
Abstract
Background
Head and neck squamous cell carcinoma (HNSCC) is a significant health concern with a variable global incidence and is linked to regional lifestyle factors and HPV infections. Despite treatment advances, patient prognosis remains variable, necessitating an understanding of its molecular mechanisms and the identification of reliable prognostic biomarkers.
Methods
We analyzed 959 HNSCC samples and employed batch correction to obtain consistent transcriptomic data across cohorts. We examined 79 disulfidptosis-related genes to determine consensus clusters and utilized high-throughput sequencing to identify genetic heterogeneity within tumors. We established a disulfidptosis prognostic signature (DSPS) using least absolute shrinkage and selection operator (LASSO) regression and developed a prognostic nomogram integrating the DSPS with clinical factors. Personalized chemotherapy prediction was performed using the "pRRophetic" R package.
Results
Batch corrections were used to harmonize gene expression data, revealing two distinct disulfidptosis subtypes, C1 and C2, with differential gene expression and survival outcomes. Subtype C1, characterized by increased expression of the MYH family genes ACTB, ACTN2, and FLNC, had a mortality rate of 48.4%, while subtype C2 had a mortality rate of 38.7% (HR = 0.77, 95% CI: 0.633–0.934, P = 0.008). LASSO regression identified 15 genes that composed the DSPS prognostic model, which independently predicted survival (HR = 2.055, 95% CI: 1.420–2.975, P < 0.001). The prognostic nomogram, which included the DSPS, age, and tumor stage, predicted survival with AUC values of 0.686, 0.704, and 0.789 at 3, 5, and 8 years, respectively, indicating strong predictive capability. In the external validation cohort (cohort B), the DSPS successfully identified patients at greater risk, with worse overall survival outcomes in the high-DSPS subgroup (HR = 1.54, 95% CI: 1.17–2.023, P = 0.002) and AUC values of 0.601, 0.644, 0.636, and 0.748 at 3, 5, 8, and 10 years, respectively, confirming the model's robustness.
Conclusion
The DSPS provides a robust prognostic tool for HNSCC, underscoring the complexity of this disease and the potential for tailored treatment strategies. This study highlights the importance of molecular signatures in oncology, offering a step toward personalized medicine and improved patient outcomes in HNSCC management.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is recognized as a significant global public health concern, representing a diverse group of cancers that affect various regions, including the oral cavity, pharynx, hypopharynx, larynx, nasal cavity, and salivary glands, and is characterized by high incidence and mortality rates. The incidence of HNSCC varies worldwide and is influenced by regional factors such as tobacco and alcohol use, dietary habits, and the prevalence of human papillomavirus (HPV) infections (Barsouk et al. 2023; Kumar et al. 2015; Auguste et al. 2020). The elevated incidence of HNSCC in Southeast Asia and Australia correlates with the intake of particular products containing carcinogens. Concurrently, the increasing incidence of oropharyngeal infections due to HPV has been linked to the increased incidence of HNSCC in the USA and Western Europe (Mehanna et al. 2013; Kanwal et al. 2019; Su et al. 2016).
Despite advancements in diagnosis and treatment, the prognosis for HNSCC patients remains variable and often depends on the stage at diagnosis, the tumor's location, and the patient's overall health. HNSCC constitutes approximately 95% of all head and neck cancers, contributes to more than 316,000 deaths annually worldwide (Burden et al. 1990), and is the sixth most prevalent malignancy (Mody et al. 2021). Predictive analyses indicate a projected 30% increase in the risk of HNSCC by the year 2030, with an anticipated 1.08 million new cases annually (Antra 2022). Moreover, survival rates for HNSCC patients have improved moderately over the last 30 years; for instance, the 5-year survival rate increased from 55% in 1992–1996 to 66% in 2002–2006, as per the Surveillance, Epidemiology, and End Results registry data encompassing all age groups and anatomical locations (Pulte and Brenner 2010).
The mainstay treatment strategies for HNSCC include surgery, radiation therapy, and chemotherapy. Despite advancements in the therapeutic landscape, the prognosis for patients with HNSCC continues to be challenging, primarily due to delayed diagnosis, frequent recurrence at the primary site, and lymphatic metastasis. (Anderson et al. 2021). These complications have curtailed any marked improvement in long-term survival rates. While early-stage oral cavity cancers may be treated with surgery alone and laryngeal cancers with surgery or radiation, the treatment for the majority of HNSCC patients typically necessitates a combination of modalities, requiring collaborative multidisciplinary management. The overexpression of epidermal growth factor receptor (EGFR) in more than 90% of HNSCC patients led to the approval of cetuximab (Erbitux), a monoclonal antibody targeting EGFR, in combination with radiation therapy for advanced local/regional carcinoma. However, the response rate to cetuximab in HNSCC patients is less than 20% (Specenier and Vermorken 2013). Current immunotherapeutic approaches being investigated include immune checkpoint inhibitors, costimulatory agonists, vaccines targeting specific antigens, oncolytic virus therapy, adoptive T-cell transfer, and therapies targeting EGFR (Yu et al. 2022).
