
Abstract
Toxoplasma gondii is an opportunistic protozoan parasite that is highly prevalent in the human population and can lead to adverse health consequences in immunocompromised patients and pregnant women. Noncoding RNAs, such as microRNAs (miRNAs) and circular RNAs (circRNAs), play important regulatory roles in the pathogenesis of many infections. However, the differentially expressed (DE) miRNAs and circRNAs implicated in the host cell response during the lytic cycle of T. gondii are unknown. In this study, we profiled the expression of miRNAs and circRNAs in human foreskin fibroblasts (HFFs) at different time points after T. gondii infection using RNA sequencing (RNA-seq). We identified a total of 7, 7, 27, 45, 70, 148, 203, and 217 DEmiRNAs and 276, 355, 782, 1863, 1738, 6336, 1229, and 1680 DEcircRNAs at 1.5, 3, 6, 9, 12, 24, 36, and 48 h post infection (hpi), respectively. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses revealed that the DE transcripts were enriched in immune response, apoptosis, signal transduction, and metabolism-related pathways. These findings provide new insight into the involvement of miRNAs and circRNAs in the host response to T. gondii infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Toxoplasmosis is caused by Toxoplasma gondii, an apicomplexan protozoan parasite infecting approximately one-third of the world’s human population (Hill and Dubey 2002). Human infection mainly occurs via ingesting food or water contaminated with the parasite cyst or oocyst stage. Healthy people usually exhibit no clinical symptoms in response to T. gondii infection. However, individuals with immune deficiency, such as those with AIDS, can exhibit serious symptoms, such as headache and seizures (Jones-Brando et al. 2003; Elsheikha et al. 2020). Following the initial infection, T. gondii establishes a lifelong latent infection which can be reactivated in the immunocompromised hosts (Elsheikha et al. 2020). Pregnant women can transmit this parasite to their unborn offspring, which may lead to abortion or stillbirth (Elsheikha 2008; Pappas et al. 2009). Given the limitations of the commercially available medications and the lack of a vaccine to prevent infection in humans (Wang et al. 2019), there is a pressing need to improve the understanding of the interaction between T. gondii and its hosts and elucidate the precise mechanisms underlying T. gondii pathogenesis during T. gondii lytic cycle (Black and Boothroyd 2000).
To address the current unmet needs and establish a pipeline of new innovative treatment solutions for effective control of T. gondii infection, it is crucial to identify the molecules that underpin the host response to T. gondii infection, since these molecules may serve as therapeutic targets. Previous analysis using global transcriptomics (Jia et al. 2013; Tanaka et al. 2013; Radke et al. 2018; Wang et al. 2022; Hu et al. 2020; Xie et al. 2022) and proteomics (Sahu et al. 2014; Lv et al. 2017; Yang et al. 2018; Zhou et al. 2021) revealed some mechanisms by which T. gondii can alter gene expression and protein abundance in various tissues in the affected hosts. T. gondii infection can also alter the expression of non-coding RNA in vivo and in vitro (Hunter and Sibley 2012; Liu et al. 2018; Wang et al. 2018). This parasite hijacks host gene expression and modulates immune response pathways, including those involved in apoptosis and cytokine production (Judice et al. 2016). Despite the significant progress that has been achieved in understanding the relationship between T. gondii infection and host cells, the functional involvement of certain molecules, such as microRNAs (miRNAs) and circular RNAs (circRNAs), in the parasite pathogenesis and their contribution to anti-T. gondii immune response remain largely unexplored. Better understanding of the function of miRNAs and circRNAs implicated in T. gondii infection can reveal new insights into the interaction between the host and T. gondii.
microRNAs and circular RNAs play a vital role in post-transcriptional regulation of gene expression and protein activity (Zhang et al. 2022). circRNAs are a subclass of non-coding RNAs involved in the regulation of many biological processes, such as regulation of immune responses. They can compete with and influence the ability of miRNAs to regulate gene expression through a sponge-like mechanism (Granados-Riveron and Aquino-Jarquin 2016). Various circRNAs are involved in regulating or modulating viral infection and antiviral innate immunity (Li et al. 2017; Lu et al. 2020). Additionally, miRNAs and circRNAs can act as regulatory factors in the pathogenesis of neurological diseases, such as epilepsy, Parkinson’s disease (PD), and Alzheimer’s disease (AD) (Shao and Chen 2016; Awuson-David et al. 2023; Dong et al. 2023). T. gondii infection also alters the expression of host miRNAs and circRNAs in vivo to promote parasite replication (Xu et al. 2013; He et al. 2016; Cong et al. 2017; Hu et al. 2018; Zou et al. 2022).
Host-parasite interactions operate at the transcriptional and epigenetic levels and are critical for establishing a latent or lytic T. gondii infection. However, the functional involvement of miRNAs and circRNAs in the immunity and pathogenesis of T. gondii lytic cycle remains largely unknown. In the present study, we employed RNA sequencing (RNA-seq) analysis to investigate the changes in the expression of miRNAs and circRNAs in human foreskin fibroblasts (HFFs) during T. gondii infection lytic cycle. Another objective of the study was to identify the differentially expressed (DE) miRNAs and circRNAs and predict the miRNA-circRNA interactions. Our data provides new insight into the roles of miRNAs and circRNAs in regulating anti-T. gondii immunity.
