gene superfamily. a Overview of genetic variants across 401 SLC genes based on the Next-Generation Sequencing data of 141,456 individuals from seven major populations. Of the 204,287 identified exonic SNVs, the majority resulted in amino acid exchanges. In addition, we identified 3688 copy-number variations (CNVs) of SLC genes. b 99.8% of all exonic SLC variants were rare with minor allele frequencies < 1% and 57.6% were only found in a single individual. c Box and whisker plot depicting the number of variants that affect the amino acid sequence of the respective gene product (missense, frameshift, start-lost, stop-gain, indels, and splicing variants). Note that the number of such variants differs drastically between genes and SLC subfamilies. The middle line depicts the median and the edges of the boxes depict the 25th and 75th percentiles. d Violin plot of total exonic SLC variants per gene, classified by endogenous transporter substrate. White dots represent the median number of variants per gene, with the ends of the white boxes indicating the 25th and 75th percentiles. Polygons represent density estimates of the data and extend to extreme values. e Stacked bar plot showing the fraction of genes under high evolutionary constraint, with genes classified by protein substrate. Evolutionary constraint was estimated using the pLI score (Lek et al. 2016), with scores < 0.5 defined as little constraint (“Null”), scores 0.5 ≤ x ≤ 0.9 for genes for which homozygous loss-of-function results in a deleterious phenotype (“Recessive”), and scores > 0.9 defined for haploinsufficient genes
Next, we focused specifically on variants that affected the amino acid sequence of the encoded gene product. Among the subfamilies, variability was highest in the cholesterol transporter family SLC65 with a median of 809 variants per gene (n = 2 genes), followed by SLC12 chloride cotransporters (604 variants per gene; n = 9) and the bicarbonate transporter family SLC4 (556 variants per gene; n = 10; Fig. 1c). By contrast, four subfamilies harbored less than 100 variants per gene (SLC31, SLC48, SLC54, and SLC57). When stratifying SLC genes by substrate, variability was highest in genes coding for inorganic ion transporters (718 ± 228 s.d. variants per gene) and fatty acids (646 ± 187 variants per gene), whereas pyruvate transporters (230 ± 213 variants per gene) and metal transporters (433 ± 150) harbored substantially fewer variants (Fig. 1d). Loss-of-function variants were depleted in transporters of neurotransmitters, hormones, choline, inorganic ions, and metals with 19–25% of genes being classified as haploinsufficient, suggesting high evolutionary constraints and reduced functional redundancy in these gene families (Fig. 1e). In contrast, none of the genes encoding transporters of cofactors, carboxylates, fatty acids, protons, oligopeptides, and urea cycle metabolites were found to be haploinsufficient.
Population-specific frequencies of clinically important SLC variants and haplotypes
SLC transporters mediate the transport of a plethora of drugs, and multiple SLC variants can impact disposition, efficacy, or toxicity of various medications (Table 1 and Supplementary Table 1). Here, we analyzed the population-specific frequencies of 31 SLC variants with clinically relevant pharmacogenomic associations, mostly to antidiabetics, analgesics, anticoagulants, and various chemotherapeutics. Importantly, variant prevalence was highly population-specific and 42% of these variants (n = 13/31) differed more than fivefold between populations. Common variants in SLC22A1 have been repeatedly linked to altered drug disposition and efficacy of metformin (Todd and Florez 2014), imatinib (Watkins et al. 2015), and various opioids (Tzvetkov 2017). Multiple variations with functional consequences, including M420del (SLC22A1*2), R61C (SLC22A1*3), G401S (SLC22A1*4), and G465R (SLC22A1*5), are absent in East Asians, whereas they can reach frequencies up to 21.9% in other populations (Table 1). In contrast, L160F, which is associated with altered imatinib pharmacokinetics and an increased risk of resistance to imatinib (Cargnin et al. 2018; Di Paolo et al. 2014; Makhtar et al. 2018), is common in East Asians (MAF = 14.2%) but lowest in Africans (3.8%).
While pharmacogenetically important polymorphisms in the nucleoside transporter genes SLC28A1 and SLC28A2 were common worldwide, their frequencies differed drastically between populations. Rs2242046 in SLC28A1, as well as the highly linked variants rs1060896 and rs11854484 in SLC28A2 (R2 = 0.9), were least common in East Asian and African populations (MAF = 7.6–17.6%), whereas they were consistently more prevalent in all other populations (MAF = 20.8–65.4%). In contrast, Africans and East Asians were among the populations with the highest frequency of rs56350726, a variant in SLC28A3 implicated in improved outcomes and reduced toxicity of antiviral hepatitis C virus (HCV) therapy (Doehring et al. 2011; Rau et al. 2013), suggesting potentially important implications for toxicity risk of nucleoside analogs used in the treatment of viral infections and various cancers. Pronounced tenfold differences between populations were furthermore observed for the missense variant rs17235409 in SLC11A1 that is implicated in treatment failure of patients with pulmonary tuberculosis to isoniazid, rifampicin, pyrazinamide, and ethambutol combination therapy (Salinas-Delgado et al. 2015).
The variant rs1529927 in the renal sodium and chloride reabsorption transporter SLC12A3 (NCC) is associated with increased efficacy of diuretics (Vormfelde et al. 2007) and was common in Europeans (MAF = 3.1–3.6%), but rare in all other populations studied. Similar population specificity was observed for the reduced function variant rs11568482 in SLC22A8 (OAT3), which was exclusively found in East Asians with frequencies of 5.9%, with important implications for the renal clearance of the OAT3 substrate cefotaxime (Yee et al. 2013). Moreover, rs16889462 in SLC30A8, a variant associated with increased response to repaglinide (Huang et al. 2010), was common in Africans (MAF = 10.5%) and East Asians (MAF = 8%), but rare in all other populations analyzed (MAF < 1%).
Genetic variants in human SLC genes are predicted to substantially contribute to inter-individual differences in transporter function
To gauge the functional effects arising from the observed genetic variability beyond variants with known pharmacogenetic associations, we employed an array of 13 partly orthogonal computational algorithms. Of the 116,300 identified missense variants, 53,642 (46%) were predicted to alter the functionality of the respective gene product (Fig. 2a). Furthermore, we considered all 14,157 variants that caused frameshifts, the loss of a start or the premature gain of a stop codon or variants affecting canonical splice sites as putative loss-of-function variants. The highest median numbers of functional variants were identified in transporters of inorganic ions (239 variants), fatty acids (238 variants), and oligopeptides (216 variants; Fig. 2b). Per gene, most putatively deleterious variants were found in SLC12A4 (577 variants), SLC12A3 (544 variants), and SLC65A2 (531 variants), whereas less than 25 variants were found in pyruvate transporters.

Rare genetic variants contribute considerably to the genetically encoded functional variability of SLC transporters. a Of the 116,300 identified missense variants, 53,642 were predicted to alter the functionality of the transporter protein. Furthermore, 14,157 variants that caused frameshifts, the loss of a start or the premature gain of a stop codon, or variants that affecting canonical splice sites were expected to result in a loss of protein function. b Box and whisker plot of all these deleterious variants (n = 67,799) per gene demonstrates that the complexity of genetically encoded functional variability differs drastically between SLC substrate classes. c When aggregating variant numbers per individual, most variants were identified in transporters of organic ions and amino acids. Common variants (MAF > 1%) are shown in light red, while rare variants (MAF < 1%) are shown in dark red. Percentage values within or above stacked columns indicate the fraction of the genetically encoded functional variability allotted to rare variants. Inlet dot plots depict the total rare deleterious SLC variants per individual per gene, with the median represented by the dark bar. The gene with the highest number of rare deleterious variants per substrate class is indicated above the inlet. d The aggregated frequency of variants that affect transporter function is plotted for the top 10 and bottom 10 SLC genes. Note that differences between the most and least variable genes exceed 1000-fold. e Across the entire SLC superfamily, each individual was found to harbor 29.7 variants that are predicted to affect the functionality of the encoded transporter protein. Of this genetically encoded functional variability, 18.7% is attributed to rare variants
Each individual was found to harbor on average 6.2 and 5.8 variants with functional effects in organic ion and amino acid transporters, respectively (Fig. 2c). In contrast, the average diploid human genome contained less than 0.1 variants in transporters of bile acids, oligopeptides, choline, protons, heme, pyruvate, and various cofactors. The contribution of rare variants differed considerably between substrate classes. While rare variants accounted for 6%, 12%, and 12% of the genetically encoded functional variability in urea cycle, organic ion, and amino acid transporters, no common variants with functional effects were identified in transporters of pyruvate, heme, or various other substrates and, thus, rare genetic variants were the only cause of genetically encoded functional effects in these transporters (Fig. 2c). SLC26A11, SLC10A2, and SLC26A10 harbored most rare functional variants per individual, whereas least were found in SLC54A1, SLC54A2, and SLC51B. When integrating rare and common variant data, most deleterious variants were identified in the putative ammonium transporter SLC42A2 (RhBG; 3 deleterious variants per individual), the poorly understood ion transporter SLC22A10 (OAT5; 2 variants per individual), and the highly clinically relevant drug transporter SLC22A1 (OCT1; 1.3 variants per individual; Fig. 2d). In contrast, less than 1 in 2000 individuals harbored a deleterious variant in SLC54A3 (0), SLC38A3 (0.0001), SLC54A1 (0.0005), SLC51B (0.0007), SLC25A51 (0.0007), and SLC30A1 (0.0009). Strikingly, when aggregating information about genetically encoded functional variability across the entire SLC superfamily of genes, each individual was found to harbor on average 29.7 variants with putative functional consequences in SLC transporters of which rare variants accounted for 18% (5.4 rare variants per individual; Fig. 2e).