Recent years have seen pivotal advancements in understanding the molecular mechanisms driving HNSCC. High-throughput sequencing technologies have revealed considerable genetic heterogeneity within HNSCC tumors, identifying key alterations in tumor suppressor genes such as TP63 and oncogenes such as EGFR and PIK3CA (Alsahafi et al. 2019). TP63 is detected in approximately 80% of HNSCCs; ΔNp63 is predominantly implicated in the pathogenesis of HNSCC, modulates critical pathways such as cell survival and renewal, inhibits senescence by repressing p16/INK4A, and governs growth factor signaling (Si et al. 2016; Rocco et al. 2006). Additionally, a genomic analysis by the Cancer Genome Atlas in 2015 revealed inactivating mutations in the NOTCH1-3 gene in 17% of HPV-positive and 26% of HPV-negative HNSCC patients (Cancer Genome Atlas 2015). Moreover, PI3K/Akt/mTOR pathway disruptions are commonly observed in HNSCC, with mutations in the PIK3CA gene present in approximately 16% of cases, suggesting that this gene is a potential target for therapeutic intervention (Kang et al. 2015). Recent days, the prognostic model based on varies omics data for HNSCC were also generated. Increasing evidence has demonstrated that small nucleolar RNAs (snoRNAs) play an important role in tumorigenesis; a risk model based on SNORD114-17, SNORA36B, SNORD78, U3: ENSG00000212182, and U3: ENSG00000212195 is reported as a prognostic marker for HNSCC (Xing et al. 2020a), as well as the five-pseudogene signature (Xing et al. 2020b) (LILRP1, RP6-191P20.5, RPL29P19,TAS2R2P, and ZBTB45P1) and the six-MPS model (Xing et al. 2020c) (hsa00290, hsa01230, hsa00430, hsa00380, hsa00232, and hsa00534).
The current academic discourse around HNSCC is focused on improving the understanding of the disease pathogenesis, optimizing existing treatment protocols, and developing novel therapeutic strategies to improve patient outcomes. Aberrant accumulation of intracellular disulfides, such as cystine, induces disulfide stress and can be highly toxic to cells (Liu et al. 2020; Joly et al. 2020). Liu et al. (2023) reported that aberrant accumulation of intracellular disulfides in SLC7A11-high cells under glucose starvation induces a previously uncharacterized form of cell death distinct from apoptosis and ferroptosis, termed as disulfidptosis, which might be promoted by 90 proteins reflected by proteomic analysis. In the current study, we aim to evaluate the potential prognostic value of disulfidptosis-associated genes in HNSCC, to provide novel insights.
Methods
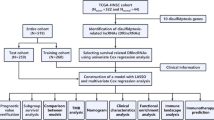
Cohort information
In this study, we collected 959 HNSCC samples from four cohorts, most of which were from the larynx, oral cavity, oropharynx, and tongue. The HNSCC cohort, comprising 509 patients, had an average age of 60.8 years, with the majority being male (74.3%). Additionally, 58.2% of the patients were alive, with an average overall survival of 30.7 months. In the EMTAB8588 cohort of 83 patients, the average age was slightly lower at 58.5 years, with a significant male majority (90.4%). A total of 63.9% of patients died, and the mean overall survival time was 58.5 months. In the GSE41613 cohort, 97 HNSCC patients, including 31 females and 66 males, met the end-of-life criteria, and the average survival time was approximately 44.1 months. Finally, the GSE65858 cohort included 270 patients, with an average age of 60.1 years and a high male predominance (82.6%). The mean overall survival was the lowest at 29.0 months, with 34.8% of the patients dying. The baseline information for patients in both cohorts is presented in Table 1.
Mitigating batch effects
Batch effects represent the nonbiological discrepancies observed across multiple datasets. To ensure analytical consistency and mitigate biases introduced by such effects, we employed the ComBat algorithms from the "sva" package. This methodology was instrumental in harmonizing the transcriptional profiles of all the enrolled cohorts, thus effectively offsetting the intrinsic batch differences among them. Subsequently, all four cohorts were used for consensus clustering, and in the prediction of the prognostic model, the training cohort, called Cohort A, was combined with the EMTAB8588, GSE41613 and GSE65858 cohorts, while the TCGA-HNSCC cohort was used as validation Cohort B.
Comparison of genes associated with disulfidptosis
Liu and colleagues delineated a novel classification system for disulfidptosis. They observed that cells with elevated levels of SLC7A11, when exposed to glucose deficiency, experienced increased uptake of cystine. This surge, concomitant with an insufficient supply of NADPH, precipitates the exhaustion of NADPH stores, leading to improper disulfide bond formation in actin cytoskeletal proteins, disassembly of the actin network, and subsequent cellular collapse (Liu et al. 2023). A total of 90 cysteine residues exhibited a 1.5-fold increase in disulfide bonding after the cells were subjected to glucose deficit. These residues were encoded by 77 distinct genes, with their identifiers collated from Supplementary Table 2 in the study by Liu et al.