Materials and methods
Parasites and cell culture
The HFF cell line was cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 U/mL streptomycin and maintained in an incubator at 37 °C and 5% CO2. Tachyzoites of T. gondii RH strain were maintained in HFF monolayers as previously described (Zhou et al. 2016).
T. gondii infection and sample collection
Monolayers of HFF cells, at 80% confluence in 25-cm2 tissue culture flasks, were infected by T. gondii RH strain at a multiplicity of infection (MOI) of 5. The mock-infected (control) HFFs were maintained under the same culture conditions except that cells were not infected by T. gondii. The cell samples were collected at 0, 1.5, 3, 6, 9, 12, 24, 36, and 48 h post infection (hpi). The RNA-seq samples were composed of 3 separate biological replicates for each time point. The collected cell samples were stored at − 80 °C until use.
RNA extraction
Total RNA was extracted using Trizol method (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The quantity and quality of the extracted RNA were determined by using the NanoDrop spectrophotometer and Agilent 2100 Bioanalyzer (Thermo Fisher Scientific, MA, USA), respectively.
RNA-seq
The RNA integrity number (RIN) was used to evaluate the quality and integrity of the isolated RNA and only samples with RIN ≥ 8.0 were used for RNA-seq. The construction of the sequencing libraries and sequencing were performed at Beijing Genomics Institute in Shenzhen. Briefly, small RNA sequencing libraries were constructed and 50 bp single-end reads were generated using HiSeq 2500 sequencing platform. Raw data of fastq format were firstly processed using in-house perlscripts. Q20, Q30, and GC-content of the clean data were calculated. To analyze the small RNA expression and distribution on the genome, small RNA tags were mapped against the reference sequence by using Bowtie (Langmead et al. 2009). For the construction of circRNA library, 5 μg RNA per sample was used as input material. Ribosomal RNA was removed by Epicentre Ribozero™ rRNA Removal Kit (Epicentre, USA) followed by the linear RNA digestion with RNase R (Epicentre, USA). The sequencing libraries were constructed by NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA) and 150 bp paired-end reads were generated by Illumina HiSeq 4000 sequencing platform. The reference Homo sapiens genome was downloaded from the NCBI genome website. Then, index of the H. sapiens genome was built using Bowtie2 v2.2.8 and paired-end clean reads were aligned to the reference genome (Langmead et al. 2009).
Data processing and differential expression profiling
The raw sequencing data was filtered with SOAPnuke (v1.5.2) (Kim et al. 2015) to obtain high-quality reads. HISAT2 was used to align the clean reads to the Homo sapiens reference genome (GRCh38.p14) in the NCBI (Langmead and Salzberg 2012), followed by the application of Bowtie2 (v2.2.5) (Li and Dewey 2011) to align the clean reads to database, novel, coding, and noncoding transcripts, established by Beijing Genomic Institute (BGI, Shenzhen, China). RSEM (v1.2.12) was used to calculate the gene expression levels (Love et al. 2014), and differential expression analysis was performed using DESeq2 R package (v1.4.5) with Q value ≤ 0.05 (Abdi 2007). The Venn diagrams were constructed using the VennDiagram R package (v1.7.3). The hierarchical clustering using heatmaps were generated using the ComplexHeatmap R package (v2.16.0).
Prediction of miRNAs and circRNAs
The data of miRBase20.0 were used as miRNA reference and the modified software miRDeep2 (Friedländer et al. 2012) was used to identify the known miRNAs. The software miREvo (Wen et al. 2012) and miRDeep2 (Friedländer et al. 2012) were combined to predict novel miRNA by exploring the secondary structure, the Dicer cleavage site, and the minimum free energy of the small RNA tags unannotated in the former steps. CircRNAs were detected and identified using the find_circ script (Memczak et al. 2013) and CIRI2 tool (Gao et al. 2018), and circRNA sequences were predicted based on junction reads and GT-AG cleavage signals (Gao et al. 2015). The miRNA target sites in the circRNAs were predicted using miRanda (version 3.3a) (Enright et al. 2003). The miRanda, PITA, and RNAhybrid softwares were used to predict potential target genes of the differentially expressed miRNAs.
Gene ontology and KEGG enrichment analysis
The DEmiRNAs and DEcircRNAs host genes were subjected to Gene Ontology (GO) enrichment analysis using web-based GO software (http:/www.geneontology.org) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using KEGG database (http://www.genome.jp/kegg/). Significant levels of enriched GO terms and pathways were corrected by Q value with a threshold (Q value ≤ 0.05) by Bonferroni method (Hu et al. 2020). Sankey diagrams were constructed using the ggsankey R package to display the common GO terms and KEGG pathways at different time points after infection.
Quantitative real-time PCR analysis
To validate the expression of miRNAs and circRNAs obtained by RNA-seq, quantitative real-time PCR (qRT-PCR) was performed on a LightCycler 480 (Roche, Basel, Switzerland) using TB Green® Premix Ex Taq™ II (Tli RNaseH Plus) (Takara, Shuzo, Kyoto, Japan). The reactions were performed at 95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s and 60 °C for 30 s. Three replicate reactions were performed for each gene, and the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene and U6 gene were included as internal controls for circRNA and miRNA, respectively. The 2−∆∆CT method was used to calculate gene expression. The primer sequences used in this study are listed in Table 1.