Genetic variability in SLC genes is highly population specific with important consequences for the predisposition to Mendelian disease
When we stratified the identified SLC variants that were predicted to affect transporter function by ancestry, we found that the distribution varied drastically between populations, with 83% of variants (n = 56,273) restricted to a single population (Fig. 3a). Most population-specific variants were identified in Europeans, whereas the lowest numbers were found in Finns and Ashkenazi Jews, at least in part due to unequal cohort sizes. Interestingly, after adjusting for cohort size, we found that East Asians had the largest number of population-specific variants with predicted functional consequence, suggesting that this population might benefit most from population-adjusted genotyping strategies (Fig. 3b). In contrast, overall genetically encoded functional variability differed only moderately between populations with individuals of African (34.6 variants/ individual) and European (28.6 variants/ individual) ancestry carrying on average the most and least deleterious variants, respectively (Fig. 3c).

Genetic SLC transporter variability is highly population-specific. a Of all putatively deleterious variants (n = 67,799), 83% were only detected in a single population. The pie chart depicts the total number of deleterious, population-specific variants for each population. Values in brackets indicate the size of the cohort for the population in question. b The number of population-specific, deleterious SLC variants per individual differed considerably across major human populations. c In contrast, only minor differences in overall genetically encoded functional SLC variability per individual were observed across populations. d, e Dot plots depicting the cumulative frequency of putative loss-of-function (LoF) variants (frameshifts, start-lost, and stop-gain variations, as well as variants affecting canonical splice sites) per gene for SLC genes associated with Mendelian diseases (d) as well as for non-disease-associated genes (e). Note that LoF frequency of disease-associated genes is much lower than of non-disease-associated genes
Next, we focused specifically on genes for which loss-of-function mutations are associated with Mendelian disorders. Notably, the cohorts which we analyzed were sampled from the general population and individuals with severe congenital diseases were excluded. However, we hypothesized that quantification of loss-of-function carrier frequencies in the general population could be used as a proxy for Mendelian disease risk with recessive mode of inheritance. To this end, we aggregated frequencies of frameshifts and stop-gain variations, as well as variants affecting canonical splice sites removed variants previously reported to not cause disease (see “Materials and methods”). Overall, 109 out of 401 human SLC genes were found to have known associations with genetic diseases, of which 84 were autosomal recessive (Supplementary Table 2).
As expected, the frequency of loss-of-function variants was substantially lower in disease-associated SLC genes compared to SLC genes that were not associated with genetic disease (Fig. 3d, e). To evaluate whether this approach was indeed suitable to identify population-specific disease risk, we focused on Mendelian disorders with well-established ethnogeographic variation. Loss-of-function of the amino acid transporter SLC3A1 has been associated with cystinuria (OMIM 220100). The disease has a worldwide prevalence of around 1 in 7000 neonates and has been reported to be most common among Jews with frequencies up to 1:2500 individuals in certain subpopulations (Eggermann et al. 2012). Interestingly, we found highest loss-of-function frequencies of SLC3A1 in Ashkenazi Jews (0.9%), in agreement with previous reports (Pras et al. 1995). Accordingly, one in 12,345 Ashkenazim individuals can be expected to be homozygous for an SLC3A1 loss-of-function variant. Carrier rates in other populations were > tenfold lower. Similarly, variability profiles of the aspartate transporter SLC25A13 recapitulated increased prevalence of type II citrullinemia (OMIM 605814) in East Asians (Lu et al. 2005), with aggregated loss-of-function frequencies of 0.8%, corresponding to 1 in 15,625 homozygous East Asian carriers.
Lysinuric protein intolerance (OMIM 222700) is most prevalent in the Finnish population and has been associated with mutations in SLC7A7 (Torrents et al. 1999). Importantly, loss-of-function frequencies of SLC7A7 in Finns were more than fivefold higher than in other populations. Similarly, our data aligned with reported population differences in the genetic basis of Pendred syndrome (OMIM 274600), the most common form of syndromic genetic deafness. While genetic variation in SLC26A4 is a major cause of these disorders in Asia, mutations in different genes have been reported to be the most important factors in Western populations (Park et al. 2003). In agreement with these genetic roots, frequencies of SLC26A4 loss-of-function variants in East Asian populations were approximately sixfold higher than in Europeans. Based on the results, we conclude that the analysis of loss-of-function frequencies in the general population can be a powerful resource to inform about disease risk and population-specific genetic complexity underlying recessive Mendelian diseases.
Structural consequences of SLC variability
To obtain mechanistic insights into the effects of SLC variability, we mapped the genetic variants to the corresponding 3D structures of the transporter proteins. To this end, we focused on transporters with important roles in human physiology and pharmacology for which high-resolution crystal structures were either available or could be modeled with high confidence.
The glucose transporter GLUT1 encoded by SLC2A1 facilitates glucose uptake into erythrocytes and is the major glucose transporter in the human blood–brain barrier. Variations in GLUT1 can cause GLUT1 deficiency syndrome with an autosomal dominant inheritance pattern, which presents as neurological problems, developmental delays, complex movement disorders, and, occasionally, hemolytic anemia (De Giorgis and Veggiotti 2013). GLUT1 belongs to the major facilitator superfamily (MFS) of transporters and consists of two discretely folded domains, termed N- and C-domain, each consisting of six transmembrane helices, that are connected by an intracellular helical bundle (ICH) (Deng et al. 2014). To translocate glucose, GLUT1 undergoes structural changes and alternates between inwards and outwards facing confirmations and the ICH has been shown to play essential roles in this process (Yan 2013). The ICH interacts with multiple transmembrane domains (TMDs) of GLUT1, thereby acting as a latch that, in the absence of a ligand, stabilizes GLUT1 in the outward facing confirmation (Deng et al. 2014). Upon ligand binding, interactions between the N and C domains are altered, resulting in a transition towards the inward-open state.
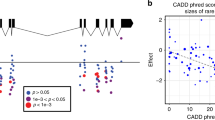
In total, 181 variants in GLUT1 were identified that were distributed across all domains of the protein, including the ICH (Fig. 4a). Notably, we identified rare variations in R400, which participates in stabilization of the interaction between the N- and C-terminal domains (Park 2015), as well as in R92, R93, R232, and E209, which form a tightly connected salt bridge network that controls GLUT1 state transitions (Galochkina et al. 2019). In contrast, no variants were observed in the glucose entry pocket (N34, V69, R126, and Y292) or in the amino acids lining the central glucose cavity (S73, Q279, Q282, Q283, N288, N411, and N415). As the analyzed cohort was depleted of patients with congenital diseases, these findings suggest that GLUT1 function can be permissive to variations that modulate salt bridges involved in state transitions, whereas residues directly involved in glucose translocation appear more conserved.