Elucidating distinct disulfidptosis phenotypes
To discern molecular subtypes, consensus clustering was performed using the "ConsensusClusterPlus" package in R (Wilkerson and Hayes 2010). This process involved 50 iterations of k-means clustering, with each iteration incorporating at least 80% of the sample set. The cumulative distribution function (CDF) score—representative of the area beneath the CDF curve—was computed to ascertain the optimal number of clusters. Both principal component analysis (PCA) and t-distributed stochastic neighbor embedding (t-SNE) were utilized to validate the integrity of the consensus clusters.
Gene set enrichment analysis
The identification of differentially expressed genes (DEGs) is pivotal for revealing distinct mechanistic pathways across subgroups. The 'limma' package in R facilitated the compilation of DEGs, setting an adjusted P value of less than 0.01 as the threshold for selection. Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and HALLMARK enrichment analyses were conducted using the 'org.Hs.eg.db' and 'msigdbr' packages (Ashburner et al. 2000; Liberzon et al. 2015), while 'ClusterProfiler' elucidated key signaling pathways (Yu et al. 2012).
Formulating the prognostic index
Prognostic genes were initially identified through univariate Cox regression in Cohorts A and B. A Venn diagram was then constructed to highlight the prognostic genes across both cohorts for further examination. The 'glmnet' package in R facilitated the implementation of LASSO regression analysis to distill and regularize input variables, thereby deriving a prognostic model of superior predictive power and interpretability (Friedman et al. 2010). LASSO is a regression analysis method that performs both variable selection and regularization to enhance the prediction accuracy and interpretability of the resulting statistical model. Genes identified through LASSO were instrumental in computing each patient's risk score, and the disulfidptosis prognostic signature (DSPS) was established through the aggregation of gene expression levels and corresponding coefficients, where DSPS = sum (gene expression × corresponding coefficient).
Construction of a predictive nomogram
Comprehensive multivariate Cox regression analysis revealed the preliminary phase in identifying independent prognostic factors. Following this, the DSPR signature was incorporated into the model. A nomogram illustrating the independent prognostic factors to consolidate the forecasted outcomes was subsequently developed using the "rms" and "regplot" R packages. Calibration plots, decision curve analysis (DCA), and clinical impact curve assessments substantiated the precision and dependability of our prognostic data.
Personalized chemotherapy prediction
The "pRRophetic" R package predicted individual chemotherapeutic responses by harnessing drug sensitivity and phenotypic data from GDSC 2016 (Geeleher et al. 2014a). Ridge regression yielded estimated IC50 values for each specimen in response to specific chemotherapeutic agents, with lower IC50 values indicating greater susceptibility to treatment (Arthur 2000), while the prediction accuracy was verified through a tenfold cross-validation method (Geeleher et al. 2014b).
Statistical analysis
The t test is a parametric test used for comparing the means of two groups, assuming a normal distribution and equal variances. Moreover, the Mann‒Whitney test is a nonparametric test used to compare the distributions of two groups without assuming a specific data distribution and is more robust to outliers. K‒M survival analysis and log-rank tests were used to generate survival curves, while univariate Cox regression models were used to calculate hazard ratios (HRs) and 95% confidence intervals (CIs) for selected genes. Multivariate Cox regression confirmed the independent prognostic significance of the risk score after adjusting for various clinical parameters. Statistical significance was determined using a two-tailed P value of less than 0.05, with all the statistical analyses performed in R software (version 4.2.2).
Results
Unveiling subclasses within the definition of disulfidptosis
Our study included 959 HNSCC patient samples from four cohorts, as outlined in “Methods”. To ensure the consistency of the transcriptomic data prior to further analysis, we initially performed batch correction on the gene expression profiles from all four cohorts. Prior to this correction, principal component analysis (PCA) graphs exhibited notable differences between the four cohorts (Fig. 1A). However, postcorrection, batch effects on gene expression distribution across all cohorts were effectively mitigated (Fig. 1B).

Batch correction and consensus clustering in HNSCC cohorts. A Raw distribution of gene expression of samples in the enrolled cohorts before batch correction. B Distribution of samples in the combined expression profile after batch correction. C Consensus cumulative distribution function (CDF) for k = 2 to 6 clusters. D Delta area under the CDF curve for different k values. E Consensus matrix for k = 2 indicating two distinct clusters
As detailed in “Methods”, we first collated 79 disulfidptosis-related genes, 68 of which were uniformly distributed across the included cohorts. Hence, the gene expression profiles of these 68 genes were gathered to serve as the input matrix for clustering. Consensus matrices for k values from 2 to 6 were tested (Fig. 1C). The cumulative distribution function (CDF) curves revealed that the area under the CDF did not change drastically with the increase in the number of k after 2 (Fig. 1D), and the relative change in the CDF was maximal between k = 2 and k = 3. Therefore, we determined that k = 2 was the optimal number of clusters for subsequent analysis (Fig. 1E).
