
Abstract
Background
Acute myeloid leukemia (AML) is a hematological malignancy derived from the accumulation of abnormal proliferation of infantile leukocytes in the hematopoietic system. DNA-damage-inducible transcript 4 (DDIT4) acting as a negative regulator of rapamycin inhibitor is involved in various cellular functions. Many studies have suggested that DDIT4 plays a key role in tumorigenesis. However, the role of DDIT4 in AML has been poorly studied.
Method
In this study, we analyzed the expression of DDIT4 in AML patients using The Cancer Genome Atlas and real-time polymerase chain reaction. The Chi-square test was used to assess the correlation between DDIT4 and clinical characters in AML patients. Loss-of-function experiments were implemented to investigate the role of DDIT4 in AML carcinogenesis. The R package was applied to evaluate the correlation between DDIT4 expression and immune cells.
Results
Results showed that the expression of DDIT4 was associated with Age, Cytogenetic risk, Cytogenetics and OS event. Moreover, high expression of DDIT4 led to a terrible prognosis. KEGG analysis showed that differently expressed genes (DEGs) were involved in the PI3-Akt signaling pathway. GSEA enrichment analysis displayed DEGs were correlated with apoptosis. Functional experiments presented that knocking down DDIT4 suppressed cell cycle transition/proliferation and facilitated apoptosis. In addition, DDIT4 is associated with immune infiltration.
Conclusion
Our research verified that DDIT4 can be used as a prognostic marker and a potential therapeutic target for AML.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The incidence of AML is approximately 4.1 per 100,000 women and men per year. The mortality rate of AML is 2.7 per 100,000 per year (2021). AML is a genetically heterogenous blood tumor, characterized by the uncontrolled clonal overexpansion of undifferentiated myeloid progenitor cells (Döhner et al. 2015). Chemotherapy is the main treatment for AML, although most patients can achieve complete remission with chemotherapy, many patients finally relapse or are resistant to chemotherapy (Reville and Kadia 2020). Improving the ability to predict treatment response, duration of remission, and likelihood of recurrence can optimize the prognosis of AML (Small et al. 2022). The newest AML classification system, published in 2022, incorporates new pathological findings and emphasizes the integration of molecular analysis into the diagnosis of AML (Shimony et al. 2023). As the prognosis of AML remains dismal, there is a strong need for advances in biomarkers to guide prognostic assessment and risk stratification and to guide better treatment strategies.
DDIT4, also known as REDD1, regulates cell growth by negatively regulating the mammalian target of rapamycin (mTOR), which is widely expressed in tissues and highly induced by hypoxia and various stressors (Shoshani et al. 2002). DDIT4 is also highly expressed in various cancer tissues, and the prognosis of patients with high DDIT4 expression is worse than those with low DDIT4 expression (Liu et al. 2019). Previous studies have shown that high level of DDIT4 is strongly associated with poorer outcomes in patients with AML (Ley et al. 2013; Zhao et al. 2018). The prognostic value and function of DDIT4 in AML have not been thoroughly studied. Our goal was to explore the relevance between DDIT4 expression and clinical characters in AML and its specific prognostic value. We also explored the impact of DDIT4 on AML cell proliferation, apoptosis and cell cycle.
Materials and methods
Data source
The TCGA data corresponding to pan-cancer and the normal tissue data corresponding to GTEx were extracted from UCSC XENA (https://xenabrowser.net/datapages/) by the Toil process (Vivian et al. 2017) unified handling TCGA and GTEx FPKM RNAseq data format, included 7568 GTEx normal controls, 727 TCGA para-carcinoma tissues, and 9807 TCGA tumor tissues. The 150 AML patients’ clinical data was downloaded from the TCGA database (https://portal.gdc.cancer.gov).
Expression analysis of DDIT4 in tumor and normal tissues
To analyze the expression of DDIT4 in different cancers and their normal counterparts, t-test or Wilcoxon rank sum test was applied. The data were displayed by the ggplot2 package. The Human Protein Atla (HPA) (Colwill and Gräslund 2011) were used to investigate the intensity of immunohistochemical staining of DDIT4 in lung adenocarcinoma, renal chromophobe carcinoma and prostate cancer tumor tissues.