Structural mapping of GLUT1, ENT1, and OCT1 variability. Schematic topology models and experimentally derived 3D protein structures viewed from both sides of the membrane plane are shown for human GLUT1 (a) and ENT1 (b). Detailed 3D structures of key protein domains with functionally relevant variants (sticks) and substrates (sticks in dark blue) are shown as insets under the respective topology models. Red segments of the 3D models represent residues with putatively deleterious variants, blue segments represent residues with putatively neutral variants, and gray segments represent residues for which no associated variants have been identified in 141,456 individuals. Deleterious and neutral common variants are depicted as red and blue spheres, respectively. ICH = intracellular helical bundle; c schematic representation of the secondary structure of human OCT1, with all deleterious (shades of red) and neutral (shades of blue) variants mapped to their respective residues. Color intensity of each residue indicates variant frequencies. Population-specific frequencies of the common variants R61C, L160F, P341L, G401, M408 V, M420del, V464I, and G465R are shown for Africans (AFR; yellow), Ashkenazi Jews (AJ; purple), Non-Finnish Europeans (NFE; dark blue), Latinos (LAT; green), East Asians (EAS; light blue), and South Asians (SAS; red) in inlets. d The predicted 3D model of OCT1 viewed from both sides of the membrane plane is shown. Red segments of the 3D models represent residues with putatively deleterious variants, blue segments represent residues with putatively neutral variants, and gray segments represent residues for which no associated variants have been identified in 141,456 individuals. Deleterious and neutral common variants are depicted as red and blue spheres, respectively. Note that R61C is not depicted, because the corresponding fold could not be accurately modeled
ENT1 encoded by SLC29A1 is an essential uptake transporter of nucleosides and nucleoside analogs. As such, ENT1 is of tremendous pharmacological importance for the disposition of various antiviral and antineoplastic medications, and is itself the pharmacological target of multiple antiarrhythmic and antihypertensive medications (Boswell-Casteel and Hays 2017). The organization in N- and C-terminal pseudo-symmetric domains as well as the transport cycle of state transitions are overall similar between ENT1 and GLUT1. However, the C-domain of ENT1 only comprises five instead of six TMDs (Wright and Lee 2019). In total, our data set contained 263 variants that affected the ENT1 amino acid sequence. The only common SLC29A1 variant was rs45573936, with a frequency of 1.8%, resulting in an I216T amino acid substitution in TMD6 that alters binding affinity of the adenosine analog inhibitor NBMPR and has been implicated in neurological symptoms upon alcohol withdrawal (Kim et al. 2011). We identified rare variants in multiple critical residues of the central cavity of the transporter that likely impact substrate affinity and binding kinetics. These include a substitution of the hydrophobic methionine M33 that lines the narrowest constriction point at the extracellular side (Wright and Lee 2019) with a charged lysine, as well as amino acid exchanges affecting various residues that define the intracellular gate, such as R111, I304, and T429 (Fig. 4b). Furthermore, multiple variants affected the residues M89, Q158, and R345 that have been shown to directly interact with the structurally heterogeneous ENT1 inhibitors dilazep and NBMPR (Wright and Lee 2019). We conclude that the genetic variability in SLC29A1 is extensive and multiple variants are highly likely to affect ENT1 pharmacology.
In contrast to GLUT1 and ENT1, no crystal structure of human OCT1 has yet been presented. However, structure–function relationships have been inferred from crystal structures of homologous fungal transporters (Pedersen et al. 2013) and mutagenesis studies using rat OCT1 (Gorbunov et al. 2008; Popp et al. 2005). Here, we used the computational tool Phyre2 (Kelley et al. 2015) to predict the structure of human OCT1 based on multiple sequence alignments and homologous experimentally determined models. We could derive a high confidence model (100% confidence score) covering 82% of the human OCT1 protein sequence, which aligned well with the putative structure of rat OCT1 (Supplementary Fig. 1).
SLC22A1 harbors eight common variants that affect OCT1 amino acid sequence, of which R61C, G401S, and G465R resulted in strongly reduced OCT1 function in vitro, whereas substrate-specific results have been reported for M420del and P341L (Choi and Song 2012; Shu et al. 2003; Tzvetkov et al. 2011, 2012). The function of OCT1 isoforms carrying V464I, L160F or M408 V was not found to be altered in vitro (Shu et al. 2003). Notably, M420del occurs exclusively together with M408 V, whereas M408 V can occur in isolation (D′ = 1, R2 = 0.061) (Tzvetkov et al. 2014). The frequencies of these common variants varied greatly across populations (Fig. 4c) with MAFs of the M240del variant ranging from 0.09% in East Asians to 21.9% in Latinos and M408 V from 20.2% in Latinos to 45.4% in Finns in accordance with previous reports on smaller cohorts (Seitz et al. 2015). In total, we identified nine OCT1 variants with a global MAF > 0.1%, half of which were localized to TMDs (G38D, M420del, M440I, and G465R localized to TM1, TM9, TM10, and TM11, respectively) (Fig. 4c). In addition to these well-characterized variants, we found 445 additional variants that alter OCT1 amino acid sequence (Fig. 4c, d). Rare variations were found to affect the mechanistically important residues S358, R439, I446, Q447, and C450, which are directly involved in the coordination of cationic substrates, as well as F485, a residue essential for state transition during substrate translocation (Gorbunov et al. 2008; Pedersen et al. 2013; Volk et al. 2009). Combined, structural mapping of the genetic variability in physiologically and pharmacologically important SLC transporters supports the conclusion that rare, as of yet uncharacterized variants, are likely to have important functional impacts on transporter structure and function.
Discussion
SLC transporters play pivotal roles in diverse physiological processes, including the uptake and disposition of nutrients, maintenance of acid-base homeostasis, neurotransmission, and the elimination of metabolic products. In addition, they are involved in the disposition of a multitude of clinically relevant medications, ranging from chemotherapeutics to antidiuretics. Their biological importance and pharmacological relevance, as well as their roles in numerous human diseases, render SLC transporters attractive drug targets. Current clinical applications include the targeted treatment of hypertension (inhibition of SLC12A1/NKCC2 by diuretics), diabetes (inhibition of SLC5A2/SGLT2 by gliflozins), gout (inhibition of SLC22A12/URAT1 by lesinurad), schizophrenia (inhibition of SLC6A9/GLYT1 by bitopertin), and depression (inhibition of SLC6 transporters by serotonin-selective reuptake inhibitors) (Lin et al. 2015). Our analyses revealed that almost half of all SLC alleles associated with altered drug response or toxicity had frequencies that differed more than fivefold between populations. These findings have important implications for the treatment with SLC transporter substrates in an ethnogeographic context, and incentivize the adoption of population-adjusted genotyping strategies to optimize patient outcomes.
In addition to the previously described SLC alleles, we identified a surprising extent of genetic complexity within SLC transporters. Importantly, less than 0.2% of all identified variants were found in more than 1% of alleles and more than half of all variants were singletons. To estimate the overall contribution of this plethora of rare variants to functional SLC variability, we used an array of 13 partly orthogonal computational algorithms that leverage sequence information, evolutionary conservation, structural considerations, and functional genomics data in the prediction process, and have been found to perform reasonably well on both disease-associated and pharmacogenomic data sets (Li et al. 2018; Zhou et al. 2019). These analyses revealed that each individual genome harbors on average 29.7 putatively functional SLC variants, with rare variants accounting for 18% of this genetically encoded functional variability. Notably, nearly half of all putatively deleterious SLC variants in an individual affected transporters of amino acids, organic ions, and urea cycle intermediates. Structural mapping of the portfolio of genetic variants on available crystal structures of the encoded proteins revealed that rare variants affect multiple residues that have been shown to be essential for transporter function, thus further corroborating the important functional roles of rare genetic variability. Besides variations that are directly involved in substrate coordination or translocation, a variety of missense variants in SLC transporters are known to affect transporter function by altering subcellular trafficking or localization. Prominent examples include variants in SLC22A1 (Chen et al. 2010), SLC12A6 (Salin-Cantegrel et al. 2011) and SLC30A5 (Thornton et al. 2011). Notably, while these effects are difficult to infer by structural mapping, variant effect predictors, such as those used in this study, faithfully predicted localization defects and even outperformed specialized subcellular localization tools (Orioli and Vihinen 2019).
