
Abstract
Avian blood parasites have been preliminarily studied in East Asia, but no data are available from long-term monitoring. The aim of this study was to evaluate the prevalence, genetic diversity, and temporal dynamics of Plasmodium, Haemoproteus, and Leucocytozoon in two passerine communities (one forest and one urban) in north China from 2008 to 2013, as well as the association between infected lineages and host specificities. Out of 633 birds from 40 species, 157 individuals (24.8 %) were infected; overall prevalence was 26.7 % and 16.8 % in two sites, respectively. The dominant avian blood parasite genus in the forest park changed yearly between Plasmodium and Haemoproteus, while the Leucocytozoon maintained a low infection level. Forty-four haplotypes were identified by sequencing a 432-bp fragment of the cytochrome b (cyt b) gene; more than 70 % were novel (six Plasmodium lineages, 16 Haemoproteus lineages, and nine Leucocytozoon lineages). Based on our data gathered over consecutive years, we found that the highly observed lineages of Haemoproteus showed higher host diversities than those of Plasmodium, and the most infected lineage EMEL01 (100 % identity with SGS1) take on the highest host diversity but low temporal diversity of the two genera, implying that this lineage infected a great diversity of species in certain years, but maintained a lower infection level or even disappeared in other years. The results suggest that genetic diversity of avian blood parasites in East Asia is high and provides scope for further research. In addition, compared with overall analysis, yearly prevalence monitoring is important in uncovering the temporal dynamic and host specificity variations over time.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Understanding the prevalence and evolutionary pattern of parasites is of fundamental importance in basic research on both wildlife epidemic diseases and host–parasite co-evolution. Avian blood parasites, which include Plasmodium, Haemoproteus, and Leucocytozoon, are widespread, abundant, and easily sampled, making them excellent models for exploring the ecological and evolutionary dynamics of host–parasite associations (Valkiūnas 2004). Research on genetic diversity and infection patterns of avian blood parasites is of great importance when predicting wildlife diseases; therefore, it has gained considerable attention in recent decades (Riper et al. 1986; Scheuerlein and Ricklefs 2004; Valkiūnas 2004; Woodworth et al. 2005).
Long-term research on blood parasites in wild birds, which was mainly conducted in Europe, America, and Oceania, was helpful in detecting the key traits and variations in pathogens (Imura et al. 2012). Research on the temporal dynamic of avian blood parasites in the wild bird community can help to understand local changes and how lineages affect each other in the natural environment (Synek et al. 2013).
However, most long-term studies have so far aimed at revealing genetic diversity and infection features of samples collected in different years of one or a few host species, and limited data are available on temporal dynamics or lineage-based phylogenies. Temporal dynamics of lineage diversity can reflect interspecific interactions between parasites (de Roode et al. 2005). In previous research, the prevalence of three lineages (two Plasmodium and one Haemoproteus) showed significant variations over years in great reed warblers (Acrocephalus arundinaceus) (Bensch et al. 2007). Furthermore, a non-parallel annual variation in prevalence of two Plasmodium parasites, Plasmodium relictum and Plasmodium circumflexum, was reported in a population of blue tits (Cyanistes caeruleus) (Knowles et al. 2011).
Compared with the traditional method of blood smear screening, molecular-based screening has become much more popular in recent studies due to its high efficacy, accuracy, and ease of use (Møller et al. 2010; Valkiūnas et al. 2006). The use of molecular-based screening methods, combining with the traditional morphological characters (Valkiunas et al. 2007b, 2008), has enabled greater insight into avian parasite species diversity, enabling identification of species number from less than 200 (Valkiūnas 2004) to several thousand species (Beadell et al. 2006; Bensch et al. 2004). Although there is a great amount of avian diversity in China, the background information on avian blood parasites is very limited (Zehtindjiev et al. 2013; Zhang et al. 2014).
In order to detect the prevalence and temporal dynamic patterns of blood parasites, using molecular genetic screening techniques, we (1) present the genetic diversity and (2) detect the inter-annual change of the parasite lineages in passerine birds collected during 2008–2013 in Beijing, north China.