Prediction of circRNA-miRNA interactions
We constructed the circRNA-miRNA interaction network using miRanda software, based on the miRNA binding sites on circRNAs and the targeted relationship of DEcircRNAs and their downstream target miRNAs, as previously described (Enright et al. 2003). The competing endogenous RNA (ceRNA) network was visualized based on the potential target relationships between DEmiRNAs and DEcircRNAs using Cytoscape v3.6.0.
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 7.0 (GraphPad Software Inc, San Diego, CA, USA). All data represent the mean ± standard deviation of triplicate. A p value of < 0.05 was considered statistically significant.
Results
Identification of the differentially expressed miRNAs
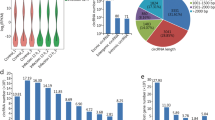
To investigate the dynamic expression and function of miRNAs in the lytic cycle of T. gondii infection, samples were collected for transcriptome sequencing at 0, 1.5, 3, 6, 9, 12, 24, 36, and 48 hpi. Q value < 0.05 and |log2 Fold change|> 1 were used as thresholds to identify the DE genes. As shown in Fig. 1, the dominant small RNAs (sRNAs) were 20–24 nt in length, and most sRNAs were 22 nt, which is consistent with the typical size of miRNA. Expression analysis identified 7 DEmiRNAs (4 upregulated and 3 downregulated), 7 DEmiRNAs (2 upregulated and 5 downregulated), 27 DEmiRNAs (19 upregulated and 8 downregulated), 45 DEmiRNAs (29 upregulated and 16 downregulated), 70 DEmiRNAs (44 upregulated and 26 downregulated), 148 DEmiRNAs (119 upregulated and 29 downregulated), 203 DEmiRNAs (177 upregulated and 26 downregulated), and 217 DEmiRNAs (168 upregulated and 49 downregulated) at 1.5, 3, 6, 9, 12, 24, 36, and 48 hpi, respectively (Fig. 2A). Two DEmiRNAs (hsa-miR-1246 and hsa-miR-663b) were commonly expressed during 1.5 to 9 hpi (Fig. 2B). Additionally, 41 DEmiRNAs were co-expressed during 12 to 48 hpi (Fig. 2C). The hierarchical clustering of miRNA expression profiles between control and infected samples is shown in Fig. 2D. A list of DEmiRNAs identified in this study is provided in Supplementary Table S1.

Basic information of microRNAs (miRNAs) showing miRNA length distribution (x-axis) and the frequency of miRNA of that length (y-axis) identified at different time points after T. gondii infection

Global changes in miRNA expression after T. gondii infection. (A) DEmiRNAs detected in HFF cells at different time points after T. gondii infection. The x-axis shows the time points after T. gondii infection and the y-axis shows the number of DEmiRNAs. Red and green colors represent the upregulated and downregulated DEmiRNAs, respectively. Four-ellipse Venn diagram outlining the common and unique DEmiRNAs at 1.5 to 9 hpi (B) and 12 to 48 hpi (C). Hierarchical clustering heatmap of the DEmiRNAs at different time points after T. gondii infection. Clustering was performed as log10 (FPKM + 1) values, and red color indicates highly expressed genes and blue color indicates low expressed genes (D). The values of miRNA expression are indicated by the color scale; from red to blue indicating highly expressed and low expressed miRNAs, respectively. Each column represents a time point after infection, and each row represents one miRNA
Enrichment analysis of the DEmiRNAs during T. gondii infection
There was a clear difference in the number of DEmiRNAs at different time points after T. gondii infection, which reflects temporal differences in the host response to T. gondii infection. The DEmiRNAs were significantly enriched in biological process (Supplementary Fig. 1), and most DEmiRNAs were enriched in the regulation of RNA polymerase II (GO:0045944), cell differentiation (GO:0030154), multicellular organism development (GO:0007275), apoptotic process (GO:0006915), and phosphorylation (GO:0016310). The KEGG pathway enrichment was used to analyze the significantly enriched pathways of the DEmiRNAs. The top 20 pathways are shown in Fig. 3. Most DEmiRNAs were involved in metabolic-related pathways, such as metabolic pathways (ko01100), glycolysis/gluconeogenesis (ko00010), and citrate cycle (TCA cycle) (ko00020).

KEGG pathway analysis of the DEmiRNA host genes in HFF cells at different times after T. gondii infection. Sankey diagram shows the top 20 predominant pathways at 1.5 hpi, 3 hpi, 6 hpi, 9 hpi, 12 hpi, 24 hpi, 36 hpi, and 48 hpi, respectively
Identification of DEcircRNAs
Our analysis identified 276 DEcircRNAs (44 upregulated and 232 downregulated), 355 DEcircRNAs (95 upregulated and 260 downregulated), 782 DEcircRNAs (302 upregulated and 480 downregulated), 1863 DEcircRNAs (986 upregulated and 877 downregulated), 1738 DEcircRNAs (1122 upregulated and 616 downregulated), 6336 DEcircRNAs (6004 upregulated and 332 downregulated), 1229 DEcircRNAs (1187 upregulated and 42 downregulated), and 1680 DEcircRNAs (1642 upregulated and 38 downregulated) at 1.5, 3, 6, 9, 12, 24, 36, and 48 hpi, respectively (Fig. 4A). Eight DEcircRNAs (hsa-circ_0004662, hsa-circ_0005472, hsa-circ_0039467, hsa-circ_0083081, hsa-circ_0083618, hsa-circ_0115893, hsa-circ_0133744, and hsa-circ_0133745) were co-expressed at different time points. Fifteen DEcircRNAs were co-expressed during 1.5 to 9 hpi (Fig. 4B) and 69 DEcircRNAs were co-expressed during 12 to 48 hpi (Fig. 4C). The hierarchical clustering of miRNA expression profiles between control and infected samples is shown in Fig. 4D. A list of the DEcircRNAs is provided in Supplementary Table S2.