Strikingly, we found that 83% of all variants that were predicted to affect SLC function were population-specific. This degree of inter-ethnic variability is similar to other highly variable pharmacogene families, such as CYPs (Fujikura et al. 2015) and UGTs (Kaniwa et al. 2005), as well as to the related SLCO family of transporters (Zhang and Lauschke 2019). While individuals of African ancestry harbored most functional SLC variants in agreement with previous findings of greater levels of genetic diversity in Africans compared to non-African populations (Campbell and Tishkoff 2008; Tishkoff et al. 2009), the largest number of population-specific SLC variants was identified in East Asians. Interestingly, when focusing on SLC genes associated with Mendelian disease, we found that population-specific carrier frequency in the general population recapitulated the ethnogeographic variation of various Mendelian disorders with a recessive mode of inheritance, including cystinuria in Jewish individuals, type II citrullinemia in East Asians, and lysinuric protein intolerance in Finns. We thus conclude, in agreement with previous studies (Fujikura 2016), that NGS data of the general population can provide a suitable tool for the analysis of the genetic variability underlying inherited disorders. Furthermore, we argue that the presented data can serve as a unique large-scale resource for clinical geneticists to inform about population-specific prevalence and allelic composition of risk alleles associated with Mendelian diseases of SLC transporters. Importantly, the approach is likely not suitable for the analyses of diseases following a dominant mode of inheritance, as individuals with severe congenital diseases were excluded from the analyzed cohorts, resulting in an underestimation of dominant disease allele frequencies in our data set. Notably, the relatively high frequencies of loss-of-function variants in SLC28A1, which are associated with autosomal dominant uridine-cytidineuria (OMIM 618477), might be explained by its putatively benign nature (Wevers et al. 2019).
NGS is already widely and successfully applied in the diagnosis of rare monogenic diseases (Boycott et al. 2013; Fernandez-Marmiesse et al. 2018). However, while targeted sequencing panels that include multiple SLC transporters have been developed (Gordon et al. 2016; Gulilat et al. 2019; Klein et al. 2019), the incorporation of these genetic data into personalized pharmacogenomic recommendations and clinical decision-making is lagging behind. In the absence of feasible experimental strategies to characterize the functional impact of the plethora of rare SLC variants, computational methods have to be used. While such in silico interpretations of pharmacogenetic variants do not yet have sufficient accuracy to warrant direct clinical implementation (Zhou et al. 2018), these tools can already be used to flag patients with suspicious variants in key pharmacogenes for closer monitoring to anticipate detrimental drug response as early as possible. However, whether NGS coupled with computational pharmacogenomic analyses can indeed facilitate informed decision-making and provide a cost-effective measure to improve patient care, remains to be evaluated in prospective trials (Lauschke and Ingelman-Sundberg 2016, 2018).
In summary, by leveraging consolidated NGS data from 141,456 individuals, we comprehensively assessed the genetic variability of the human SLC transporter superfamily on an unprecedented scale. We demonstrate that SLC genes are highly variable and each individual genome is estimated to contain around 30 variants that affect SLC transporter function. The vast majority of variants were rare, and computational analyses based on evolutionary, structural, and functional genomics data indicate that these rare variants contribute approximately 20% to the genetically encoded functional variability of SLC transporters. Thus, these data serve as a powerful resource for the worldwide pattern of SLC variability and motivate the integration of comprehensive NGS-based genotyping into personalized predictions of SLC substrate disposition and precision public health.
References
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. https://doi.org/10.1038/nmeth0410-248
Ampuero J, Del Campo JA, Rojas L, Calleja JL, Cabezas J, Lens S, Crespo J, Forns X, Andrade RJ, Fernández I, Buti M, Millán R, Romero-Gómez M (2015) Role of ITPA and SLC28A2 genes in the prediction of anaemia associated with protease inhibitor plus ribavirin and peginterferon in hepatitis C treatment. J Clin Virol 68:56–60. https://doi.org/10.1016/j.jcv.2015.05.010
Anderson CM, Stahl A (2013) SLC27 fatty acid transport proteins. Mol Aspects Med 34:516–528. https://doi.org/10.1016/j.mam.2012.07.010
Bohanec Grabar P, Leandro-García LJ, Inglada-Pérez L, Logar D, Rodríguez-Antona C, Dolzan V (2012) Genetic variation in the SLC19A1 gene and methotrexate toxicity in rheumatoid arthritis patients. Pharmacogenomics 13:1583–1594. https://doi.org/10.2217/pgs.12.150
Boswell-Casteel RC, Hays FA (2017) Equilibrative nucleoside transporters—a review. Nucleosides Nucleotides Nucleic Acids 36:7–30. https://doi.org/10.1080/15257770.2016.1210805
Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE (2013) Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet 14:681–691. https://doi.org/10.1038/nrg3555
Bray J, Sludden J, Griffin MJ, Cole M, Verrill M, Jamieson D, Boddy AV (2010) Influence of pharmacogenetics on response and toxicity in breast cancer patients treated with doxorubicin and cyclophosphamide. Br J Cancer 102:1003–1009. https://doi.org/10.1038/sj.bjc.6605587
Bush WS, Crosslin DR, Owusu-Obeng A, Wallace J, Almoguera B, Basford MA, Bielinski SJ, Carrell DS, Connolly JJ, Crawford D, Doheny KF, Gallego CJ, Gordon AS, Keating B, Kirby J, Kitchner T, Manzi S, Mejia AR, Pan V, Perry CL, Peterson JF, Prows CA, Ralston J, Scott SA, Scrol A, Smith M, Stallings SC, Veldhuizen T, Wolf W, Volpi S, Wiley K, Li R, Manolio T, Bottinger E, Brilliant MH, Carey D, Chisholm RL, Chute CG, Haines JL, Hakonarson H, Harley JB, Holm IA, Kullo IJ, Jarvik GP, Larson EB, McCarty CA, Williams MS, Denny JC, Rasmussen-Torvik LJ, Roden DM, Ritchie MD (2016) Genetic variation among 82 pharmacogenes: the PGRNseq data from the eMERGE network. Clin Pharmacol Ther 100:160–169. https://doi.org/10.1002/cpt.350
Cai L-L, Huang WQ, Su Z-Y, Ye H-M, Wang L-S, Wu Y, Zhang Z-Y, Zhang W, Tzeng C-M (2017) Identification of two novel genes SLC15A2 and SLCO1B3 associated with maintenance dose variability of warfarin in a Chinese population. Sci Rep 7:17379. https://doi.org/10.1038/s41598-017-17731-1
Campbell MC, Tishkoff SA (2008) African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet 9:403–433. https://doi.org/10.1146/annurev.genom.9.081307.164258
Cargnin S, Ravegnini G, Soverini S, Angelini S, Terrazzino S (2018) Impact of SLC22A1 and CYP3A5 genotypes on imatinib response in chronic myeloid leukemia: a systematic review and meta-analysis. Pharmacol Res 131:244–254. https://doi.org/10.1016/j.phrs.2018.02.005
Carter H, Douville C, Stenson PD, Cooper DN, Karchin R (2013) Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14(Suppl 3):S3. https://doi.org/10.1186/1471-2164-14-S3-S3