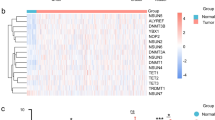
Thus, the 959 patients from all cohorts were classified into two subgroups, C1 and C2, based on the expression of 68 genes (Fig. 2A). Patients in subgroup C1 exhibited increased expression of genes from the MYH family, ACTB, ACTN2, and FLNC, whereas patients in subgroup C2 displayed increased expression of other genes, particularly AAAS, ARMC6, SART3, and CHCHD3. A random forest classification based on the expression profiles of the 68 genes also confirmed the distinction between the two groups (Fig. 2B). The prognosis of patients from different subtypes was examined. There were 411 individuals in subtype C1, with 199 deaths, representing a mortality rate of 48.4%; subtype C2 comprised 548 individuals, with 212 deaths, representing a mortality rate of 38.7%, indicating a better prognosis for patients in subtype C2 (HR = 0.77, 95% CI: 0.633–0.934, P = 0.008; Fig. 2C).

Subclassification of HNSCC based on disulfidptosis-related genes. A Heatmap displaying the expression profiles of 68 genes in the two subgroups. B The results of random forest showing the separation of the C1 and C2 subgroups. C Kaplan‒Meier survival curves comparing the prognosis of patients in subgroups C1 and C2
Key genes and pathways impacted by disulfidptosis
Due to the significant differences in OS between subtypes C1 and C2, we compared the DEGs between these subtypes. Using a P value threshold of less than 0.05, we identified 4943 DEGs. Moreover, to investigate the potential functional mechanisms of these genes, we conducted pathway enrichment analyses, which suggested that disulfidptosis may affect signaling pathways related to extracellular matrix structure, cellular development and differentiation, cell adhesion, myeloid leukocyte chemotaxis, migration, and mitochondrial function (Fig. 3A). Additionally, HALLMARK terms indicated that these genes were involved in signaling pathways related to epithelial–mesenchymal transition, cell cycle-associated E2F targets, MYC targets, and the G2M checkpoint (Fig. 3B).

Pathway enrichment analysis and prognostic gene isolation. A Gene Ontology term enrichment analysis for identified differentially expressed genes. B HALLMARK pathway enrichment analysis highlighting key biological processes. C Volcano plot of prognostic genes in Cohort A. D Volcano plot of prognostic genes in Cohort B. E Venn diagram showing overlap of risk-related and protective genes across cohorts
Disulfidptosis-related genes associated with patient prognosis
We assessed the prognostic significance of these 4943 genes in Cohort A and Cohort B. In Cohort A, we identified 659 risk-related genes (HR > 1, P < 0.05) and 468 protective genes (HR < 1, P < 0.05) (Fig. 3C), and in Cohort B, we found 98 risk-related genes (HR > 1, P < 0.05) and 87 protective genes (HR < 1, P < 0.05) (Fig. 3D). After filtering for genes that exhibited similar trends in both cohorts, we identified 13 risk-related genes and 9 protective genes associated with the prognosis of HNSCC patients (Fig. 3E).
Prognostic model based on disulfidptosis-related genes
We included the 22 genes identified via LASSO regression to further filter HNSCC prognosis-related genes and establish a prognostic prediction signature. In the LASSO pathway diagram (Fig. 4A), each line represents a gene in the model. With the smallest lambda value at 0.0269, the partial likelihood deviation also met the minimum, reflecting the optimal formula and preventing overfitting (Fig. 4B). Ultimately, the LASSO regression model selected 15 out of the 22 input genes, constructing a prognostic feature known as the DSPS. DSPS = (MRPS17 × 0.076) + (LPL × 0.075) + (CHCHD2 × 0.133) + (FAM98A × 0.235) + (CXCL1 × 0.006) + (SURF4 × 0.375) + (PNMA1 × 0.112) + (RAI14 × 0.04) + (HSD17B12 × 0.146) + (C4orf19 × − 0.007) + (IP6K2 × − 0.224) + (PTPN13 × − 0.208) + (STAR × − 0.021) + (PITX1 × − 0.035) + (ZBTB48 × − 0.013). Subsequently, we calculated a risk score for each patient in Cohort A based on the DSPS, with the dot plot on the left displaying each patient's calculated DSPS, the scatter plot in the middle depicting each patient's status, and the heatmap on the right showing the expression of the 15 genes between the two groups (Fig. 4C).

Development of the disulfidptosis prognostic signature (DSPS). A LASSO coefficient profiles of the disulfidptosis-related genes. B Selection of the optimal lambda value in the LASSO model. C Risk score distribution, patient status, and gene expression heatmap for the DSPS model. D Kaplan‒Meier curves for the high- and low-DSPS groups. E ROC curves evaluating the prognostic accuracy of the DSPS
Patients were then divided into low- and high-score groups, with those in the high-DSPS group exhibiting poorer OS than those in the low-DSPS group, with a hazard ratio of 2.44 and 95% confidence interval ranging from 1.818 to 3.283 (Fig. 4D). The ROC curve was then used to assess the predictive value of the overall clinical outcomes, showing 3-year AUCs of 0.676, 5-year AUCs of 0.683, 8-year AUCs of 0.782, and 10-year AUCs of 0.766 (Fig. 4E). After excluding the potential influences of other clinical parameters (such as patient age, sex, primary tumor location, tumor stage, smoking history, and alcohol consumption history), the DSPS was determined to be an independent prognostic indicator (HR = 2.055, 95% CI: 1.420–2.975, P < 0.001), as was tumor stage (stage IVB: HR = 3.13, 95% CI: 1.196–8.193, P = 0.02; stage IVC: HR = 2.785, 95% CI: 1.001–7.748, P = 0.05; Table 2).