Global changes in circRNA expression after T. gondii infection. (A) DEcircRNAs detected in HFF cells at different time points after T. gondii infection. The x-axis shows the time points after T. gondii infection and the y-axis shows the number of DEcircRNAs. Red and green colors represent the upregulated and downregulated DEcircRNAs, respectively. Four-ellipse Venn diagram outlining the common and unique DEcircRNAs at 1.5 to 9 hpi (B) and 12 to 48 hpi (C). Hierarchical clustering heatmap of the DEcircRNAs at different time points after T. gondii infection. Clustering was performed as log10 (FPKM + 1) values. The values of circRNA expression are indicated by the color scale; from red to blue indicating highly expressed and low expressed circRNAs, respectively. Each column represents a time point after infection, and each row represents one circRNA (D)
Functions of DEcircRNA host genes
GO enrichment analysis was performed to elucidate the biological function of the target host genes of DEcircRNAs and their role in T. gondii infection. According to GO functional annotation, the DEcircRNA host genes were classified into three categories: biological process, cellular component, and molecular function. The GO terms are shown in Supplementary Fig. 2. The DEcircRNAs were most significantly enriched in the biological process category, and most DEcircRNAs were enriched in cellular process (GO:0009987), metabolic process (GO:0008152), biological regulation (GO:0065007), regulation of biological process (GO:0050789), and response to stimulus (GO:0050896). The KEGG pathway enrichment was used to analyze the significantly enriched pathways of the DEcircRNAs. The top 20 pathways are shown in Fig. 5. Most DEcircRNAs were involved in immune-related signaling pathways, such as NOD-like receptor signaling pathway, apoptosis, TNF signaling pathway, and metabolic-related pathway, such as TCA cycle pathway and glutathione metabolism.

KEGG pathway analysis of the DEcircRNA host genes in HFF cells at different times after T. gondii infection. Sankey diagram shows the top 20 predominant pathways at 1.5 hpi, 3 hpi, 6 hpi, 9 hpi, 12 hpi, 24 hpi, 36 hpi, and 48 hpi, respectively
Validation of the deep sequencing results by qRT-PCR
To confirm the differential gene expression obtained from the transcriptome sequencing data, a subset of unigenes co-expressed during T. gondii infection were randomly selected for qRT-PCR verification. These included three DEmiRNAs (hsa-miR-671-5p, hsa-miR-663b, and hsa-miR-1246) (Fig. 6A–C) and three DEcircRNAs (hsa-circ_0081083, hsa-circ_0133744, and hsa-circ_0083081) (Fig. 6D–F). The results obtained by qRT-PCR analysis were consistent with that of the high-throughput sequencing, suggesting the reliability and accuracy of the RNA-seq-based transcriptomic analysis.

A qRT-PCR-based validation of the expression of representative EDmiRNAs (hsa-miR-671-5p, hsa-miR-663b, and hsa-miR-1246) (A–C) and DEcircRNAs (hsa-circ_0081083, hsa-circ_0133744, and hsa-circ_0083081) (D–F). The x-axis shows the time points after T. gondii infection and the y-axis shows the relative expression levels. The U6 gene and GAPDH gene were used for miRNA and circRNA normalization, respectively
Analysis of DEcircRNAs-DEmiRNAs interaction
CircRNAs can act as miRNA decoys or sponges to regulate host gene expression. To reveal correlations between DEmiRNAs and DEcircRNAs and elucidate the possible role of miRNAs and circRNAs in T. gondii infection, the ceRNA network was constructed. In the ceRNA network, one miRNA appears to be associated with one or more upstream circRNAs (Fig. 7). These circRNAs could play a role as biological targets of T. gondii infection.

The competing endogenous RNA (ceRNA) network of miRNAs and circRNAs in HFF cells infected by T. gondii. Cyan rectangles represent the circRNAs and orange nodes represent the miRNAs
Discussion
In this study, we used sequencing technology to investigate the dynamic and differential expression profiles of miRNAs and circRNAs in HFF cells in the presence or absence of T. gondii infection at 0, 1.5, 3, 6, 9, 12, 24, 36, and 48 hpi. Our aim was to identify transcripts with an essential role in the host response to T. gondii during the parasite infection lytic cycle. Our study showed that T. gondii infection caused significant alterations in the expression of circRNAs involved in immune-related pathways, such as NOD-like receptor signaling pathway, apoptosis, and NF-kappa B signaling pathway at the early stage of infection. These results are consistent with previous transcriptomics studies showing that T. gondii infection deregulates many genes involved in immune regulation and activation (Hunter and Sibley 2012; Liu et al. 2018; Wang et al. 2018).