César-Razquin A, Snijder B, Frappier-Brinton T, Isserlin R, Gyimesi G, Bai X, Reithmeier RA, Hepworth D, Hediger MA, Edwards AM, Superti-Furga G (2015) A call for systematic research on solute carriers. Cell 162:478–487. https://doi.org/10.1016/j.cell.2015.07.022
Chen L, Takizawa M, Chen E, Schlessinger A, Segenthelar J, Choi JH, Sali A, Kubo M, Nakamura S, Iwamoto Y, Iwasaki N, Giacomini KM (2010) Genetic polymorphisms in organic cation transporter 1 (OCT1) in Chinese and Japanese populations exhibit altered function. J Pharmacol Exp Ther 335:42–50. https://doi.org/10.1124/jpet.110.170159
Choi M-K, Song I-S (2012) Genetic variants of organic cation transporter 1 (OCT1) and OCT2 significantly reduce lamivudine uptake. Biopharm Drug Dispos 33:170–178. https://doi.org/10.1002/bdd.1783
Choi J-H, Yee SW, Ramirez AH, Morrissey KM, Jang GH, Joski PJ, Mefford JA, Hesselson SE, Schlessinger A, Jenkins G, Castro RA, Johns SJ, Stryke D, Sali A, Ferrin TE, Witte JS, Kwok P-Y, Roden DM, Wilke RA, McCarty CA, Davis RL, Giacomini KM (2011) A common 5′-UTR variant in MATE2-K is associated with poor response to metformin. Clin Pharmacol Ther 90:674–684. https://doi.org/10.1038/clpt.2011.165
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the functional effect of amino acid substitutions and indels. PLoS One 7:e46688. https://doi.org/10.1371/journal.pone.0046688
Chun S, Fay JC (2009) Identification of deleterious mutations within three human genomes. Genome Res 19:1553–1561. https://doi.org/10.1101/gr.092619.109
Corrigan A, Walker JL, Wickramasinghe S, Hernandez MA, Newhouse SJ, Folarin AA, Lewis CM, Sanderson JD, Spicer J, Marinaki AM (2014) Pharmacogenetics of pemetrexed combination therapy in lung cancer: pathway analysis reveals novel toxicity associations. Pharmacogenomics J 14:411–417. https://doi.org/10.1038/tpj.2014.13
Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S (2010) Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP ++. PLoS Comput Biol 6:e1001025-13. https://doi.org/10.1371/journal.pcbi.1001025
De Giorgis V, Veggiotti P (2013) GLUT1 deficiency syndrome 2013: current state of the art. Seizure 22:803–811. https://doi.org/10.1016/j.seizure.2013.07.003
Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N (2014) Crystal structure of the human glucose transporter GLUT1. Nature 510:121–125. https://doi.org/10.1038/nature13306
Di Paolo A, Polillo M, Capecchi M, Cervetti G, Baratè C, Angelini S, Guerrini F, Fontanelli G, Arici R, Ciabatti E, Grassi S, Bocci G, Hrelia P, Danesi R, Petrini M, Galimberti S (2014) The c.480C > G polymorphism of hOCT1 influences imatinib clearance in patients affected by chronic myeloid leukemia. Pharmacogenomics J 14:328–335. https://doi.org/10.1038/tpj.2014.7
Doehring A, Hofmann WP, Schlecker C, Zeuzem S, Sarrazin C, Berg T, Müller T, Herrmann E, Geisslinger G, Lötsch J (2011) Role of nucleoside transporters SLC28A2/3 and SLC29A1/2 genetics in ribavirin therapy: protection against anemia in patients with chronic hepatitis C. Pharmacogenet Genomics 21:289–296. https://doi.org/10.1097/FPC.0b013e32834412e7
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X (2015) Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 24:2125–2137. https://doi.org/10.1093/hmg/ddu733
Drögemöller BI, Monzon JG, Bhavsar AP, Borrie AE, Brooks B, Wright GEB, Liu G, Renouf DJ, Kollmannsberger CK, Bedard PL, Aminkeng F, Amstutz U, Hildebrand CA, Gunaretnam EP, Critchley C, Chen Z, Brunham LR, Hayden MR, Ross CJD, Gelmon KA, Carleton BC (2017) Association between SLC16A5 genetic variation and cisplatin-induced ototoxic effects in adult patients with testicular cancer. JAMA Oncol 3:1558–1562. https://doi.org/10.1001/jamaoncol.2017.0502
Dujic T, Zhou K, Donnelly LA, Tavendale R, Palmer CNA, Pearson ER (2015) Association of organic cation transporter 1 with intolerance to metformin in type 2 diabetes: a GoDARTS study. Diabetes 64:1786–1793. https://doi.org/10.2337/db14-1388
Eggermann T, Venghaus A, Zerres K (2012) Cystinuria: an inborn cause of urolithiasis. Orphanet J Rare Dis 7:19. https://doi.org/10.1186/1750-1172-7-19
Fernandez-Marmiesse A, Gouveia S, Couce ML (2018) NGS technologies as a turning point in rare disease research, diagnosis and treatment. Curr Med Chem 25:404–432. https://doi.org/10.2174/0929867324666170718101946
Fredriksson R, Nordström KJV, Stephansson O, Hägglund MGA, Schiöth HB (2008) The solute carrier (SLC) complement of the human genome: phylogenetic classification reveals four major families. FEBS Lett 582:3811–3816. https://doi.org/10.1016/j.febslet.2008.10.016
Fujikura K (2016) Global carrier rates of rare inherited disorders using population exome sequences. PLoS One 11:e0155552. https://doi.org/10.1371/journal.pone.0155552
Fujikura K, Ingelman-Sundberg M, Lauschke VM (2015) Genetic variation in the human cytochrome P450 supergene family. Pharmacogenet Genomics 25:584–594. https://doi.org/10.1097/FPC.0000000000000172
Fukuda T, Chidambaran V, Mizuno T, Venkatasubramanian R, Ngamprasertwong P, Olbrecht V, Esslinger HR, Vinks AA, Sadhasivam S (2013) OCT1 genetic variants influence the pharmacokinetics of morphine in children. Pharmacogenomics 14:1141–1151. https://doi.org/10.2217/pgs.13.94
Galochkina T, Ng Fuk Chong M, Challali L, Abbar S, Etchebest C (2019) New insights into GluT1 mechanics during glucose transfer. Sci Rep 9:998. https://doi.org/10.1038/s41598-018-37367-z
Gorbunov D, Gorboulev V, Shatskaya N, Mueller T, Bamberg E, Friedrich T, Koepsell H (2008) High-affinity cation binding to organic cation transporter 1 induces movement of helix 11 and blocks transport after mutations in a modeled interaction domain between two helices. Mol Pharmacol 73:50–61. https://doi.org/10.1124/mol.107.040170
Gordon AS, Fulton RS, Qin X, Mardis ER, Nickerson DA, Scherer S (2016) PGRNseq: a targeted capture sequencing panel for pharmacogenetic research and implementation. Pharmacogenet Genom. https://doi.org/10.1097/FPC.0000000000000202
Gulilat M, Lamb T, Teft WA, Wang J, Dron JS, Robinson JF, Tirona RG, Hegele RA, Kim RB, Schwarz UI (2019) Targeted next generation sequencing as a tool for precision medicine. BMC Med Genomics 12:81. https://doi.org/10.1186/s12920-019-0527-2
Henrie A, Hemphill SE, Ruiz-Schultz N, Cushman B, DiStefano MT, Azzariti D, Harrison SM, Rehm HL, Eilbeck K (2018) ClinVar Miner: demonstrating utility of a Web-based tool for viewing and filtering ClinVar data. Hum Mutat 39:1051–1060. https://doi.org/10.1002/humu.23555
Höglund PJ, Nordström KJV, Schiöth HB, Fredriksson R (2011) The solute carrier families have a remarkably long evolutionary history with the majority of the human families present before divergence of Bilaterian species. Mol Biol Evol 28:1531–1541. https://doi.org/10.1093/molbev/msq350
Huang Q, Yin J-Y, Dai X-P, Wu J, Chen X, Deng C-S, Yu M, Gong Z-C, Zhou H-H, Liu Z-Q (2010) Association analysis of SLC30A8 rs13266634 and rs16889462 polymorphisms with type 2 diabetes mellitus and repaglinide response in Chinese patients. Eur J Clin Pharmacol 66:1207–1215. https://doi.org/10.1007/s00228-010-0882-6