Molecular mechanisms and suitable chemotherapeutic drugs for the DSPS subgroups
To understand the underlying molecular characteristics of patients divided into high-DSPS and low-DSPS subgroups, or to explore the molecular mechanisms underlying the different clinical outcomes of the two groups, we evaluated the activation levels of 50 cancer-related pathways from the HALLMARK cohort to assess the unique molecular mechanism features of the two subgroups. We observed that in the high-DSPS subgroup, pathways related to epithelial–mesenchymal transition, apical junction, and immune-related pathways exhibited increased activation, while in the low-DSPS subgroup, pathways related to estrogen response, fatty acid metabolism, and KRAS signaling exhibited increased activation (Fig. 5A).

Molecular mechanisms and drug susceptibility of the DSPS subgroups. A Pathway activation levels in the high- vs. low-DSPS subgroups. B Differential drug sensitivity analysis between the DSPS subgroups
Further studies aimed at guiding clinical treatment for HNSCC utilized the "pRRophetic" R package to analyze the differences in the efficacy of 86 potential chemotherapeutic drugs between the different subgroups. Patients in the low-DSPS group benefited more from treatment with drugs such as 5-fluorouracil, A-443654, BI-2536, BAY 61-3606, GNF-2, GSK-650394, GW-2580, GW843682X, erlotinib, crizotinib, CP466722, KIN001-135, lapatinib, PAC-1, paclitaxel, PHA-665752, phenformin, and rapamycin, while patients with a high-DSPS score might benefit more from treatment with AUY922, bortezomib, HG-6-64-1, GSK269962A, FTI-277, embelin, CHIR-99021, bexarotene, AZ628, JNK-9 L, JQ12, linsitinib, MG-132, midostaurin, Obatoclax mesylate, thapsigargin, WH-4-023, and TGX221 (all P < 0.05, Fig. 5B).
Clinical characteristics and the DSPS used to construct the prognostic nomogram
Multivariate Cox regression analysis revealed that age, tumor stage, and the DSPS are independent factors for the prognosis of HNSCC patients. Hence, we constructed a prognostic prediction nomogram based on these three indicators (Fig. 6). For instance, as marked with red lines and dots, a 70-year-old male HNSCC patient with a postoperative tumor stage of IV and a DSPS of 8.3 had a cumulative score of 193, corresponding to a 1-year mortality risk of 0.524, a 3-year mortality risk of 0.885, and a 5-year mortality risk of 0.981 (Fig. 6).

Prognostic prediction nomogram incorporating the clinical characteristics and the DSPS
Based on the aforementioned nomogram, we calculated points for all patients and found that those who died had significantly more points than those who still survived at the last follow-up (P < 0.01, Fig. 7A). Dividing the patients according to the median points into high and low groups revealed that patients with higher points had a worse prognosis (HR = 2.31, 95% CI: 1.641–3.256, P < 0.001; Fig. 7B). The predictive accuracy of the nomogram was further improved, with 3-year AUC values of 0.686,5-year AUC values of 0.704, and 8-year AUC values of 0.789 (Fig. 7C). Calibration curves also indicated a high consistency between the nomogram's prognostic prediction and the actual outcomes, with Hosmer–Lemeshow P values at 1 year, 3 years, and 5 years of 0.199, 0.262, and 0.245, respectively, confirming the nomogram's commendable ability to predict clinical outcomes (Fig. 7D). Clinical impact curves suggested that treating all patients with a predicted clinical death probability greater than 70% could yield the most significant clinical benefit (Fig. 7E). Decision curve analysis (DCA) demonstrated that using the DSPS feature to predict the risk of death provided an equal or greater benefit than treating all or no patients when the threshold probability exceeded 20% (Fig. 7F).

Validation and clinical utility of the prognostic nomogram. A Boxplot showing the nomogram point distribution by survival status. B Kaplan‒Meier curves for risk stratification based on nomogram points. C ROC curve analysis of the nomogram's prediction accuracy. D Calibration curves for the nomogram at 1, 3, and 5 years. E Clinical impact curves demonstrating the benefit of nomogram-based risk stratification. F DCA showing the net benefit of the nomogram across different risk thresholds
The DSPS successfully identified high-risk patients in the validation cohort
Using the DSPS formula derived from the LASSO analysis based on Cohort A, we calculated the risk score for each patient in the validation cohort, Cohort B (Fig. 8A). Patients were divided into low and high-DSPS groups, with the high-DSPS group showing worse overall survival outcomes than the low-DSPS group (HR = 1.54, 95% CI: 1.17–2.023; P = 0.002; Fig. 8B). Furthermore, the AUC values demonstrated satisfactory results, with 3-year survival at 0.601, 5-year survival at 0.644, 8-year survival at 0.636, and 10-year survival at 0.748 (Fig. 8C). After conducting multifactorial Cox regression analysis to eliminate the impact of other factors on prognosis, after adjusting for factors such as sex, age, tumor stage, and history of alcohol consumption, the DSPS could serve as an independent prognostic indicator (HR = 1.542, 95% CI: 1.137–2.091, P = 0.005; Table 3).