Apoptosis is a form of programmed cell death that can limit the proliferation of intracellular pathogens by depriving them of their intracellular niche (Wallach and Kang 2018). To counter apoptosis, T. gondii disrupts the mitochondrial and death receptor-mediated apoptotic pathway (Vutova et al. 2007; Wallach and Kang 2018) and inhibits the expression of caspases-8, -9, -3, and -7 in different cell lines (Hwang et al. 2010; Graumann et al. 2015; Afshar-Khamseh et al. 2021). T. gondii can delay apoptosis of infected human neutrophils by deactivating apoptotic caspases via activating cytosolic proliferating cell nuclear antigen (Wallach and Kang 2018). In the present study, we detected an increase in the expression of miR-142a-3p which plays a role in immune regulation and apoptosis in many cells by targeting p53, TNFAIP2, and GLUT3 (Lu et al. 2015). These results add more evidence to the literature showing the ability of T. gondii to evade and manipulate host immune response to establish an optimal replicative niche (Seeber and Steinfelder 2016; Lima et al. 2021).
T. gondii infection is involved in several neuropsychiatric disorders (Elsheikha and Zhu 2016; Elsheikha et al. 2016; Liang et al. 2021). However, the mechanisms underlying this connection remain inconclusive. Interestingly, our study revealed that T. gondii can activate DEmiRNAs associated with nervous system-related pathways, such as axon guidance (ko04360), neurotrophin signaling pathway (ko04722), and neuroactive ligand-receptor interaction (ko04080). We also detected a decreasing expression trend of miRNA-146a-5p during T. gondii infection, suggesting that T. gondii may contribute to brain dysfunction via altering miRNAs expression. This agrees with the literature reporting the involvement of miRNAs in the pathogenesis of diseases of the nervous system (Hutchison et al. 2009). The miRNA-146a can switch microglial phenotypes to resist the pathological processes and cognitive degradation associated with Alzheimer’s disease (MacRae et al. 2012).
Previous studies revealed how the metabolism of T. gondii and the host is altered after infection (Blume et al. 2009, 2015; Ma et al. 2019, 2020, 2021; Olson et al. 2020). In the present study, we have shown that T. gondii infection changes the host cells’ metabolism by altering the expression of circRNAs involved in TCA cycle, metabolic pathways, fatty acid metabolism, glutathione metabolism, and glycolysis/gluconeogenesis. The TCA cycle is essential for T. gondii growth (Blume et al. 2009). In the absence of glucose or under genetically ablated glycolysis, T. gondii can utilize an intrinsic protein, gluconeogenic enzyme fructose bisphosphatase 2, to perform glutaminolysis and generate carbon for gluconeogenesis (Blume et al. 2009, 2015). The exact impact of miRNAs and circRNAs on the regulation of host metabolism during T. gondii infection merit further investigated.
CircRNAs act as miRNA decoys or sponges to influence miRNA function and regulate the expression of target mRNAs (Panda 2018). To better understand the regulatory effects of DEcircRNA, we predicted the putative circRNA-miRNA interaction networks based on the DEcircRNAs and DEmiRNAs. The results showed that DEmiRNAs (hsa-miR-149-5p, hsa-miR-370-3p, hsa-miR-449a, and hsa-miR-150-5p) were regulated by DEcircRNAs (hsa-circ_0081574, hsa-circ_0081567, hsa-circ_0081569, and hsa-circ_0081575). The miR-149-5p serves as a negative regulator of inflammatory and immune responses (Hübner et al. 2020; Li et al. 2023) and regulates the transcription levels of IL-10, TREM-1, and PIM1, which have an anti-inflammatory activity (Neamah et al. 2019; Wang et al. 2023; Yue et al. 2023), highlighting the role of the identified DEcircRNAs in regulating the immune-inflammatory response during T. gondii infection.
Conclusion
This study revealed changes in the transcriptional patterns of miRNAs and circRNAs in HFF cells during T. gondii lytic cycle. Many DEmiRNAs and DEcircRNA were identified at different time points post T. gondii infection. The DEmiRNAs and DEcircRNAs were mainly involved in key physiopathological processes, including apoptosis, metabolism, signal transduction, and immune responses. Our data adds more insight into the role of miRNAs and circRNAs in the pathogenesis of T. gondii infection. The mechanism by which these DEmiRNAs and DEcircRNAs regulate antiparasitic responses requires further investigation.
Data availability
The RNA-seq data presented in this study were deposited in the NCBI repository under accession numbers PRJNA721229 and PRJNA769531.