Huang L, Zhang T, Xie C, Liao X, Yu Q, Feng J, Ma H, Dai J, Li M, Chen J, Zang A, Wang Q, Ge S, Qin K, Cai J, Yuan X (2013) SLCO1B1 and SLC19A1 gene variants and irinotecan-induced rapid response and survival: a prospective multicenter pharmacogenetics study of metastatic colorectal cancer. PLoS One 8:e77223. https://doi.org/10.1371/journal.pone.0077223
Ingelman-Sundberg M, Mkrtchian S, Zhou Y, Lauschke VM (2018) Integrating rare genetic variants into pharmacogenetic drug response predictions. Human Genomics 12:26. https://doi.org/10.1186/s40246-018-0157-3
Johnson DC, Corthals SL, Walker BA, Ross FM, Gregory WM, Dickens NJ, Lokhorst HM, Goldschmidt H, Davies FE, Durie BGM, Van Ness B, Child JA, Sonneveld P, Morgan GJ (2011) Genetic factors underlying the risk of thalidomide-related neuropathy in patients with multiple myeloma. J Clin Oncol 29:797–804. https://doi.org/10.1200/JCO.2010.28.0792
Kaniwa N, Kurose K, Jinno H, Tanaka-Kagawa T, Saito Y, Saeki M, Sawada J-i, Tohkin M, Hasegawa R (2005) Racial variability in haplotype frequencies of UGT1A1 and glucuronidation activity of a novel single nucleotide polymorphism 686C>T (P229L) found in an African–American. Drug Metab Dispos 33:458–465. https://doi.org/10.1124/dmd.104.001800
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. https://doi.org/10.1038/nprot.2015.053
Kim J-H, Karpyak VM, Biernacka JM, Nam HW, Lee MR, Preuss UW, Zill P, Yoon G, Colby C, Mrazek DA, Choi D-S (2011) Functional role of the polymorphic 647 T/C variant of ENT1 (SLC29A1) and its association with alcohol withdrawal seizures. PLoS One 6:e16331. https://doi.org/10.1371/journal.pone.0016331
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Rev Drug Discov 46:310–315. https://doi.org/10.1038/ng.2892
Klein K, Tremmel R, Winter S, Fehr S, Battke F, Scheurenbrand T, Schaeffeler E, Biskup S, Schwab M, Zanger UM (2019) A new panel-based next-generation sequencing method for ADME genes reveals novel associations of common and rare variants with expression in a human liver cohort. Front Genet 10:56. https://doi.org/10.3389/fgene.2019.00007
Koepsell H (2013) The SLC22 family with transporters of organic cations, anions and zwitterions. Mol Aspects Med 34:413–435. https://doi.org/10.1016/j.mam.2012.10.010
Kozyra M, Ingelman-Sundberg M, Lauschke VM (2017) Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet Med 19:20–29. https://doi.org/10.1038/gim.2016.33
Kwon JS, Joo YH, Nam HJ, Lim M, Cho E-Y, Jung MH, Choi J-S, Kim B, Kang D-H, Oh S, Park T, Hong KS (2009) Association of the glutamate transporter gene SLC1A1 with atypical antipsychotics-induced obsessive-compulsive symptoms. Arch Gen Psychiatry 66:1233–1241. https://doi.org/10.1001/archgenpsychiatry.2009.155
Lanvers-Kaminsky C, Sprowl JA, Malath I, Deuster D, Eveslage M, Schlatter E, Mathijssen RH, Boos J, Jürgens H, am Zehnhoff-Dinnesen AG, Sparreboom A, Ciarimboli G (2015) Human OCT2 variant c.808G>T confers protection effect against cisplatin-induced ototoxicity. Pharmacogenomics 16:323–332. https://doi.org/10.2217/pgs.14.182
Lauschke VM, Ingelman-Sundberg M (2016) Precision medicine and rare genetic variants. Trends Pharmacol Sci 37:85–86. https://doi.org/10.1016/j.tips.2015.10.006
Lauschke VM, Ingelman-Sundberg M (2018) How to consider rare genetic variants in personalized drug therapy. Clin Pharmacol Ther 103:745–748. https://doi.org/10.1002/cpt.976
Lee Y-S, Kim BH, Kim BC, Shin A, Kim JS, Hong S-H, Hwang J-A, Lee JA, Nam S, Lee SH, Bhak J, Park J-W (2015) SLC15A2 genomic variation is associated with the extraordinary response of sorafenib treatment: whole-genome analysis in patients with hepatocellular carcinoma. Oncotarget 6:16449–16460. https://doi.org/10.18632/oncotarget.3758
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won H-H, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Consortium EA (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. https://doi.org/10.1038/nature19057
Li J, Zhao T, Zhang Y, Zhang K, Shi L, Chen Y, Wang X, Sun Z (2018) Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res 46:7793–7804. https://doi.org/10.1093/nar/gky678
Lima A, Bernardes M, Sousa H, Azevedo R, Costa L, Ventura F, Seabra V, Medeiros R (2014) SLC19A1 80G allele as a biomarker of methotrexate-related gastrointestinal toxicity in Portuguese rheumatoid arthritis patients. Pharmacogenomics 15:807–820. https://doi.org/10.2217/pgs.13.244
Lin L, Yee SW, Kim RB, Giacomini KM (2015) SLC transporters as therapeutic targets: emerging opportunities. Nat Rev Drug Discovery 14:543–560. https://doi.org/10.1038/nrd4626
Lu YB, Kobayashi K, Ushikai M, Tabata A, Iijima M, Li MX, Lei L, Kawabe K, Taura S, Yang Y, Liu T-T, Chiang S-H, Hsiao K-J, Lau Y-L, Tsui L-C, Lee DH, Saheki T (2005) Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency. J Hum Genet 50:338–346. https://doi.org/10.1007/s10038-005-0262-8
Machiela MJ, Chanock SJ (2015) LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31:3555–3557. https://doi.org/10.1093/bioinformatics/btv402
Makhtar SM, Husin A, Baba AA, Ankathil R (2018) Genetic variations in influx transporter gene SLC22A1 are associated with clinical responses to imatinib mesylate among Malaysian chronic myeloid leukaemia patients. Journal of Genetics 97:835–842. https://doi.org/10.1007/s12041-018-0978-9
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TFC, McCarroll SA, Visscher PM (2009) Finding the missing heritability of complex diseases. Nature 461:747–753. https://doi.org/10.1038/nature08494
Moncrieffe H, Hinks A, Ursu S, Kassoumeri L, Etheridge A, Hubank M, Martin P, Weiler T, Glass DN, Thompson SD, Thomson W, Wedderburn LR (2010) Generation of novel pharmacogenomic candidates in response to methotrexate in juvenile idiopathic arthritis: correlation between gene expression and genotype. Pharmacogenet Genomics 20:665–676. https://doi.org/10.1097/FPC.0b013e32833f2cd0
Mueckler M, Thorens B (2013) The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med 34:121–138. https://doi.org/10.1016/j.mam.2012.07.001
Ng PC, Henikoff S (2001) Predicting deleterious amino acid substitutions. Genome Res 11:863–874. https://doi.org/10.1101/gr.176601
Orioli T, Vihinen M (2019) Benchmarking subcellular localization and variant tolerance predictors on membrane proteins. BMC Genomics 20:547. https://doi.org/10.1186/s12864-019-5865-0
Park M-S (2015) Molecular dynamics simulations of the human glucose transporter GLUT1. PLoS One 10:e0125361. https://doi.org/10.1371/journal.pone.0125361
Park HJ, Shaukat S, Liu X-Z, Hahn SH, Naz S, Ghosh M, Kim H-N, Moon S-K, Abe S, Tukamoto K, Riazuddin S, Kabra M, Erdenetungalag R, Radnaabazar J, Khan S, Pandya A, Usami S-I, Nance WE, Wilcox ER, Griffith AJ (2003) Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet 40:242–248. https://doi.org/10.1136/jmg.40.4.242
Pedersen BP, Kumar H, Waight AB, Risenmay AJ, Roe-Zurz Z, Chau BH, Schlessinger A, Bonomi M, Harries W, Sali A, Johri AK, Stroud RM (2013) Crystal structure of a eukaryotic phosphate transporter. Nature 496:533–536. https://doi.org/10.1038/nature12042
Pellicer M, García-González X, García MI, Robles L, Grávalos C, García-Alfonso P, Pachón V, Longo F, Martínez V, Blanco C, Iglesias I, Sanjurjo M, López-Fernández LA (2017) Identification of new SNPs associated with severe toxicity to capecitabine. Pharmacol Res 120:133–137. https://doi.org/10.1016/j.phrs.2017.03.021