Validation of the DSPS in an external cohort. A Distribution of risk scores and survival status in the validation cohort. B Kaplan‒Meier curves for the high- and low-DSPS groups in the validation cohort. C ROC curves evaluating the prognostic accuracy of the DSPS
Discussion
The notion of disulfidptosis and its link to HNSCC prognosis has been substantiated by our extensive cohort analysis, underscoring the use of the DSPS as a robust prognostic indicator. This study, encompassing diverse patient datasets, has meticulously accounted for batch effects, thus bolstering the integrity of the gene expression data that underpins our signature. We firstly identified two disulfidptosis subtypes by consensus clustering, and then 4943 differentially expressed genes between the above two subtypes were selected and their prognostic value evaluated in two independent clinical cohorts. Thirdly, 13 risk-related genes and 9 protective genes associated with the prognosis of HNSCC patients were chosen. Finally, the prognostic DPSP signature was generated by the LASSO regression analysis.
Our findings indicate that subtype C2, characterized by lower mortality rates, may represent a less aggressive form of HNSCC. In contrast, the higher mortality of subtype C1 could indicate a more invasive disease trajectory and greater expression of genes from the MYH family, ACTB, ACTN2, and FLNC. Several studies have reported the prognostic value of the MYH family genes MYL1, MYL2, MYH1, MYH2, and MYH7, which are unfavorable prognostic markers in HNSCC and might promote CD4 + T-cell activation (Li et al. 2023; Ju et al. 2023). The level of phosphorylated MYH2-Y1381 is reportedly significantly lower in recurrent HNSCC patients than in primary patients (Kaneko et al. 2022). Hub genes might be pivotal components that accelerate the progression of HNSCC. Zhang et al. (2018) identified ten hub genes by weighted gene coexpression network analysis, indicating the influence of MMP1, TNFRSF12A, PLAU, FSCN1, PDPN, KRT78, EVPL, GGT6, SMIM5, and CYSRT1. Focusing on the hub genes might support molecular targeted therapeutic drug development.
The constructed DSPS, derived from LASSO regression analysis, encapsulates the prognostic importance of disulfidptosis-related genes, including MRPS17, LPL, CHCHD2, FAM98A, CXCL1, SURF4, PNMA1, RAI14, HSD17B12, C4orf19, IP6K2, PTPN13, STAR, PITX1, and ZBTB48. CXCL1 plays a role in the etiology of HNSCC, with its overexpression observed in fibroblasts within oral submucous fibrosis—a precancerous condition. This chemokine facilitates oncogenic activities in oral submucous fibrosis by inducing keratinocyte proliferation and migration and enhancing the stem-like properties of these cells (Ye et al. 2018). FAM98A was shown to promote the progression of endometrial carcinoma (Li et al. 2019), non-small cell lung cancer (Zheng et al. 2018), and breast cancer (Liu et al. 2021), and we also revealed the role of FAM98A in the risk of HNSCC in the present study. SURF4 interacts with the ERGIC53 and p25 proteins and engages with STIM1 within the endoplasmic reticulum (ER) lumens. It plays a regulatory role in STIM1-mediated store-operated calcium entry (SOCE), a fundamental mechanism for calcium influx in cells (Fujii et al. 2012). In tumors, SURF4 reportedly promotes tumorigenesis in breast cancer (Zhai et al. 2022), ovarian cancer (Yue et al. 2020), and myeloid leukemia (Kim et al. 2023).
Furthermore, the DSPS has proven to be an independent prognostic factor, suggesting that it could be used alongside traditional staging systems to more accurately predict patient outcomes. The construction of a prognostic nomogram that integrates the DSPS with clinical characteristics such as age and tumor stage represents a significant step toward personalized medicine. The nomogram's predictive accuracy, as indicated by the area under the curve (AUC) values and calibration curves, enhances its clinical utility, potentially aiding clinicians in making informed decisions about patient management.
The molecular pathways associated with disulfidptosis revealed in our study shed light on potential mechanisms that could drive the observed prognostic differences. For instance, the activation of pathways related to epithelial–mesenchymal transition and immune response in the high-DSPS subgroup points to a more aggressive disease phenotype, which is corroborated by their poorer prognosis. These pathways may serve as targets for novel therapies that could mitigate the aggressive nature of HNSCC in these patients. Moreover, our study's personalized chemotherapy prediction suggested that the DSPS can inform treatment choices. Patients in the low-DSPS group could benefit from a range of drugs, including well-established agents such as 5-fluorouracil, indicating the potential for individualized treatment plans based on the molecular profile of the tumor.
Our study, however, is not without limitations. Although the DSPS holds promise, its application in clinical practice requires further validation in prospective studies. The cohorts used in this study, although diverse, may not capture all the nuances of the global HNSCC population. Additionally, the mechanisms underlying disulfidptosis and its impact on HNSCC progression need to be elucidated through functional studies.
Conclusion
In summary, the DSPS has emerged as a potent prognostic tool that captures the complexity of HNSCC. This study provides insight into the future of oncology, where molecular signatures could guide therapeutic decisions and improve patient outcomes. As we move toward a more tailored approach to cancer treatment, the DSPS stands as a testament to the power of genomic medicine in improving the prognosis and management of HNSCC.