Clinical correlation analysis
To explore the prognostic ability of DDIT4 and its relationship with clinical features, we conducted survival analysis and ROC analysis. R software (version 4.2.1) was used to analyze the prognostic significance of DDIT4 in different cancers. Survival package was used to test the proportional risk hypothesis and to fit survival regression. The results were visualized using survminer package and the ggplot2 package. Clinical features were analyzed by univariate and multivariate Cox regression using forest plot. Nomograph was established to assess the survival of AML patients based on the results of multivariate logistic model. Calibration curve was used to evaluate the accuracy and clarity of Nomograph.
Differential expressed genes analysis
AML patients in the TCGA database were divided into two groups with DDIT4high expression and DDIT4low expression according to the median DDIT4 expression. Limma package was used to analyze the differentially expressed genes in the two groups and draw the volcano map. The filtering condition was that log2 fold change (|log2 FC|) > 1.0 with an adjusted P-value < 0.05.
Functional enrichment analysis and PPI analysis
We used the ClusterProfiler (version 3.14.2) package to complete Gene Ontology (GO) and Kyoto Encyclopedia of genes and Genomes (KEGG) pathway enrichment analyses on that are differently expressed with DDIT4 in AML. Gene Set Enrichment Analysis (GSEA) is a computing method that estimates whether an a priori defined set of genes shows statistically significant, consistency between two states. GSEA database (www.broadinstitute.org/gsea) was used to analyze screened differently expressed genes and identify enrichment signaling pathways associated with AML. P-value < 0.05 was considered statistically significant.
The STRING database (https://string-db.org/) was used for PPI network analysis. We investigated the correlation between DEGs with a confidence score than 0.4 to establish the PPI network. The visualization of the PPI network was accomplished with the Cytoscape software plugin MCODE (Shannon et al. 2003). Finally, seven important core genes (THBS1, HGF, IGF1, IL-6, CCL25, MMP7, and IL-10) were obtained through screening of the PPI-related genes and prognostic genes.
Immunoinfiltration analysis
Based on the ssGSEA algorithm provided by R package –GSVA [1.46.0] (Hänzelmann et al. 2013), this paper utilizes Immunity article (Bindea et al. 2013) provided markers for 24 immune cells to calculate immune infiltration. Spearman correlation analysis was performed between principal variables and immune infiltration matrix data in the data, and the analysis results were visualized with the lollipop chart in ggplot2 package. Spearman correlation analysis was used to analyze the relationship between DDIT4 and 6 classic immune checkpoint molecules (TIGIT, PDCD1, LAG3, CD274, HAVCR2 and CTLA4), and the analysis results were visualized by ggplot2 package coexpression heat map.
Cell culture and small interfering RNA transfection
Human leukemia cell lines THP1 and NB4 were obtained from the Cell Bank of Chinese Academy of Science and cultured in RPMI-1640 medium with 10% fetal bovine serum and penicillin–streptomycin at 37 ℃. DDIT4 small interfering RNA and control siRNA were synthesized by GenePharma (Shanghai, China). Transfection was performed using RFECT reagents (Baidai biotechnology, Changzhou, China) according to the manufacturer’s protocol.
Real-time quantitative polymerase chain reaction (RT-qPCR)
RNA was extracted using Trizol reagent (Sigma, Aldrich), and then reversed into cDNA for RT-qPCR reaction. Primers are as follows: DDIT4, 5′-TGCATTGGGGACACATACCC-3′ and 5′-CCCAAGTGATCCCTGACACC-3′; Actin, 5′-GTGGCCGAGGACTTTGATTG-3′ and 5′-CCTGTAACAACGCATCTCATATT-3′.
Cell counting kit (CCK) 8 assay
After transfected with DDIT4 siRNA, THP1 and NB4 cells were seeded in 96-well plates, and 10 µl CCK-8 solution (7Sea Pharmatech, China) was added to each well at 0 h, 24 h, 48 h and 72 h, respectively. After incubation for 4 h, absorbance at 450 nm was measured.
EdU, cell apoptosis and cell cycle assay
After transfection of DDIT4 siRNA, cell proliferation, apoptosis and cell cycle were detected by flow cytometry. Cell proliferation was detected using EdU assay kit (Beyotime, Shanghai, China). Cells were stained with Annexin V-PE/RedNucleus (Beyotime, Shanghai, China) and apoptosis rate was detected. The cells were fixed with cold 70% alcohol for 24 h and then stained with PI solution (Beyotime, Shanghai, China) and Rnase A (Beyotime, Shanghai, China) for cell cycle detection.