Popp C, Gorboulev V, Müller TD, Gorbunov D, Shatskaya N, Koepsell H (2005) Amino acids critical for substrate affinity of rat organic cation transporter 1 line the substrate binding region in a model derived from the tertiary structure of lactose permease. Mol Pharmacol 67:1600–1611. https://doi.org/10.1124/mol.104.008839
Pras E, Raben N, Golomb E, Arber N, Aksentijevich I, Schapiro JM, Harel D, Katz G, Liberman U, Pras M (1995) Mutations in the SLC3A1 transporter gene in cystinuria. Am J Hum Genet 56:1297–1303
Qiu H-B, Zhuang W, Wu T, Xin S, Lin C-Z, Ruan H-L, Zhu X, Huang M, Li J-L, Hou X-Y, Zhou Z-W, Wang XD (2018) Imatinib-induced ophthalmological side-effects in GIST patients are associated with the variations of EGFR, SLC22A1, SLC22A5 and ABCB1. Pharmacogenomics J 18:460–466. https://doi.org/10.1038/tpj.2017.40
Quang D, Chen Y, Xie X (2015) DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31:761–763. https://doi.org/10.1093/bioinformatics/btu703
Rau M, Stickel F, Russmann S, Manser CN, Becker PP, Weisskopf M, Schmitt J, Dill MT, Dufour J-F, Moradpour D, Semela D, Müllhaupt B, Geier A, (SCCS) SHCCSG (2013) Impact of genetic SLC28 transporter and ITPA variants on ribavirin serum level, hemoglobin drop and therapeutic response in patients with HCV infection. J Hepatol 58:669–675. https://doi.org/10.1016/j.jhep.2012.11.027
Reva B, Antipin Y, Sander C (2011) Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 39:e118. https://doi.org/10.1093/nar/gkr407
Romero MF, Chen A-P, Parker MD, Boron WF (2013) The SLC4 family of bicarbonate (HCO3 −) transporters. Mol Aspects Med 34:159–182. https://doi.org/10.1016/j.mam.2012.10.008
Salinas-Delgado Y, Galaviz-Hernández C, Toral RG, Ávila Rejón CA, Reyes-Lopez MA, Martínez AR, Martínez-Aguilar G, Sosa-Macías M (2015) The D543N polymorphism of the SLC11A1/NRAMP1 gene is associated with treatment failure in male patients with pulmonary tuberculosis. Drug Metab Personal Ther 30:211–214. https://doi.org/10.1515/dmpt-2015-0019
Salin-Cantegrel A, Rivière J-B, Shekarabi M, Rasheed S, Dacal S, Laganière J, Gaudet R, Rochefort D, Lesca G, Gaspar C, Dion PA, Lapointe J-Y, Rouleau GA (2011) Transit defect of potassium-chloride Co-transporter 3 is a major pathogenic mechanism in hereditary motor and sensory neuropathy with agenesis of the corpus callosum. J Biol Chem 286:28456–28465. https://doi.org/10.1074/jbc.M111.226894
Santos M, Niemi M, Hiratsuka M, Kumondai M, Ingelman-Sundberg M, Lauschke VM, Rodríguez-Antona C (2018) Novel copy-number variations in pharmacogenes contribute to interindividual differences in drug pharmacokinetics. Genet Med 20:622–629. https://doi.org/10.1038/gim.2017.156
Schlessinger A, Yee SW, Sali A, Giacomini KM (2013) SLC classification: an update. Clin Pharmacol Ther 94:19–23. https://doi.org/10.1038/clpt.2013.73
Seitz T, Stalmann R, Dalila N, Chen J, Pojar S, dos Santos Pereira JN, Krätzner R, Brockmöller J, Tzvetkov MV (2015) Global genetic analyses reveal strong inter-ethnic variability in the loss of activity of the organic cation transporter OCT1. Genome Med 7:56. https://doi.org/10.1186/s13073-015-0172-0
Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, Edwards KJ, Day INM, Gaunt TR (2012) Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 34:57–65. https://doi.org/10.1002/humu.22225
Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day INM, Gaunt TR, Campbell C (2015) An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31:1536–1543. https://doi.org/10.1093/bioinformatics/btv009
Shin S-W, Park BL, Chang H, Park JS, Bae D-J, Song H-J, Choi IS, Kim M-K, Park H-S, Kim LH, Namgoong S, Kim JO, Shin HD, Park CS (2014) Exonic variants associated with development of aspirin exacerbated respiratory diseases. PLoS One 9:e111887. https://doi.org/10.1371/journal.pone.0111887
Shu Y, Leabman MK, Feng B, Mangravite LM, Huang CC, Stryke D, Kawamoto M, Johns SJ, DeYoung J, Carlson E, Ferrin TE, Herskowitz I, Giacomini KM, Investigators POMT (2003) Evolutionary conservation predicts function of variants of the human organic cation transporter, OCT1. Proc Natl Acad Sci 100:5902–5907. https://doi.org/10.1073/pnas.0730858100
Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, Ianculescu AG, Yue L, Lo JC, Burchard EG, Brett CM, Giacomini KM (2007) Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Investig 117:1422–1431. https://doi.org/10.1172/JCI30558
Song IS, Shin HJ, Shim EJ, Jung IS, Kim WY, Shon JH, Shin JG (2008) Genetic variants of the organic cation transporter 2 influence the disposition of metformin. Clin Pharmacol Ther 84:559–562. https://doi.org/10.1038/clpt.2008.61
Soo RA, Wang LZ, Ng SS, Chong PY, Yong WP, Lee SC, Liu JJ, Choo TB, Tham LS, Lee HS, Goh BC, Soong R (2009) Distribution of gemcitabine pathway genotypes in ethnic Asians and their association with outcome in non-small cell lung cancer patients. Lung Cancer 63:121–127. https://doi.org/10.1016/j.lungcan.2008.04.010
Stocker SL, Morrissey KM, Yee SW, Castro RA, Xu L, Dahlin A, Ramirez AH, Roden DM, Wilke RA, McCarty CA, Davis RL, Brett CM, Giacomini KM (2013) The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther 93:186–194. https://doi.org/10.1038/clpt.2012.210
Sundelin E, Gormsen LC, Jensen JB, Vendelbo MH, Jakobsen S, Munk OL, Christensen M, Brøsen K, Frøkiaer J, Jessen N (2017) Genetic polymorphisms in organic cation transporter 1 attenuates hepatic metformin exposure in humans. Clin Pharmacol Ther 102:841–848. https://doi.org/10.1002/cpt.701
Suthandiram S, Gan G-G, Zain SM, Bee P-C, Lian L-H, Chang K-M, Ong T-C, Mohamed Z (2014) Effect of polymorphisms within methotrexate pathway genes on methotrexate toxicity and plasma levels in adults with hematological malignancies. Pharmacogenomics 15:1479–1494. https://doi.org/10.2217/pgs.14.97
Tarasova L, Kalnina I, Geldnere K, Bumbure A, Ritenberga R, Nikitina-Zake L, Fridmanis D, Vaivade I, Pirags V, Klovins J (2012) Association of genetic variation in the organic cation transporters OCT1, OCT2 and multidrug and toxin extrusion 1 transporter protein genes with the gastrointestinal side effects and lower BMI in metformin-treated type 2 diabetes patients. Pharmacogenet Genomics 22:659–666. https://doi.org/10.1097/FPC.0b013e3283561666
Tecza K, Pamula-Pilat J, Lanuszewska J, Butkiewicz D, Grzybowska E (2018) Pharmacogenetics of toxicity of 5-fluorouracil, doxorubicin and cyclophosphamide chemotherapy in breast cancer patients. Oncotarget 9:9114–9136. https://doi.org/10.18632/oncotarget.24148
Thornton JK, Taylor KM, Ford D, Valentine RA (2011) Differential subcellular localization of the splice variants of the zinc transporter ZnT5 is dictated by the different C-terminal regions. PLoS One 6:e23878. https://doi.org/10.1371/journal.pone.0023878
Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, Hirbo JB, Awomoyi AA, Bodo J-M, Doumbo O, Ibrahim M, Juma AT, Kotze MJ, Lema G, Moore JH, Mortensen H, Nyambo TB, Omar SA, Powell K, Pretorius GS, Smith MW, Thera MA, Wambebe C, Weber JL, Williams SM (2009) The genetic structure and history of Africans and African Americans. Science 324:1035–1044. https://doi.org/10.1126/science.1172257
Todd JN, Florez JC (2014) An update on the pharmacogenomics of metformin: progress, problems and potential. Pharmacogenomics 15:529–539. https://doi.org/10.2217/pgs.14.21