Materials and methods
Sample collection
A total of 633 passerines were captured with mist nets between September and October, from 2009 to 2013 at Xiaolongmen Forest Park (39° 57′ 54″ N 115° 26′ 00″ E) and 2008, 2012, and 2013 in the campus of Beijing Normal University campus (BNU, 39° 57′ 46″ N 116° 21′ 28″ E), Beijing. Blood samples were collected from the brachial vein and preserved in ethanol for subsequent processing in the laboratory. DNA was extracted using a TIANamp DNA kit (Tiangen, Beijing) according to the manufacturer’s protocol.
Blood parasite identification
Blood parasites were identified using a nested PCR protocol based on the amplification of a 479-bp fragment of the mitochondrial cytochrome b (cyt b) gene (Hellgren et al. 2004). The first reaction included use of the primers HaemNFI (5′-CATATATTAAGAGAAITATGGAG-3′) and HaemNR3 (5′-ATAGAAAGATAAGAAATACCATTC-3′), performed in a 25 μL reaction mixture containing 12.5 μL 2× ExTaq buffer (Takara, Japan), 0.5 μL of each primer, and 1 μL template DNA. The second PCR reaction was carried out using 1 μL of the products amplified during the first reaction as template and included the primers HaemNF (5′-ATGGTGCTTTCGATATATGCATG-3′) and HaemNR2 (5′-GCATTATCTGGATGTGATAATGGT-3′). Positive or negative samples were determined by 1.5 % agarose gels stained with SYBR Green I in an ultraviolet trans-illuminator (GDS-8000PC, GENE, USA).
Nested PCR was conducted at least twice on each sample in order to avoid false-negative samples. Negative control (ddH2O) was used in each reaction. The PCR products of positive samples were sequenced in both directions using a 3730XL automatic sequencer (ABI, USA), and sequences were assembled by CodonCode Aligner 5.1.5 (CodonCode Corporation, USA).
Phylogenetic analysis
Haplotypes identified by DnaSP 5.10.01 (Librado and Rozas 2009) and their most similar morphospecies, identified by the BLAST module in the MalAvi database (Bensch et al. 2009) were aligned by MEGA 5.04 (Tamura et al. 2011) for phylogenetic analysis. In addition, a fragment of cyt b sequence of the human malaria parasite Plasmodium falciparum (GenBank No.: M76611.1) was used as an out-group due to its close genetic relationship with avian haemosporidia (Outlaw and Ricklefs 2011). In total of four closely related morphospecies lineages to our results, SW3 (Haemoproteus belopolskyi) (Valkiunas et al. 2007a), ROBIN1 (Haemoproteus attenuatus) (Valkiūnas et al. 2006), YWT3 (Haemoproteus motacillae) (Dimitrov et al. 2013), and GRW06 (Plasmodium elongatum) (Valkiunas et al. 2008), identified by BLAST module in MalAvi database (Table 1), were employed as inner-group of phylogenetic analysis. Haplotypes with even one base difference from lineages in the MalAvi database were defined as new lineages and all sequences were submitted to Genbank (No.KT757541-KT757584).
Nucleotide substitution models were tested using jModelTest 2.1.4 (Darriba et al. 2012), and corrected Akaike Information Criterion (AICc) was adopted so as to determine the best-fit model. BEAST v1.8.0 (Drummond and Rambaut 2007) was used for Bayesian phylogenetic inference with default parameters except for strict molecular clock and Yule process for tree prior. Markov chain Monte Carlo (MCMC) was run for different steps, with the length of the chain set as 1 × 109 and log parameters as every 1 × 105 generation. The maximum credibility tree was searched by TreeAnnotator v1.8.0 after the first 1000 trees were discarded as burn-in. The selected tree was then adjusted in FigTree v1.3.1 (Andrew Rambaut, University of Edinburgh, UK; http://tree.bio.ed.ac.uk/software/figtree/).