Western blotting
The cells were lysed with RIPA reagent (Xianfeng Biotechnology, Xi’an, China) and quantified by BCA Protein Assay Kit (Beyotime, Shanghai, China). 40 μg of total protein was added to 10% SDS-PAGE and then transferred to 0.2 mm PVDF membrane (Millipore). Incubate 5% skim milk at room temperature for 1 h and then add the corresponding primary antibody (Proteintech, 1:1000 dilution) for overnight incubation at 4 ℃. After TBST cleaning, add the secondary antibody (Proteintech, 1:10,000) and incubate at room temperature for 1 h. Finally, the protein signal was detected by the ECL system.
Statistical analysis
The results were shown as mean ± SD. GraphPad Prism software 9.0 and SPSS software 18.0 were adopted for data processing. Chi-square test were performed to explore the correlation between DDIT4 and clinical features of categorical variables. Student t-test or ANOVA test were used for continuous variables. Furthermore, Welch t’test, Kruskal–Wallis test and Mann–Whitney U test were applied for comparison between two groups. P value < 0.05 was considered statistically significant.
Results
Pan-cancer analysis of DDIT4
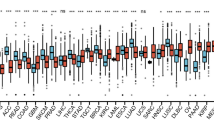
We analyzed DDIT4 expression in 33 tumor tissues and corresponding normal tissues in the TCGA and GTEx databases. As shown in Fig. 1A, DDIT4 was up-regulated in multiple tumors compared with normal controls. As shown in the survival curves in Fig. 1B, we also found that patients with high DDIT4 expression in LUAD (p = 0.003), KICH (p = 0.047), PAAD (p = 0.040) and HNSC (p = 0.010) had worse prognosis. In ESCA (p = 0.030), patients with high DDIT4 expression had a better prognosis than those with low DDIT4 expression. At the same time, we used GEPIA2.0 website to investigate DDIT4 expression in different tumor stages. The results showed that DDIT4 expression was gradually increased with the staged in LUAD, KICH, THCA, ACC and HNSC (Fig. 1C). Immunohistochemical staining results of HPA database showed that DDIT4 expression decreased in KICH and PARD tumor tissues and increased in LUAD tumor tissues (Fig. 1D), this is consistent with the results of pan-cancer analysis.

High DDIT4 expression was associated with adverse clinical outcomes. AThe expression of DDIT4 between tumor tissues and normal tissues in TCGA and GTEx databases. B The level of DDIT4 is related to patients’ overall survival. C DDIT4 was expressed differently in different stages of the tumor. D Immunohistochemistry detected DDIT4 expression in tumor and normal tissues. (ns p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001)
DDIT4 expression and correlation with clinical features in AML
We compared DDIT4 mRNA expression levels in AML patients with normal controls in the GEPIA 2.0 database. DDIT4 expression was downregulated in AML (p < 0.05) (Fig. 2A). In addition, the AUC value of the ROC analysis was 0.974 (95% confidence interval, CI 0.953–0.996, Fig. 2B). We then analyzed the relationship between DDIT4 expression level and clinical features of AML patients. In this study, we analyzed 151 AML patients with clinical information from the TCGA database. As shown in Fig. 2C–G, high DDIT4 expression was associated with unfavorable cytogenetic risk (p < 0.001, Fig. 2C), FLT3 negative mutation (p < 0.05, Fig. 2D), older age (p < 0.01, Fig. 2E), death (p < 0.001, Fig. 2F), and IDH1 R132 positive mutation (p < 0.05, Fig. 2G) in AML patients, with no association with French-American-British (FAB) classifications (Fig. 2H). As shown in Table 1, 151 AML patients were further divided into low and high expression of DDIT4 according to the median value of DDIT4 mRNA expression. Correlation analysis results showed that DDIT4 expression was correlated with Age (p < 0.001), Cytogenetic risk (p < 0.001), Cytogenetics (p = 0.002) and OS event (p = 0.001).