References
Abdi H (2007) The Bonferonni and Sˇidák corrections for multiple comparisons. Encycl Meas Stat 1:1–9
Afshar-Khamseh R, Javeri A, Taha MF (2021) MiR-146a suppresses the expression of CXCR4 and alters survival, proliferation and migration rate in colorectal cancer cells. Tissue Cell 73:101654
Awuson-David B, Williams AC, Wright B, Hill LJ, Di Pietro V (2023) Common microRNA regulated pathways in Alzheimer’s and Parkinson’s disease. Front Neurosci 17:1228927
Black MW, Boothroyd JC (2000) Lytic cycle of Toxoplasma gondii. Microbiol Mol Biol Rev 64(3):607–623
Blume M, Rodriguez-Contreras D, Landfear S, Fleige T, Soldati-Favre D, Lucius R, Gupta N (2009) Host-derived glucose and its transporter in the obligate intracellular pathogen Toxoplasma gondii are dispensable by glutaminolysis. Proc Natl Acad Sci U S A 106(31):12998–13003
Blume M, Nitzsche R, Sternberg U, Gerlic M, Masters SL, Gupta N, McConville MJ (2015) A Toxoplasma gondii gluconeogenic enzyme contributes to robust central carbon metabolism and is essential for replication and virulence. Cell Host Microbe 18(2):210–220
Cong W, Zhang XX, He JJ, Li FC, Elsheikha HM, Zhu XQ (2017) Global miRNA expression profiling of domestic cat livers following acute Toxoplasma gondii infection. Oncotarget 8(15):25599–25611
Dong X, Bai Y, Liao Z, Gritsch D, Liu X, Wang T, Borges-Monroy R, Ehrlich A, Serrano GE, Feany MB, Beach TG, Scherzer CR (2023) Circular RNAs in the human brain are tailored to neuron identity and neuropsychiatric disease. Nat Commun 14:5327
Elsheikha HM (2008) Congenital toxoplasmosis: priorities for further health promotion action. Public Health 122(4):335–353
Elsheikha HM, Zhu XQ (2016) Toxoplasma gondii infection and schizophrenia: an inter-kingdom communication perspective. Curr Opin Infect Dis 29(3):311–318
Elsheikha HM, Büsselberg D, Zhu XQ (2016) The known and missing links between Toxoplasma gondii and schizophrenia. Metab Brain Dis 31(4):749–759
Elsheikha HM, Marra CM, Zhu XQ (2020) Epidemiology, pathophysiology, diagnosis, and management of cerebral toxoplasmosis. Clin Microbiol Rev 34(1):e00115–e119
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS (2003) MicroRNA targets in drosophila. Genome Biol 5(1):R1
Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N (2012) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40(1):37–52
Gao Y, Wang J, Zhao F (2015) CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 16(1):4
Gao Y, Zhang J, Zhao F (2018) Circular RNA identification based on multiple seed matching. Brief Bioinform 19(5):803–810
Granados-Riveron JT, Aquino-Jarquin G (2016) The complexity of the translation ability of circRNAs. Biochim Biophys Acta 1859:1245–1251
Graumann K, Schaumburg F, Reubold TF, Hippe D, Eschenburg S, Lüder CG (2015) Toxoplasma gondii inhibits cytochrome c-induced caspase activation in its host cell by interference with holo-apoptosome assembly. Microb Cell 2(5):150–162
He JJ, Ma J, Wang JL, Xu MJ, Zhu XQ (2016) Analysis of miRNA expression profiling in mouse spleen affected by acute Toxoplasma gondii infection. Infect Genet Evol 37:137–142
Hill D, Dubey JP (2002) Toxoplasma gondii: transmission, diagnosis and prevention. Clin Microbiol Infect 8(10):634–640
Hu RS, He JJ, Elsheikha HM, Zhang FK, Zou Y, Zhao GH, Cong W, Zhu XQ (2018) Differential brain microRNA expression profiles after acute and chronic infection of mice with Toxoplasma gondii oocysts. Front Microbiol 9:2316
Hu RS, He JJ, Elsheikha HM, Zou Y, Ehsan M, Ma QN, Zhu XQ, Cong W (2020) Transcriptomic profiling of mouse brain during acute and chronic infections by Toxoplasma gondii oocysts. Front Microbiol 11:570903
Hübner K, Karwelat D, Pietsch E, Beinborn I, Winterberg S, Bedenbender K, Benedikter BJ, Schmeck B, Vollmeister E (2020) NF-κB-mediated inhibition of microRNA-149-5p regulates chitinase-3-like 1 expression in human airway epithelial cells. Cell Signal 67:109498
Hunter CA, Sibley LD (2012) Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol 10(11):766–778
Hutchison ER, Okun E, Mattson MP (2009) The therapeutic potential of microRNAs in nervous system damage, degeneration, and repair. Neuromolecular Med 11(3):153–161
Hwang IY, Quan JH, Ahn MH, Ahmed HA, Cha GH, Shin DW, Lee YH (2010) Toxoplasma gondii infection inhibits the mitochondrial apoptosis through induction of Bcl-2 and HSP70. Parasitol Res 107(6):1313–1321
Jia B, Lu H, Liu Q, Yin J, Jiang N, Chen Q (2013) Genome-wide comparative analysis revealed significant transcriptome changes in mice after Toxoplasma gondii infection. Parasit Vectors 6:161
Jones-Brando L, Torrey EF, Yolken R (2003) Drugs used in the treatment of schizophrenia and bipolar disorder inhibit the replication of Toxoplasma gondii. Schizophr Res 62(3):237–244
Judice CC, Bourgard C, Kayano AC, Albrecht L, Costa FT (2016) MicroRNAs in the host-apicomplexan parasites interactions: a review of immunopathological aspects. Front Cell Infect Microbiol 6:5
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4):357–360