Statistical analysis
Both the host species diversity index (SH) and temporal diversity index (TS) were calculated using the Shannon–Wiener index algorithm for lineages that infected at least four individuals. Considering the discontinuous sample collection in the campus, only samples collected in Xiaolongmen were utilized for analysis in this part.
where H is the number of infected host species and H i the proportion of infected individuals of species i (0 < i < H); then, supposing the number of infected individuals of the certain lineages is N, and in bird i is n i , then, H i = n i /N.
S represents the sample year, while P i is the proportion of infected individuals in year i (0 < i < S). Supposing the number of infected individuals of the certain lineage is N, and in year i is n i , then, P i = n i /N.
Results
Prevalence of avian blood parasites
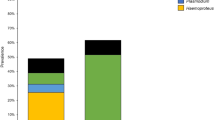
A total of 633 birds belonging to 40 passerine species were sampled (Table S1), of which 157 individuals (24.8 %) were infected: 62 birds by 12 lineages of Plasmodium, 75 birds by 16 lineages of Haemoproteus; 19 birds by 14 lineages of Leucocytozoon, and two birds by two undefined lineages with low similarity (<95 %) to the reported lineages in MalAvi database. Only one willow tit, Parus montanus, showed an evidence of dual infection (both Plasmodium and Haemoproteus were detected in this sample). In Xiaolongmen Forest Park, 26.7 % (132/494) of the sampled individuals were infected including one dual infection. Among them, 35 lineages (10 Plasmodium, 12 Haemoproteus, and 13 Leucocytozoon) were detected. While in the campus, the overall prevalence was 16.8 % (25/139) with 14 lineages (four Plasmodium, seven Haemoproteus, one Leucocytozoon, and two undefined lineages).
The total prevalence was not considerably varied among years in Xiaolongmen Forest Park (χ 2 = 8.01, P = 0.09), but the dominant parasite genus changed yearly between Plasmodium and Haemoproteus (Fig. 1) with significant variation among years (Plasmodium χ 2 = 43.28, P < 0.01, Haemoproteus χ 2 = 11.31, P < 0.05) and the prevalence of Leucocytozoon maintained a low level (0–4.17 %) with no significant differences over the years. In BNU campus, the significant yearly varied prevalence was observed in total samples (χ 2 = 17.22, P < 0.01) and Haemoproteus (χ 2 = 16.73, P < 0.01), but not significant in Plasmodium and Leucocytozoon.

Annual variation (2008–2013) in prevalence of avian blood parasites in passerines in Beijing. a Total samples. b Xiaolongmen Forest Park. c BNU campus. P Plasmodium, H Haemoproteus, and L Leucocytozoon
Phylogenetic tree
A total of 44 avian blood parasite lineages were detected in this study (Table 1), and 31 of them were novel. Two lineages could be designated as morphospecies according to BLAST in MalAvi database using cyt b sequences, EMEL01 (100 % with SGS1) belongs to P. relictum (Palinauskas et al. 2007) and ANHO02 (100 % with TURDUS1) belongs to P. circumflexum (Palinauskas et al. 2007). Three clades were formed in the Bayesian cyt b phylogenic tree (Fig. 2). All lineages of Plasmodium clustered together, forming a sister cluster to lineages of Haemoproteus. The lineages EMPU01 and CAPU01, which were most identified with Haemoproteus lineages according to the BLAST result, clustered together with Leucocytozoon lineages, although the node support was low (Fig. 2).

Bayesian cyt b phylogenetic tree of blood parasite haplotypes reconstructed under TN93 + I + G model. Posterior probabilities over 0.7 are listed above nodes. Novel lineages are indicated in bold. The lineages only detected in Xiaolongmen Forest Park were underlined, and only detected in BNU campus were double underlined. Morphospecies were labeled after the lineage in brackets
Most lineages that belong to Plasmodium are exactly the same as those reported ones. Morphospecies were combined with their most similar lineages respectively. The situation observed in Haemoproteus was much more complex with lineages differing from each other by only one or two bases and combined with the same morphospecies. Leucocytozoon lineages detected here are mostly novel.