A The expression of DDIT4 was lower than normal control (n = 70) in AML (n = 173) in the TCGA database. B ROC of DDIT4 in AML. C–G The expression of DDIT4 between different genetic risk stratification (C), FLT3 mutation status (D), age distribution (E), overall survival event (F), IDH1 R132 mutation status (G) and AML subtypes (H). ROC, Receiver operating characteristic analysis (ns p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001)
DDIT4 is an independent prognostic factor in patients with AML
Kaplan–Meier analysis was used to determine whether the expression level of DDIT4 was correlated with patient’s prognosis. The results showed that AML patients with high expression of DDIT4 had shorter overall survival (OS) compared to low expression DDIT4 cohort (HR = 2.28, p < 0.001)(Fig. 3A). To explore the prognostic value of DDIT4 in AML patients, ROC curves were constructed for 1,3, and 5 year OS. The 1-year, 3-year and 5-year AUCs were 0.657, 0.677, 0.717, respectively (Fig. 3C). Univariate and multivariate regression analyses were used to further explore the predictive value of DDIT4 for survival. Univariate analysis showed age, cytogenetic risk, and DDIT4 expression were associated with prognosis (p < 0.001, p < 0.001, p < 0.001, respectively) in AML patients (Fig. 3B). Meanwhile, multivariate regression revealed that age was an independent prognostic factor of poor OS (HR, 2.890; 95%CI, 1.848–4.519; p < 0.001) (Fig. 3D). Furthermore, according to these prediction factors, we constructed a nomogram plot including age, cytogenetic risk, and DDIT4 expression to predict the survival probability at 1,3, and 5 years of AML patients (Fig. 3E). The observation results were consistent with those predicted by nomogram calibration curve (Fig. 3F). In summary, high DDIT4 expression in AML patients is associated with poor prognosis.

Prognostic value of DDIT4 expression in AML. A DDIT4’s predictive ability on survival for AML, KM curve dispayed OS between high and low DDIT4 expression groups. B Forest plot for univariate analysis results of AML patients. C Diagnostic ability of DDIT4 in AML with 1-,3-, and 5-year. D Forest plot for multivariate regression analysis results of AML patients. E Nomograph for predicting 1-year, 3-year and 5-year OS in AML patients. F Calibration curve of Nomogram. OS overall survival
Differentially expressed genes correlated with DDIT4 enrichment analysis
To further elucidate the mechanism of DDIT4 in the pathogenesis of AML. We identified genes that were differentially expressed between the high DDIT4 and low DDIT4 cohorts of AML patients. A total of 1679 differentially expressed genes (DEGs) were displayed by volcano plot (Fig. 4A), where 1216 genes were up-regulated and 463 genes were down-regulated (|log2FC|> 1, adjusted p-value < 0.05). Then we completed GO and KEGG analysis of DEGs using Cluster Profilter software package. KEGG analysis showed that these DEGs were mainly involved in the PI3-Akt signaling pathway, ECM-receptor interaction, Neuroactive ligand-receptor interaction and Neuroactive ligand-receptor interaction (Fig. 4B). Next, GSEA software was used to perform GSEA enrichment analysis on DEGs, and the results showed that DDIT4 was involved in PI3K-Akt, Jak Stat, IL6, Wnt signaling pathways and Apoptosis, Oxidative Stress Induced Senescence (Fig. 4C–H).

A Volcano plot for DEGs among DDIT4high and DDIT4low groups. B GO terms and KEGG pathways analysis of DEGs. C–H GSEA enrichment analysis of DEGs. C Enriched genes of Pi3k akt pathways. D Enriched genes in Oxidative Stress induced Senecence. E Enriched genes of Jak Stat Signaling pathway. F Enriched genes of IL6 signaling pathway. G Enriched genes of Wnt signaling pathway. H Enriched genes in apoptosis pathway. DEGs different expression genes
PPI networks of DDIT4-related partners
To identify the hub genes related to DDIT4 in AML, the PPI network of 287 encoding DEGs was constructed using the online bioinformatics tool STRING. The results were visualized using Cytoscape software (Fig. 5A). The PPI network was then analyzed using MCODE, a plugin of Cytoscape software, to search for hub genes. The top 15 hub genes were determined by maximum neighborhood component (DMNC), density of maximum neighborhood component (MNC), and maximal clique centrality (MCC) screening (Fig. 5B–D).

PPI network of 287 encoding DEGs. (B–D) Hub genes filtrated by DMNC (B), MNC(C), and MCC (D) analysis. DEGs different expression genes
Among these core genes, THBS1 and HGF are highly expressed in AML (Fig. 6A, B), IGF1 and IL-6 are low expressed in AML (Fig. 6C, D), and CCL25, MMP7 and IL-10 are not different from healthy controls (Fig. 6E–G). CCL25 and IL-10 were positively correlated with DDIT4 in AML (Fig. 6L, N), while THBS1, HGF, IGF1, IL-6, and MMP7 were not correlated with DDIT4 (Fig. 6H–K, M). In addition, patients with high expression of CCL25, THBS1,MMP7, IGF1 and IL-10 in AML have worse OS (Fig. 6O, Q, S-U), while patients with HGF high expression has longer OS (Fig. 6P). IL-6 expression was not correlated with OS in AML patients (Fig. 6R).