Data availability
The datasets analyzed during the current study are available from the corresponding author upon reasonable request.
References
Alsahafi E, Begg K, Amelio I, Raulf N, Lucarelli P, Sauter T, Tavassoli M (2019) Clinical update on head and neck cancer: molecular biology and ongoing challenges. Cell Death Dis 10(8):540
Anderson G, Ebadi M, Vo K, Novak J, Govindarajan A, Amini A (2021) An updated review on head and neck cancer treatment with radiation therapy. Cancers (basel) 13(19):4912
Antra, Parashar P, Hungyo H, Jain A, Ahmad S, Tandon V (2022) Unraveling molecular mechanisms of head and neck cancer. Crit Rev Oncol Hematol 178:103778
Arthur E, Hoerl RWK (2000) Ridge regression: biased estimation for nonorthogonal problems. Technometrics 42(1):80–86
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT et al (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25(1):25–29
Auguste A, Deloumeaux J, Joachim C, Gaete S, Michineau L, Herrmann-Storck C, Duflo S, Luce D (2020) Joint effect of tobacco, alcohol, and oral HPV infection on head and neck cancer risk in the French West Indies. Cancer Med 9(18):6854–6863
Barsouk A, Aluru JS, Rawla P, Saginala K, Barsouk A (2023) Epidemiology, risk factors, and prevention of head and neck squamous cell carcinoma. Med Sci (basel). https://doi.org/10.3390/medsci11020042
Global Burden of Disease Cancer C, Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R et al (2017) Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol 3(4):524–548
Cancer Genome Atlas N (2015) Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517(7536):576–582
Friedman J, Hastie T, Tibshirani R (2010) Regularization paths for generalized linear models via coordinate descent. J Stat Softw 33(1):1–22
Fujii Y, Shiota M, Ohkawa Y, Baba A, Wanibuchi H, Kinashi T, Kurosaki T, Baba Y (2012) Surf4 modulates STIM1-dependent calcium entry. Biochem Biophys Res Commun 422(4):615–620
Geeleher P, Cox N, Huang RS (2014a) pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 9(9):e107468
Geeleher P, Cox NJ, Huang RS (2014b) Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol 15(3):R47
Joly JH, Delfarah A, Phung PS, Parrish S, Graham NA (2020) A synthetic lethal drug combination mimics glucose deprivation-induced cancer cell death in the presence of glucose. J Biol Chem 295(5):1350–1365
Ju G, Yao Z, Zhao Y, Zhao X, Liu F (2023) Data mining on identifying diagnosis and prognosis biomarkers in head and neck squamous carcinoma. Sci Rep 13(1):10020
Kaneko T, Zeng PYF, Liu X, Abdo R, Barrett JW, Zhang Q, Nichols AC, Li SS (2022) Proteome and phosphoproteome signatures of recurrence for HPV(+) head and neck squamous cell carcinoma. Commun Med (lond) 2:95
Kang H, Kiess A, Chung CH (2015) Emerging biomarkers in head and neck cancer in the era of genomics. Nat Rev Clin Oncol 12(1):11–26
Kanwal M, Haider G, Zareef U, Saleem S (2019) Addiction of tobacco chewing and smoking in the patients of head and neck squamous cell carcinoma: a descriptive epidemiological study in Pakistan. Pak J Med Sci 35(6):1712–1717
Kim J, Lee H, Hong CM, Nam JH, Yeo HJ, Cho WH, Kim HS, Hong C, Kim YH, Lee D (2023) Novel endogenous endoplasmic reticulum transmembrane protein SURF4 suppresses cell death by negatively regulating the STING-STAT6 axis in myeloid leukemia. Cancer Commun (lond) 43(3):395–399
Kumar R, Rai AK, Das D, Das R, Kumar RS, Sarma A, Sharma S, Kataki AC, Ramteke A (2015) Alcohol and tobacco increases risk of high risk HPV infection in head and neck cancer patients: study from north-east region of India. PLoS ONE 10(10):e0140700
Li Z, Li N, Sun X, Wang J (2019) FAM98A promotes cancer progression in endometrial carcinoma. Mol Cell Biochem 459(1–2):131–139
Li C, Guan R, Li W, Wei D, Cao S, Chang F, Wei Q, Wei R, Chen L, Xu C et al (2023) Analysis of myosin genes in HNSCC and identify MYL1 as a specific poor prognostic biomarker, promotes tumor metastasis and correlates with tumor immune infiltration in HNSCC. BMC Cancer 23(1):840
Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P (2015) The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1(6):417–425
Liu X, Olszewski K, Zhang Y, Lim EW, Shi J, Zhang X, Zhang J, Lee H, Koppula P, Lei G et al (2020) Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat Cell Biol 22(4):476–486
Liu T, Wang Z, Dong M, Wei J, Pan Y (2021) MicroRNA-26a inhibits cell proliferation and invasion by targeting FAM98A in breast cancer. Oncol Lett 21(5):367
Liu X, Nie L, Zhang Y, Yan Y, Wang C, Colic M, Olszewski K, Horbath A, Chen X, Lei G et al (2023) Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol 25(3):404–414