Lineage diversity and temporal dynamics
A total of eight lineages were identified as the most frequent in the wild passerine community in Xiaolongmen Forest Park (infecting at least four individuals), including four Plasmodium lineages, three Haemoproteus lineages, and one Leucocytozoon lineage (Table 2).
Among the Plasmodium lineages, EMEL01 had the highest host species diversity index (S H = 2.77) but the lowest temporal diversity index (T S = 0.83), which suggested that it had an outbreak in a particular year (2009) and infected a diverse amount of bird species in the community, but then subsequently fell until disappearing in 2013. In contrast, EMEL02 had a higher temporal diversity index (T S = 2.25) than others, implying that the prevalence of this lineage had shown little variation over the years. At the same time, ACAE01 had the highest value in both indexes (S H = 2.77, T S = 2.17) in Haemoproteus, showing a comparatively stable prevalence.
Discussion
Temporal prevalence dynamics of Plasmodium and Haemoproteus
We established the temporal dynamic of avian blood parasites in wild birds in north China across 6 years. Compared with similar fauna, the overall prevalence of avian blood parasites in northern China (26.7 % in forest and 16.8 % in urban) was comparable to that reported in western (Zehtindjiev et al. 2013) and southern China (Zhang et al. 2014), but was slightly higher than that reported in Japan (Murata 2002; Imura et al. 2012). The prevalence of blood parasites in passerine birds showed a significant variation over the years, as in other studies (Piersma and van der Velde 2012).
Ceratopogonid- and culicid-transmitted parasites, i.e., Haemoproteus and Plasmodium, showed a much higher prevalence than Leucocytozoon, which is transmitted by simuliid vectors. This is the opposite of the pattern in North America (Greiner et al. 1975) and Europe (Scheuerlein and Ricklefs 2004). As vector diversity and abundance are supposed to shape the diversity and the distribution of blood parasites (Santiago-Alarcon et al. 2012), further study of vector abundance in our study site would be helpful in pinpointing the cause behind this difference.
As reported previously, Haemoproteus seems to be more prevalent than Plasmodium (Silva-Iturriza et al. 2012). The generally accepted explanation is that of pathogenicity difference between the two genera (Atkinson et al. 2000). However, we discovered that Haemoproteus showed a higher prevalence in 4 of the 6 sampled years while the prevalence of Plasmodium was higher in the other 2 years. The different pattern uncovered here may due to the fact that the previous analysis of parasite prevalence and its contributing factors was based on pooled data from different years, while much less attention has been given to temporal dynamics (Ricklefs et al. 2005). Where both the biotic and abiotic factors and the correlation between parasites and their hosts may vary over different years (Baillie et al. 2012; Sol et al. 2003), this may induce temporal dynamics of annual prevalence of the two genera.
Lineage diversity and temporal dynamics
Most of the lineages detected in this study merely infected one or two individuals (Table 1). Only six of them were found in both sites; the rest were mixed up in the phylogenetic tree, showing an unclear pattern between the forest and urban communities.
More than 70 % of the lineages were newly detected. Among them, CAPU01 and EMPU01 (both detected in Xiaolongmen) showed a large genetic divergence with lineages in the MalAvi database (only 94 and 95 % similar, respectively, with the closed reported lineages, Table 1) and held a unique position on the phylogenetic tree (Fig. 2), suggesting that they may represent a new species or new subgenus.
Within the 13 previously reported lineages, EMEL01 (SGS1), the most abundant one in Plasmodium as several previous studies in Europe, e.g., Ferrer et al. (2012), was detected almost all over the world (MalAvi database), while EMEL03 (HLW1), which was the most frequently detected Haemoproteus lineage in the present study, was only found in Asia (Bensch et al. 2000; Scordato and Kardish 2014).