Torrents D, Mykkänen J, Pineda M, Feliubadaló L, Estévez R, de Cid R, Sanjurjo P, Zorzano A, Nunes V, Huoponen K, Reinikainen A, Simell O, Savontaus ML, Aula P, Palacín M (1999) Identification of SLC7A7, encoding y + LAT-1, as the lysinuric protein intolerance gene. Nat Genet 21:293–296. https://doi.org/10.1038/6809
Tzvetkov MV (2017) OCT1 pharmacogenetics in pain management: is a clinical application within reach? Pharmacogenomics 18:1515–1523. https://doi.org/10.2217/pgs-2017-0095
Tzvetkov MV, Saadatmand AR, Lotsch J, Tegeder I, Stingl JC, Brockmöller J (2011) Genetically polymorphic OCT1: another piece in the puzzle of the variable pharmacokinetics and pharmacodynamics of the opioidergic drug tramadol. Clin Pharmacol Ther 90:143–150. https://doi.org/10.1038/clpt.2011.56
Tzvetkov MV, Saadatmand AR, Bokelmann K, Meineke I, Kaiser R, Brockmöller J (2012) Effects of OCT1 polymorphisms on the cellular uptake, plasma concentrations and efficacy of the 5-HT(3) antagonists tropisetron and ondansetron. Pharmacogenomics J 12:22–29. https://doi.org/10.1038/tpj.2010.75
Tzvetkov MV, dos Santos Pereira JN, Meineke I, Saadatmand AR, Stingl JC, Brockmöller J (2013) Morphine is a substrate of the organic cation transporter OCT1 and polymorphisms in OCT1 gene affect morphine pharmacokinetics after codeine administration. Biochem Pharmacol 86:666–678. https://doi.org/10.1016/j.bcp.2013.06.019
Tzvetkov MV, Seitz T, Bokelmann K, Mueller T, Brockmöller J, Koepsell H (2014) Does the haplotype Met408-Del420, which was apparently predictive for imatinib efficacy, really exist and how strongly may it affect OCT1 activity? Blood 123:1427–1429. https://doi.org/10.1182/blood-2013-11-535864
Urban TJ, Brown C, Castro RA, Shah N, Mercer R, Huang Y, Brett CM, Burchard EG, Giacomini KM (2008) Effects of genetic variation in the novel organic cation transporter, OCTN1, on the renal clearance of gabapentin. Clin Pharmacol Ther 83:416–421. https://doi.org/10.1038/sj.clpt.6100271
Visscher H, Rassekh SR, Sandor GS, Caron HN, van Dalen EC, Kremer LC, van der Pal HJ, Rogers PC, Rieder MJ, Carleton BC, Hayden MR, Ross CJ, consortium C (2015) Genetic variants in SLC22A17 and SLC22A7 are associated with anthracycline-induced cardiotoxicity in children. Pharmacogenomics 16:1065–1076. https://doi.org/10.2217/pgs.15.61
Volk C, Gorboulev V, Kotzsch A, Müller TD, Koepsell H (2009) Five amino acids in the innermost cavity of the substrate binding cleft of organic cation transporter 1 interact with extracellular and intracellular corticosterone. Mol Pharmacol 76:275–289. https://doi.org/10.1124/mol.109.054783
Vormfelde SV, Sehrt D, Toliat MR, Schirmer M, Meineke I, Tzvetkov M, Nürnberg P, Brockmöller J (2007) Genetic variation in the renal sodium transporters NKCC2, NCC, and ENaC in relation to the effects of loop diuretic drugs. Clin Pharmacol Ther 82:300–309. https://doi.org/10.1038/sj.clpt.6100131
Wang S-M, Sun L-L, Zeng W-X, Wu W-s, Zhang G-l (2014) Effects of a microRNA binding site polymorphism in SLC19A1 on methotrexate concentrations in Chinese children with acute lymphoblastic leukemia. Med Oncol 31:62. https://doi.org/10.1007/s12032-014-0062-0
Watkins DB, Hughes TP, White DL (2015) OCT1 and imatinib transport in CML: is it clinically relevant? Leukemia 29:1960–1969. https://doi.org/10.1038/leu.2015.170
Wevers RA, Christensen M, Engelke UFH, Geuer S, Coene KLM, Kwast JT, Lund AM, Vissers LELM (2019) Functional disruption of pyrimidine nucleoside transporter CNT1 results in a novel inborn error of metabolism with high excretion of uridine and cytidine. J Inherit Metab Dis 42:494–500. https://doi.org/10.1002/jimd.12081
Wright NJ, Lee S-Y (2019) Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat Struct Mol Biol 26:599–606. https://doi.org/10.1038/s41594-019-0245-7
Wright GEB, Carleton B, Hayden MR, Ross CJD (2018) The global spectrum of protein-coding pharmacogenomic diversity. Pharmacogenomics J 18:187–195. https://doi.org/10.1038/tpj.2016.77
Yan N (2013) Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem Sci 38:151–159. https://doi.org/10.1016/j.tibs.2013.01.003
Yee SW, Nguyen AN, Brown C, Savic RM, Zhang Y, Castro RA, Cropp CD, Choi JH, Singh D, Tahara H, Stocker SL, Huang Y, Brett CM, Giacomini KM (2013) Reduced renal clearance of cefotaxime in asians with a low-frequency polymorphism of OAT3 (SLC22A8). J Pharm Sci 102:3451–3457. https://doi.org/10.1002/jps.23581
Yoon H, Cho H-Y, Yoo H-D, Kim S-M, Lee Y-B (2013) Influences of organic cation transporter polymorphisms on the population pharmacokinetics of metformin in healthy subjects. AAPS J 15:571–580. https://doi.org/10.1208/s12248-013-9460-z
Zhang B, Lauschke VM (2019) Genetic variability and population diversity of the human SLCO (OATP) transporter family. Pharmacol Res 139:550–559. https://doi.org/10.1016/j.phrs.2018.10.017
Zhou Y, Lauschke VM (2018) Comprehensive overview of the pharmacogenetic diversity in Ashkenazi Jews. J Med Genet 55:617–627. https://doi.org/10.1136/jmedgenet-2018-105429
Zhou Y, Fujikura K, Mkrtchian S, Lauschke VM (2018) Computational methods for the pharmacogenetic interpretation of next generation sequencing data. Front Pharmacol 9:1437. https://doi.org/10.3389/fphar.2018.01437
Zhou Y, Mkrtchian S, Kumondai M, Hiratsuka M, Lauschke VM (2019) An optimized prediction framework to assess the functional impact of pharmacogenetic variants. Pharmacogenom J 19:115–126. https://doi.org/10.1038/s41397-018-0044-2
Acknowledgements
Open access funding provided by Karolinska Institute. The work was supported by the Swedish Research Council (Grant agreement numbers: 2016-01153 and 2016-01154), by the European Union’s Horizon 2020 research and innovation program U-PGx (Grant agreement number 668353), and by the Strategic Research Programme in Diabetes at Karolinska Institutet. We would like to thank the Exome Aggregation Consortium and all contributing groups for sharing their data, without which this study could not have been performed.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
VML is co-founder and owner of HepaPredict AB. LS has no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table
1. Overview of the 401 SLC genes included in the dataset, including their respective endogenous and drug substrates. Data obtained from http://slc.bioparadigms.org (XLSX 68 kb)
Supplementary Table
2.SLC genes and their known Mendelian disease associations, including OMIM identification numbers and mode of inheritance are shown. The cumulative frequency of putative loss-of-function variants (frameshifts, stop-gain and canonical splice site variants) is shown for each gene. AR = autosomal recessive; AD = autosomal dominant; XL = X-linked; XLR = X-linked recessive, XLD = X-linked dominant; U = unknown (XLSX 115 kb)
Supplementary Fig.
1. Comparison of the predicted tertiary structures of human (blue) and rat OCT1 (red) (PDF 6128 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schaller, L., Lauschke, V.M. The genetic landscape of the human solute carrier (SLC) transporter superfamily. Hum Genet 138, 1359–1377 (2019). https://doi.org/10.1007/s00439-019-02081-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-019-02081-x