A-G Expression of 7 core genes in AML patients (n = 173) versus healthy controls (n = 70). A THBS1. B HGF. C IGF1. D IL-6. E CCL25. F MMP7 (G) IL-10. (H–N) Correlation analysis between 7 core genes with DDIT4. H THBS1. I HGF. J IGF1. K IL-6. L CCL25. M MMP7. N IL-10. O–U OS analysis between high- and low- core genes expression displayed by KM curves. O THBS1. P HGF. Q IGF1. R IL-6. S CCL25. T MMP7. U IL-10. OS overall survival. (*p < 0.05)
Oncogenic function of DDIT4 in AML cells
To investigate the role of DDIT4 in AML, we used small interfering RNAs (siRNAs) to knock down DDIT4 in leukemia cell lines THP1 and NB4. The expression of DDIT4 was significantly decreased (Fig. 7A). The results of CCK-8 showed that knocking down DDIT4 significantly inhibited the activity of THP1 and NB4 cells (Fig. 7C). EdU proliferation assay confirmed the results that cell proliferation decreased after DDIT4 knocking down (Fig. 7D). Flow cytometry results showed that after DDIT4 knockdown, the apoptosis of THP-1 and NB4 cells increased (Fig. 7E), the number of G0/G1 phase cells increased, and the number of S phase cells decreased (Fig. 7F). Figure 7B showed the expression of apoptosis and cell cycle related-protein of leukemia cells, in which the expression of C-myc, Bcl-2 and Cdk2 were decreased, while the expression of p21 and Bax were increased. Together, these results suggested that DDIT4 affects the proliferation of AML by influencing cell apoptosis and cell cycle.

A DDIT4 knockdown efficiency was assessed by qRT-PCR and western blot. B Western blot measured apoptosis, proliferation and cell cycle-related proteins. C Cell viability was assessed by CCK-8 assay. D Apoptosis increased after DDIT4 knockdown. E Cell proliferation ability was tested by EdU assay. F The proportion of the G1 stage increased when DDIT4 was knocked down. (ns p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001)
Correlation analysis between DDIT4 and immune cells and immune checkpoint molecules
To explore the role of DDIT4 in the immune infiltration of leukemia, we analyzed the relationship between the expression level of DDIT4 and the expression of 24 kinds of immune cells. Spearman analysis results showed that the expression level of DDIT4 was positively correlated with Macrophages, Treg cells, Th17 cells, Cytotoxic cells, NK CD56 bright cells, TH1 cells, TFH, NK CD56 dim cells, B cells and CD8 T cells (Fig. 8A). More specific correlation analysis results are shown in Fig. 8B–K. In addition, we also analyzed the relationship between DDIT4 and 6 classic immune checkpoint genes. As shown in Fig. 8L, the expression level of DDIT4 was positively correlated with the levels of PDCD1, CD274, CTLA4, LAG3 and TIGIT, and the specific analysis was shown in Fig. 8M–Q.

Correlation analysis among DDIT4 expression and immune cells or immune checkpoint molecules. A Relationship among DDIT4 expression and 24 kinds of immune cells levels. B–K Correlativity between DDIT4 expression and immune infiltration in AML. B Macrophages. C Treg cells. D Th17 cells. E Cytotoxic cells. F NK CD56bright cells. G Th1 cells. H TFH cells. I NK CD56dim cells. J B cells. K CD8 T cells. L Heat map of the relationship between DDIT4 and 6 immune checkpoint molecules (PDCD1, CD274, CTLA4, LAG3, TIGIT, HAVCR2). M–Q Scatter plot of correlation between DDIT4 and TIGIT M, PDCD1 N, LAG3 O, CD274 P and CTLA4 Q. (*p < 0.05, **p < 0.01, ***p < 0.001)
Discussion
Acute myeloid leukemia is the most common type of acute leukemia in adults. The pathophysiology of AML remains unclear (Stanchina et al. 2020). The available research showed that chromosome aberration, DNA methylation and gene mutation in AML prevented the normal maturation of bone marrow progenitor cells and led to the proliferation of original cell clones (Ley et al. 2013; Nguyen et al. 2019). DDIT4 plays a driving role in the development of several malignant tumors and it can be induced by a variety of conditions (Pinto et al. 2017). DDIT4 mainly involves in the PI3K-Akt-mTOR signaling pathway, which regulates cell growth, proliferation and apoptosis (Sofer et al. 2005).