The different patterns inside the main clade in the phylogenetic tree may be due to a different evolutionary history of the parasite genera. As postulated generalist parasites, Plasmodium spp. continuously infect novel hosts to form their evolutionarily stable associations (Beadell et al. 2004); therefore, lineages found in different host species may be exactly the same, and different species were clearly separated from each other. In contrast, Haemoproteus spp. have strong host–family specificities (Beadell et al. 2004). It is reported that Haemoproteus parasites are able to avoid hybridization within a host species although there are ample opportunities for mixing (Pérez-Tris et al. 2007), resulting in sympatric speciation, where new species were formed with slight differences.
The Plasmodium lineage EMEL01 had a high host diversity index, but showed a contemporarily low temporal diversity, implying that this lineage infected a great diversity of bird species in a certain year, but maintained a lower infection level and even disappeared in the following years. Meanwhile, the Haemoproteus lineages ACAE01 and EMEL03 break out suddenly or after several years’ stable prevalent respectively. Previous study has shown that there is a strong effect of competitive exclusion between Haemoproteus and Plasmodium (Szöllözi et al. 2011). Hence, EMEL01 may fade away with the eruption of ACAE01 and EMEL03.
Furthermore, it is reported that in great reed warbler populations, the chronic stages of infection was correlated with parasitemia during the acute phase (Asghar et al. 2012) and varied over the years (Asghar et al. 2011). If these findings are common in other species, they may also lead to a variation in prevalence.
It is generally accepted that Plasmodium lineages have a wider host range at the family level than other blood parasites. However, in our study, they did not show a higher host diversity compared with Haemoproteus lineages. This may be because our collection mainly focused on passerines, while Haemoproteus specificity is sometimes not obvious within an order, which was considered to be its maximum theoretical possible level (Valkiūnas 2004). Further study of more taxa from different orders is needed to address this question.
References
Asghar M, Hasselquist D, Bensch S (2011) Are chronic avian haemosporidian infections costly in wild birds? J Avian Biol 42:530–537
Asghar M, Westerdahl H, Zehtindjiev P, Ilieva M, Hasselquist D, Bensch S (2012) Primary peak and chronic malaria infection levels are correlated in experimentally infected great reed warblers. Parasitology 139:1246–1252. doi:10.1017/s0031182012000510
Atkinson CT, Dusek RJ, Woods KL, Iko WM (2000) Pathogenicity of avian malaria in experimentally-infected Hawaii Amakihi. J Wildl Dis 36:197–201
Baillie SM, Gudex-Cross D, Barraclough RK, Blanchard W, Brunton DH (2012) Patterns in avian malaria at founder and source populations of an endemic New Zealand passerine. Parasitol Res 111:2077–2089. doi:10.1007/s00436-012-3055-y
Beadell JS et al (2004) Prevalence and differential host-specificity of two avian blood parasite genera in the Australo-Papuan region. Mol Ecol 13:3829–3844. doi:10.1111/j.1365-294X.2004.02363.x
Beadell JS et al (2006) Global phylogeographic limits of Hawaii’s avian malaria. Proc R Soc B-Biol Sci 273:2935–2944. doi:10.1098/rspb.2006.3671
Bensch S, Stjernman M, Hasselquist D, Örjan Ö, Hannson B, Westerdahl H, Pinheiro RT (2000) Host specificity in avian blood parasites: a study of Plasmodium and Haemoproteus mitochondrial DNA amplified from birds. Proc R Soc B-Biol Sci 267:1583–1589
Bensch S, Pérez-Tris J, Waldenström J, Hellgren O (2004) Linkage between nuclear and mitochondrial DNA sequences in avian malaria parasites: multiple cases of cryptic speciation? Evolution 58:1617–1621. doi:10.1554/04-026