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10(3):R25
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12:323
Li X, Liu CX, Xue W, Zhang Y, Jiang S, Yin QF, Wei J, Yao RW, Yang L, Chen LL (2017) Coordinated circRNA biogenesis and function with NF90/NF110 in viral infection. Mol Cell 67(2):214-227.e7
Li Q, Li S, Xu C, Zhao J, Hou L, Jiang F, Zhu Z, Wang Y, Tian L (2023) microRNA-149–5p mediates the PM2.5-induced inflammatory response by targeting TAB2 via MAPK and NF-κB signaling pathways in vivo and in vitro. Cell Biol Toxicol 39(3):703–717
Liang C, Zou T, Zhang M, Fan W, Zhang T, Jiang Y, Cai Y, Chen F, Chen X, Sun Y, Zhao B, Wang Y, Cui L (2021) MicroRNA-146a switches microglial phenotypes to resist the pathological processes and cognitive degradation of Alzheimer’s disease. Theranostics 11(9):4103–4121
Lima TS, Mallya S, Jankeel A, Messaoudi I, Lodoen MB (2021) Toxoplasma gondii extends the life span of infected human neutrophils by inducing cytosolic PCNA and blocking activation of apoptotic caspases. mBio 12(1):e02031-20
Liu W, Huang L, Wei Q, Zhang Y, Zhang S, Zhang W, Cai L, Liang S (2018) Microarray analysis of long non-coding RNA expression profiles uncovers a Toxoplasma-induced negative regulation of host immune signaling. Parasit Vectors 11(1):174
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Bio 15(12):550
Lu X, Wei Y, Liu F (2015) Direct regulation of p53 by miR-142a-3p mediates the survival of hematopoietic stem and progenitor cells in zebrafish. Cell Discov 1:15027
Lu S, Zhu N, Guo W, Wang X, Li K, Yan J, Jiang C, Han S, Xiang H, Wu X, Liu Y, Xiong H, Chen L, Gong Z, Luo F, Hou W (2020) RNA-seq revealed a circular RNA-microRNA-mRNA regulatory network in Hantaan virus infection. Front Cell Infect Microbiol 10:97
Lv L, Wang Y, Feng W, Hernandez JA, Huang W, Zheng Y, Zhou X, Lv S, Chen Y, Yuan ZG (2017) iTRAQ-based differential proteomic analysis in Mongolian gerbil brains chronically infected with Toxoplasma gondii. J Proteomics 160:74–83
Ma J, He JJ, Hou JL, Zhou CX, Zhang FK, Elsheikha HM, Zhu XQ (2019) Metabolomic signature of mouse cerebral cortex following Toxoplasma gondii infection. Parasit Vectors 12(1):373
Ma J, He JJ, Hou JL, Zhou CX, Elsheikha HM, Zhu XQ (2020) Ultra performance liquid chromatography-tandem mass spectrometry-based metabolomics reveals metabolic alterations in the mouse cerebellum during Toxoplasma gondii infection. Front Microbiol 11:1555
Ma J, He JJ, Wang M, Hou JL, Elsheikha HM, Zhu XQ (2021) Toxoplasma gondii induces metabolic disturbances in the hippocampus of BALB/c mice. Parasitol Res 120(8):2805–2818
MacRae JI, Sheiner L, Nahid A, Tonkin C, Striepen B, McConville MJ (2012) Mitochondrial metabolism of glucose and glutamine is required for intracellular growth of Toxoplasma gondii. Cell Host Microbe 12(5):682–692
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, Kocks C, le Noble F, Rajewsky N (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495(7441):333–338
Neamah WH, Singh NP, Alghetaa H, Abdulla OA, Chatterjee S, Busbee PB, Nagarkatti M, Nagarkatti P (2019) AhR activation leads to massive mobilization of myeloid-derived suppressor cells with immunosuppressive activity through regulation of CXCR2 and microRNA miR-150-5p and miR-543-3p that target anti-inflammatory genes. J Immunol 203(7):1830–1844
Olson WJ, Martorelli Di Genova B, Gallego-Lopez G, Dawson AR, Stevenson D, Amador-Noguez D, Knoll LJ (2020) Dual metabolomic profiling uncovers Toxoplasma manipulation of the host metabolome and the discovery of a novel parasite metabolic capability. PLoS Pathog 16(4):e1008432
Panda AC (2018) Circular RNAs act as miRNA sponges. Adv Exp Med Biol 1087:67–79
Pappas G, Roussos N, Falagas ME (2009) Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int J Parasitol 39(12):1385–1394
Radke JB, Worth D, Hong D, Huang S, Sullivan WJ Jr, Wilson EH, White MW (2018) Transcriptional repression by ApiAP2 factors is central to chronic toxoplasmosis. PLoS Pathog 14(5):e1007035
Sahu A, Kumar S, Sreenivasamurthy SK, Selvan LD, Madugundu AK, Yelamanchi SD, Puttamallesh VN, Dey G, Anil AK, Srinivasan A, Mukherjee KK, Gowda H, Satishchandra P, Mahadevan A, Pandey A, Prasad TS, Shankar SK (2014) Host response profile of human brain proteome in toxoplasma encephalitis co-infected with HIV. Clin Proteomics 11(1):39
Seeber F, Steinfelder S (2016) Recent advances in understanding apicomplexan parasites. F1000Res 5:F1000 Faculty Rev-1369
Shao Y, Chen Y (2016) Roles of circular RNAs in neurologic disease. Front Mol Neurosci 9:25
Tanaka S, Nishimura M, Ihara F, Yamagishi J, Suzuki Y, Nishikawa Y (2013) Transcriptome analysis of mouse brain infected with Toxoplasma gondii. Infect Immun 81(10):3609–3619
Vutova P, Wirth M, Hippe D, Gross U, Schulze-Osthoff K, Schmitz I, Lüder CG (2007) Toxoplasma gondii inhibits Fas/CD95-triggered cell death by inducing aberrant processing and degradation of caspase 8. Cellular Microbiol 9(6):1556–1570
Wallach D, Kang TB (2018) Programmed cell death in immune defense: knowledge and presumptions. Immunity 49(1):19–32