In this study, we used the public database to investigate the mRNA expression and prognostic value of DDIT4 in pan-carcinoma. We found that DDIT4 was significantly abnormally expressed in a variety of tumors. DDIT4 works differently in different cancers. High DDIT4 expression had better OS in ESCA and worse OS in LUAD, KICH, PAAD, and HNSC. Expression of DDIT4 in tumors is correlated with cancer type, but high expression is associated with worse staging in most tumors. These results suggest that DDIT4 expression and prognostic significance were highly cancer-dependent, and the unique role of DDIT4 in each cancer needs to be further identified. We also studied the clinical significance and effect of DDIT4 in AML by bioinformatics analysis and functional determination. In our study, DDIT4 was expressed at a low level in AML patients. Analysis of the clinical features of AML patients in the TCGA cohort indicated that up-regulated expression of DDIT4 was an independent prognostic biomarker for OS in AML. High expression of DDIT4 was also associated with unfavorable genetic risk, advanced age (> 60 years) and IDH1-R132 mutations in AML patients. Those results suggest that DDIT4 may play a role in the pathogenesis of AML.
Chemotherapy is the preferred treatment for acute myeloid leukemia. Relapse due to chemotherapy resistance is the leading cause of death in AML patients (Kantarjian et al. 2021). Studies have shown that drug resistance in tumor cells is partly due to the acquisition of anti-apoptotic capabilities and the failure of DNA damage response and repair (Assaraf et al. 2019). Lin Fu, et al. reported that patients with elevated DDIT4 expression who received chemotherapy alone had a worse prognosis and might benefit from allo-HSCT (Cheng et al. 2020). To explore the potential mechanism of DDIT4 affecting the prognosis of AML, patients were divided into two groups with high DDIT4 expression and low DDIT4 expression, and the differential genes in these two groups were analyzed by GO and KEGG pathways. The results showed that DEGs were related to the PI3K-Akt signaling pathway and apoptosis signaling pathway. PI3K-Akt is a key pathway that regulates cell growth, proliferation and apoptosis (Porta et al. 2014). We knocked down DDIT4 in AML cells, and the decrease of DDIT4 in THP-1 and NB4 cell lines showed increased apoptosis, decreased proliferation, and cell cycle arrest in the G0/G1 phase, indicating that DDIT4 has a carcinogenic function in AML.
The expression of DDIT4 in AML patients is lower than that in healthy controls, but patients with high expression have a poor prognosis, which should involve unknown complex mechanisms. Previous studies have shown that IL-10 strongly up-regulates DDIT4 expression and inhibits mTOR activity during macrophage activation (Ip et al. 2017). IL-6 can reduce IL-6-induced mTOR signal activation by decreasing DDIT4 expression (Pinno et al. 2016). We constructed the PPI network of differential genes and screened the core genes, including IL-6 and IL-10. IL-6 is highly expressed in AML and is not correlated with DDIT4 expression and AML prognosis; IL-10 is positively correlated with DDIT4 expression and those with high expression in AML have shorter OS. Core genes CCL25, THBS1, MMP7, IGF1 and HGF were found to have an effect on OS in AML patients. In our current study, we have not found why DDIT4 expression was reduced in AML, which may involve other mechanisms besides IL-6 and IL-10, which need to be further studied in the future.