Bensch S, Waldenström J, Jonzén N, Westerdahl H, Hansson B, Sejberg D, Hasselquist D (2007) Temporal dynamics and diversity of avian malaria parasites in a single host species. J Anim Ecol 76:112–122. doi:10.1111/j.1365-2656.2006.01176.x
Bensch S, Hellgren O, Pérez-Tris J (2009) MalAvi: a public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol Ecol Resour 9:1353–1358
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772–772
de Roode J et al (2005) Virulence and competitive ability in genetically diverse malaria infections. Proc Natl Acad Sci U S A 102:7624–7628
Dimitrov D, Valkiūnas G, Zehtindjiev P, Ilieva M, Bensch S (2013) Molecular characterization of haemosporidian parasites (Haemosporida) in yellow wagtail (Motacilla flava), with description of in vitro ookinetes of Haemoproteus motacillae. Zootaxa 3666(3):369–381
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Ferrer ES, Garcia-Navas V, Jose Sanz J, Ortego J (2012) Molecular characterization of avian malaria parasites in three Mediterranean blue tit (Cyanistes caeruleus) populations. Parasitol Res 111:2137–2142. doi:10.1007/s00436-012-3062-z
Greiner EC, Bennett GF, White EM, Coombs RF (1975) Distribution of the avian hematozoa of North America. Can J Zool 53:1762–1787
Hellgren O, Waldenström J, Bensch S (2004) A new PCR assay for simultaneous studies of Leucocytozoon, Plasmodium, and Haemoproteus from avian blood. J Parasitol 90:797–802. doi:10.1645/ge-184r1
Imura T et al (2012) Prevalence of avian haematozoa in wild birds in a high-altitude forest in Japan. Vet Parasitol 183:244–248. doi:10.1016/j.vetpar.2011.07.027
Knowles SC, Wood MJ, Alves R, Wilkin TA, Bensch S, Sheldon BC (2011) Molecular epidemiology of malaria prevalence and parasitaemia in a wild bird population. Mol Ecol 20:1062–1076
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Møller AP, Soler JJ, Vivaldi MM (2010) Spatial heterogeneity in distribution and ecology of Western Palearctic birds. Ecology 91:2769–2782. doi:10.1890/09-1300.1
Murata K (2002) Prevalence of blood parasites in Japanese wild birds. J Vet Med Sci 64:785–790
Outlaw DC, Ricklefs RE (2011) Rerooting the evolutionary tree of malaria parasites. Proc Natl Acad Sci U S A 108:13183–13187. doi:10.1073/pnas.1109153108
Palinauskas V, Kosarev V, Shapoval A, Bensch S, Valkiūnas G (2007) Comparison of mitochondrial cytochrome b lineages and morphospecies of two avian malaria parasites of the subgenera Haemamoeba and Giovannolaia (Haemosporida: Plasmodiidae). Zootaxa :39–50
Pérez-Tris J et al (2007) Within-host speciation of malaria parasites. PLoS One 2:e235. doi:10.1371/journal.pone.0000235
Piersma T, van der Velde M (2012) Dutch House Martins Delichon urbicum gain blood parasite infections over their lifetime, but do not seem to suffer. J Ornithol 153:907–912. doi:10.1007/s10336-012-0826-2
Ricklefs RE, Swanson BL, Fallon SM, Martinez-Abrain A, Scheuerlein A, Gray J, Latta SC (2005) Community relationships of avian malaria parasites in southern Missouri. Ecol Monogr 75:543–559. doi:10.1890/04-1820
Riper CV III, Riper SGV, Goff ML, Laird M (1986) The epizootiology and ecological significance of malaria in Hawaiian land birds. Ecol Monogr 56:327–344
Santiago-Alarcon D, Palinauskas V, Schaefer HM (2012) Diptera vectors of avian Haemosporidian parasites: untangling parasite life cycles and their taxonomy. Biol Rev 87:928–964. doi:10.1111/j.1469-185X.2012.00234.x
Scheuerlein A, Ricklefs RE (2004) Prevalence of blood parasites in European passeriform birds. Proc R Soc B-Biol Sci 271:1363–1370. doi:10.1098/rspb.2004.2726
Scordato ES, Kardish MR (2014) Prevalence and beta diversity in avian malaria communities: host species is a better predictor than geography. J Anim Ecol 83:1387–1397