Mehanna H, Beech T, Nicholson T, El-Hariry I, McConkey C, Paleri V, Roberts S (2013) Prevalence of human papillomavirus in oropharyngeal and nonoropharyngeal head and neck cancer—systematic review and meta-analysis of trends by time and region. Head Neck 35(5):747–755
Mody MD, Rocco JW, Yom SS, Haddad RI, Saba NF (2021) Head and neck cancer. Lancet 398(10318):2289–2299
Pulte D, Brenner H (2010) Changes in survival in head and neck cancers in the late 20th and early 21st century: a period analysis. Oncologist 15(9):994–1001
Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW (2006) p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 9(1):45–56
Si H, Lu H, Yang X, Mattox A, Jang M, Bian Y, Sano E, Viadiu H, Yan B, Yau C et al (2016) TNF-alpha modulates genome-wide redistribution of DeltaNp63alpha/TAp73 and NF-kappaB cREL interactive binding on TP53 and AP-1 motifs to promote an oncogenic gene program in squamous cancer. Oncogene 35(44):5781–5794
Specenier P, Vermorken JB (2013) Cetuximab: its unique place in head and neck cancer treatment. Biologics 7:77–90
Su YY, Chien CY, Luo SD, Huang TL, Lin WC, Fang FM, Chiu TJ, Chen YH, Lai CC, Hsu CM et al (2016) Betel nut chewing history is an independent prognosticator for smoking patients with locally advanced stage IV head and neck squamous cell carcinoma receiving induction chemotherapy with docetaxel, cisplatin, and fluorouracil. World J Surg Oncol 14:86
Wilkerson MD, Hayes DN (2010) ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26(12):1572–1573
Xing L, Zhang X, Zhang X, Tong D (2020a) Expression scoring of a small-nucleolar-RNA signature identified by machine learning serves as a prognostic predictor for head and neck cancer. J Cell Physiol 235(11):8071–8084
Xing L, Zhang X, Guo M, Zhang X, Liu F (2020b) Application of machine learning in developing a novelty five-pseudogene signature to predict prognosis of head and neck squamous cell carcinoma: a new aspect of “junk genes” in biomedical practice. DNA Cell Biol 39(4):709–723
Xing L, Guo M, Zhang X, Zhang X, Liu F (2020c) A transcriptional metabolic gene-set based prognostic signature is associated with clinical and mutational features in head and neck squamous cell carcinoma. J Cancer Res Clin Oncol 146(3):621–630
Ye MY, Chen MY, Chang YH, Huang JS, Huang TT, Wong TY, Hong TM, Chen YL (2018) Growth-regulated oncogene-alpha from oral submucous fibrosis fibroblasts promotes malignant transformation of oral precancerous cells. J Oral Pathol Med 47(9):880–886
Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16(5):284–287
Yu C, Li Q, Zhang Y, Wen ZF, Dong H, Mou Y (2022) Current status and perspective of tumor immunotherapy for head and neck squamous cell carcinoma. Front Cell Dev Biol 10:941750
Yue Y, Xia L, Xu S, Wang C, Wang X, Lu W, Xie X (2020) SURF4 maintains stem-like properties via BIRC3 in ovarian cancer cells. J Gynecol Oncol 31(4):e46
Zhai J, Han J, Li C, Guo F, Ma F, Xu B (2022) High SURF4 expression is associated with poor prognosis of breast cancer. Aging (albany NY) 14(22):9317–9337
Zhang X, Feng H, Li Z, Li D, Liu S, Huang H, Li M (2018) Application of weighted gene co-expression network analysis to identify key modules and hub genes in oral squamous cell carcinoma tumorigenesis. Onco Targets Ther 11:6001–6021
Zheng R, Liu Q, Wang T, Wang L, Zhang Y (2018) FAM98A promotes proliferation of non-small cell lung cancer cells via the P38-ATF2 signaling pathway. Cancer Manag Res 10:2269–2278
Acknowledgements
We express our gratitude for the assistance provided by ChatGPT 4.0 and AJE Curie in rectifying grammatical inaccuracies.
Funding
This study was funded by the Anhui Provincial Natural Science Foundation General Project (1808085MH251) and the Anhui Medical University First Affiliated Hospital 2021 Clinical Research Plan Project (LCYJ2021YB003).
Author information
Authors and Affiliations
Contributions
Conception and design: HX, QYS, and HQZ. Data curation and methodology: HX and HQZ. Analysis and interpretation of data: HX and QYS. Writing of the manuscript: HX. Review of the manuscript: HXH and HWX. Study supervision: HXH and HWX. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Not applicable. This study was based on publicly available data for which ethical approval was not needed. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Since the dataset is public and anonymized, informed consent was not needed.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xue, H., Sun, Q., Zhang, H. et al. Disulfidptosis features and prognosis in head and neck squamous cell carcinoma patients: unveiling and validating the prognostic signature across cohorts. J Cancer Res Clin Oncol 150, 156 (2024). https://doi.org/10.1007/s00432-024-05691-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00432-024-05691-9