Up to now, the outcome of AML is still unsatisfactory, and immune escape of tumor cells plays an important role in the cause of poor response to chemotherapy and recurrence. AML cells can cause changes in the microenvironment of bone marrow tumors to facilitate immune escape (Stanchina et al. 2020). The microenvironment of AML has an inhibitory effect on immune effector cells. Elevated Treg cell-mediated immunosuppression in AML patients allows leukemia cells to evade immune monitoring (Ustun et al. 2011). High levels of macrophage infiltration in tumors are associated with poor prognosis and progression of cancer (Gwak et al. 2015). TH17 cells can increase the expression of Treg cells in the presence of TNF-α, resulting in an immunosuppressive state that promotes the progression of AML (Wang et al. 2018). The role of DDIT4 in the immune infiltration of AML remains unclear. We analyzed the correlation between DDIT4 expression and immune infiltration in AML from the data published in the public database, and the results showed that the abundance of Cytotoxic cells, NK CD56 bright cells, TH1 cells, TFH, NK CD56 dim cells, B cells and CD8 T cells significantly increased in the DDIT4 overexpression group. These results indicated that overexpression of DDIT4 was associated with increased immune infiltration in AML. The abundance of Macrophages, TH17 cells and Treg cells also increased in the DDIT4 high expression group. The elevation of these three immune cells should be responsible for the progression of immune escape disease in the group of AML patients with high DDIT4 expression.
In summary, we found that patients with high DDIT4 expression in AML have a poor prognosis, and DDIT4 may affect the progression of AML by regulating cell proliferation, apoptosis, cell cycle and immune microenvironment. DDIT4 is expected to be a potential prognostic marker for AML.
Data availability
The orginal data displayed in this study are included in the article. Further requests can be contact to the corresponding author.
References
Assaraf YG, Brozovic A, Gonçalves AC, Jurkovicova D, Linē A, Machuqueiro M, Saponara S, Sarmento-Ribeiro AB, Xavier CPR, Vasconcelos MH (2019) The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist Updat 46:100645
Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, Bruneval P, Fridman WH, Becker C, Pagès F, Speicher MR, Trajanoski Z, Galon J (2013) Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39:782–795
Cheng Z, Dai Y, Pang Y, Jiao Y, Liu Y, Cui L, Quan L, Qian T, Zeng T, Si C, Huang W, Chen J, Pang Y, Ye X, Shi J, Fu L (2020) Up-regulation of DDIT4 predicts poor prognosis in acute myeloid leukaemia. J Cell Mol Med 24:1067–1075
Colwill K, Gräslund S (2011) A roadmap to generate renewable protein binders to the human proteome. Nat Methods 8:551–558
Döhner H, Weisdorf DJ, Bloomfield CD (2015) Acute myeloid Leukemia. N Engl J Med 373:1136–1152
Gwak JM, Jang MH, Kim DI, Seo AN, Park SY (2015) Prognostic value of tumor-associated macrophages according to histologic locations and hormone receptor status in breast cancer. PLoS ONE 10:e0125728
Hänzelmann S, Castelo R, Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14:7
Ip WE, Hoshi N, Shouval DS, Snapper S, Medzhitov R (2017) Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356:513–519
Kantarjian HM, Short NJ, Fathi AT, Marcucci G, Ravandi F, Tallman M, Wang ES, Wei AH (2021) Acute myeloid leukemia: historical perspective and progress in research and therapy over 5 decades. Clin Lymphoma Myeloma Leuk 21:580–597
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ, Laird PW, Baty JD, Fulton LL, Fulton R, Heath SE, Kalicki-Veizer J, Kandoth C, Klco JM, Koboldt DC, Kanchi KL, Kulkarni S, Lamprecht TL, Larson DE, Lin L, Lu C, Mclellan MD, Mcmichael JF, Payton J, Schmidt H, Spencer DH, Tomasson MH, Wallis JW, Wartman LD, Watson MA, Welch J, Wendl MC, Ally A, Balasundaram M, Birol I, Butterfield Y, Chiu R, Chu A, Chuah E, Chun HJ, Corbett R, Dhalla N, Guin R, He A, Hirst C, Hirst M, Holt RA, Jones S, Karsan A, Lee D, Li HI, Marra MA, Mayo M, Moore RA, Mungall K, Parker J, Pleasance E, Plettner P, Schein J, Stoll D, Swanson L, Tam A, Thiessen N, Varhol R, Wye N, Zhao Y, Gabriel S, Getz G, Sougnez C, Zou L, Leiserson MD, Vandin F, Wu HT, Applebaum F, Baylin SB, Akbani R, Broom BM, Chen K, Motter TC, Nguyen K, Weinstein JN, Zhang N, Ferguson ML, Adams C, Black A, Bowen J, Gastier-Foster J, Grossman T, Lichtenberg T, Wise L, Davidsen T, Demchok JA, Shaw KR, Sheth M, Sofia HJ, Yang L, Downing JR, Eley G (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368:2059–74
Liu C, Li Y, Wei M, Zhao L, Yu Y, Li G (2019) Identification of a novel glycolysis-related gene signature that can predict the survival of patients with lung adenocarcinoma. Cell Cycle 18:568–579
National Cancer Institute. Cancer stat facts: leukemia—acute myeloid leukemia (AML).https://seer.cancer.gov/statfacts/html/amyl.html. https://seer.cancer.gov/statfacts/html/amyl.html. 2021. Cancer Stat Facts: Leukemia—Acute Myeloid Leukemia (AML). https://seer.cancer.gov/statfacts/html/amyl.htmlhttps://seer.cancer.gov/statfacts/html/amyl.html. Accessed 21 Sep 2017.