Silva-Iturriza A, Ketmaier V, Tiedemann R (2012) Prevalence of avian haemosporidian parasites and their host fidelity in the central Philippine islands. Parasitol Int 61:650–657. doi:10.1016/j.parint.2012.07.003
Sol D, Jovani R, Torres J (2003) Parasite mediated mortality and host immune response explain age-related differences in blood parasitism in birds. Oecologia 135:542–547
Synek P, Albrecht T, Vinkler M, Schnitzer J, Votypka J, Munclinger P (2013) Haemosporidian parasites of a European passerine wintering in South Asia: diversity, mixed infections and effect on host condition. Parasitol Res 112:1667–1677. doi:10.1007/s00436-013-3323-5
Szöllözi E et al (2011) Determinants of distribution and prevalence of avian malaria in blue tit populations across Europe: separating host and parasite effects. J Evol Biol 24:2014–2024. doi:10.1111/j.1420-9101.2011.02339.x
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Valkiūnas G (2004) Avian malaria parasites and other haemosporidia. CRC Press, Boca Raton
Valkiūnas G, Bensch S, Iezhova TA, Krizanauskiene A, Hellgren O, Bolshakov CV (2006) Nested cytochrome B polymerase chain reaction diagnostics underestimate mixed infections of avian blood haemosporidian parasites: microscopy is still essential. J Parasitol 92:418–422. doi:10.1645/ge-3547rn.1
Valkiunas G, Krizanauskiene A, Iezhova TA, Hellgren O, Bensch S (2007a) Molecular phylogenetic analysis of circumnuclear hemoproteids (Haemosporida: Haemoproteidae) of sylviid birds, with a description of Haemoproteus parabelopolskyi sp. nov. J Parasitol 93:680–687. doi:10.1645/ge-1102r.1
Valkiunas G, Zehtindjiev P, Hellgren O, Ilieva M, Iezhova TA, Bensch S (2007b) Linkage between mitochondrial cytochrome b lineages and morphospecies of two avian malaria parasites, with a description of Plasmodium (Novyella) ashfordi sp. nov. Parasitol Res 100:1311–1322. doi:10.1007/s00436-006-0409-3
Valkiunas G, Zehtindjiev P, Dimitrov D, Krizanauskiene A, Iezhova TA, Bensch S (2008) Polymerase chain reaction-based identification of Plasmodium (Huffia) elongatum, with remarks on species identity of haemosporidian lineages deposited in GenBank. Parasitol Res 102:1185–1193. doi:10.1007/s00436-008-0892-9
Woodworth BL et al (2005) Host population persistence in the face of introduced vector-borne diseases: Hawaii amakihi and avian malaria. Proc Natl Acad Sci U S A 102:1531–1536
Zehtindjiev P, Ivanova K, Mariaux J, Georgiev BB (2013) First data on the genetic diversity of avian haemosporidians in China: cytochrome b lineages of the genera Plasmodium and Haemoproteus (Haemosporida) from Gansu Province. Parasitol Res 112:3509–3515
Zhang Y, Wu Y, Zhang Q, Su D, Zou F (2014) Prevalence patterns of avian Plasmodium and Haemoproteus parasites and the influence of host relative abundance in southern China. PLoS One 9:e99501
Acknowledgments
We are grateful that the project was supported by the State Key Laboratory of Earth Surface Processes and Resource Ecology, Beijing Normal University (No. 2012-ZY-08) and to the Fundamental Research Funds for the Central Universities (2013NT35) for the support to LD, the Monitering Fund for the Blood Parasites of Wild Birds in Beijing supported by the State Forestry Administration of China to Yanyun Zhang, Jie Zhou for the lab work assistance, and all volunteers from the College of Life Sciences, Beijing Normal University, for the field work assistance.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
(DOCX 25 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Huang, X., Dong, L., Zhang, C. et al. Genetic diversity, temporal dynamics, and host specificity in blood parasites of passerines in north China. Parasitol Res 114, 4513–4520 (2015). https://doi.org/10.1007/s00436-015-4695-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-015-4695-5