Wang M, Zhang FK, Elsheikha HM, Zhang NZ, He JJ, Luo JX, Zhu XQ (2018) Transcriptomic insights into the early host-pathogen interaction of cat intestine with Toxoplasma gondii. Parasit Vectors 11(1):592
Wang JL, Zhang NZ, Li TT, He JJ, Elsheikha HM, Zhu XQ (2019) Advances in the development of anti-Toxoplasma gondii vaccines: challenges, opportunities, and perspectives. Trends Parasitol 35(3):239–253
Wang SS, Zhou CX, Elsheikha HM, He JJ, Zou FC, Zheng WB, Zhu XQ, Zhao GH (2022) Temporal transcriptomic changes in long non-coding RNAs and messenger RNAs involved in the host immune and metabolic response during Toxoplasma gondii lytic cycle. Parasit Vectors 15(1):22
Wang Y, Qin J, Dong L, He C, Zhang D, Wu X, Li T, Yue H, Mu L, Wang Q, Yang J (2023) Suppression of mir-150-5p attenuates the anti-inflammatory effect of glucocorticoids in mice with ulcerative colitis. Mol Immunol 163:28–38
Wen M, Shen Y, Shi S, Tang T (2012) miREvo: an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinformatics 13:140
Xie SC, Zhou CX, Zhai BT, Zheng WB, Liu GH, Zhu XQ (2022) A combined miRNA-piRNA signature in the serum and urine of rabbits infected with Toxoplasma gondii oocysts. Parasit Vectors 15(1):490
Xu MJ, Zhou DH, Nisbet AJ, Huang SY, Fan YF, Zhu XQ (2013) Characterization of mouse brain microRNAs after infection with cyst-forming Toxoplasma gondii. Parasit Vectors 6:154
Yang J, Du F, Zhou X, Wang L, Li S, Fang R, Zhao J (2018) Brain proteomic differences between wild-type and CD44- mice induced by chronic Toxoplasma gondii infection. Parasitol Res 117(8):2623–2633
Yue C, Wang W, Gao S, Ye J, Zhang T, Xing Z, Xie Y, Qian H, Zhou X, Li S, Yu A, Wang L, Wang J, Hua C (2023) Agomir miRNA-150-5p alleviates pristane-induced lupus by suppressing myeloid dendritic cells activation and inflammation via TREM-1 axis. Inflamm Res 72(7):1391–1408
Zhang X, Liang Z, Wang C, Shen Z, Sun S, Gong C, Hu X (2022) Viral circular RNAs and their possible roles in virus-host interaction. Front Immunol 13:939768
Zhou CX, Zhou DH, Liu GX, Suo X, Zhu XQ (2016) Transcriptomic analysis of porcine PBMCs infected with Toxoplasma gondii RH strain. Acta Trop 154:82–88
Zhou CX, Gao M, Han B, Cong H, Zhu XQ, Zhou HY (2021) Quantitative peptidomics of mouse brain after infection with cyst-forming Toxoplasma gondii. Front Immunol 12:681242
Zou Y, Meng JX, Wei XY, Gu XY, Chen C, Geng HL, Yang LH, Zhang XX, Cao HW (2022) CircRNA and miRNA expression analysis in livers of mice with Toxoplasma gondii infection. Front Cell Infect Microbiol 12:1037586
Acknowledgements
We would like to thank BGI-Shenzhen for technical assistance.
Funding
Project support was provided by the National Natural Science Foundation of China (Grant No. 32172887), the NSFC-Yunnan Joint Fund (Grant No. U2202201), the Agricultural Science and Technology Innovation Program (ASTIP) of China (Grant no. CAAS-ASTIP-2016-LVRI-03), the Research Fund of Shanxi Province for Introduced High-level Leading Talents (Grant No. RFSXIHLT202101), and the Veterinary Public Health Innovation Team of Yunnan Province (Grant No. 202105AE160014). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
SSW: methodology, validation, data curation, and writing—original draft. XWW, JJH, and WBZ: validation, resources, and data curation. CXZ, HME, and XQZ: conceptualization, project administration, and writing—review and editing. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Ethical approval
This study was reviewed and approved by the Animal Research Ethics Committee of Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (Permit No. 2019–012).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Handling Editor: Julia Walochnik
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Functional enrichment analysis of the predicted target genes of the differentially expressed miRNAs detected in HFF cells after T. gondii infection. Sankey plot shows the most significantly enriched GO terms in the biological process at 1.5 hpi, 3 hpi, 6 hpi, 9 hpi, 12 hpi, 24 hpi, 36 hpi, and 48 hpi (PDF 434 KB)
Supplementary Fig. 2
Functional enrichment analysis of the predicted target genes of the differentially expressed circRNAs detected in HFF cells after T. gondii infection. Sankey plot shows the most enriched GO terms at 1.5 hpi, 3 hpi, 6 hpi, 9 hpi, 12 hpi, 24 hpi, 36 hpi, and 48 hpi (PDF 1.15 MB)
Table S1
A list of all differentially expressed miRNAs detected in HFF cells at different time points after T. gondii infection (XLS 99.5 KB)
Table S2
A list of all differentially expressed circRNAs detected in HFF cells at different time points after T. gondii infection (XLS 3.46 MB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, SS., Wang, X., He, JJ. et al. Expression profiles of host miRNAs and circRNAs and ceRNA network during Toxoplasma gondii lytic cycle. Parasitol Res 123, 145 (2024). https://doi.org/10.1007/s00436-024-08152-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00436-024-08152-x