Nguyen CH, Glüxam T, Schlerka A, Bauer K, Grandits AM, Hackl H, Dovey O, Zöchbauer-Müller S, Cooper JL, Vassiliou GS, Stoiber D, Wieser R, Heller G (2019) SOCS2 is part of a highly prognostic 4-gene signature in AML and promotes disease aggressiveness. Sci Rep 9:9139
Pinno J, Bongartz H, Klepsch O, Wundrack N, Poli V, Schaper F, Dittrich A (2016) Interleukin-6 influences stress-signalling by reducing the expression of the mTOR-inhibitor REDD1 in a STAT3-dependent manner. Cell Signal 28:907–916
Pinto JA, Rolfo C, Raez LE, Prado A, Araujo JM, Bravo L, Fajardo W, Morante ZD, Aguilar A, Neciosup SP, Mas LA, Bretel D, Balko JM, Gomez HL (2017) In silico evaluation of DNA damage inducible transcript 4 gene (DDIT4) as prognostic biomarker in several malignancies. Sci Rep 7:1526
Porta C, Paglino C, Mosca A (2014) Targeting PI3K/Akt/mTOR Signaling in cancer. Front Oncol 4:64
Reville PK, Kadia TM (2020) Maintenance therapy in AML. Front Oncol 10:619085
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Shimony S, Stahl M, Stone RM (2023) Acute myeloid leukemia: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol 98:502–526
Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanov A, Chajut A, Kalinski H, Kamer I, Rozen A, Mor O, Keshet E, Leshkowitz D, Einat P, Skaliter R, Feinstein E (2002) Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol 22:2283–2293
Small S, Oh TS, Platanias LC (2022) Role of biomarkers in the management of acute myeloid leukemia. Int J Mol Sci 23:14543
Sofer A, Lei K, Johannessen CM, Ellisen LW (2005) Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol 25:5834–5845
Stanchina M, Soong D, Zheng-Lin B, Watts JM, Taylor J (2020) Advances in acute myeloid leukemia: recently approved therapies and drugs in development. Cancers 12:3225
Ustun C, Miller JS, Munn DH, Weisdorf DJ, Blazar BR (2011) Regulatory T cells in acute myelogenous leukemia: is it time for immunomodulation? Blood 118:5084–5095
Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, Pfeil J, Narkizian J, Deran AD, Musselman-Brown A, Schmidt H, Amstutz P, Craft B, Goldman M, Rosenbloom K, Cline M, O’connor B, Hanna M, Birger C, Kent WJ, Patterson DA, Joseph AD, Zhu J, Zaranek S, Getz G, Haussler D, Paten B (2017) Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 35:314–316
Wang M, Zhang C, Tian T, Zhang T, Wang R, Han F, Zhong C, Hua M, Ma D (2018) Increased regulatory T cells in peripheral blood of acute myeloid leukemia patients rely on tumor necrosis factor (TNF)-α-TNF receptor-2 pathway. Front Immunol 9:1274
Zhao X, Li Y, Wu H (2018) A novel scoring system for acute myeloid leukemia risk assessment based on the expression levels of six genes. Int J Mol Med 42:1495–1507
Funding
This work was supported by the Natural Science Foundation of Shaanxi Province (Grant no. 2020JQ-539).
Author information
Authors and Affiliations
Contributions
Fangmei Li and Jiyu Miao launched the study; Fangmei Li and Rui Liu undertook the work of bioinformatics analyses, statistical analyses and drafted the manuscript. Ru Zhang carried out the function experiments; Aili He reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, F., Miao, J., Liu, R. et al. Pan-cancer analysis of DDIT4 identifying its prognostic value and function in acute myeloid leukemia. J Cancer Res Clin Oncol 150, 144 (2024). https://doi.org/10.1007/s00432-024-05676-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00432-024-05676-8