
Abstract
Purpose
Renal cell carcinoma belongs among the deadliest malignancies despite great progress in therapy and accessibility of primary care. One of the main unmet medical needs remains the possibility of early diagnosis before the tumor dissemination and prediction of early relapse and disease progression after a successful nephrectomy. In our study, we aimed to identify novel diagnostic and prognostic biomarkers using next-generation sequencing on a novel cohort of RCC patients.
Methods
Global expression profiles have been obtained using next-generation sequencing of paired tumor and non-tumor tissue of 48 RCC patients. Twenty candidate lncRNA have been selected for further validation on an independent cohort of paired tumor and non-tumor tissue of 198 RCC patients.
Results
Sequencing data analysis showed significant dysregulation of more than 2800 lncRNAs. Out of 20 candidate lncRNAs selected for validation, we confirmed that 14 of them are statistically significantly dysregulated. In order to yield better discriminatory results, we combined several best performing lncRNAs into diagnostic and prognostic models. A diagnostic model consisting of AZGP1P1, CDKN2B-AS1, COL18A1, and RMST achieved AUC 0.9808, sensitivity 95.96%, and specificity 90.4%. The model for prediction of early relapse after nephrectomy consists of COLCA1, RMST, SNHG3, and ZNF667-AS1 and achieved AUC 0.9241 with sensitivity 93.75% and specificity 71.07%. Notably, no combination has outperformed COLCA1 alone. Lastly, a model for stage consists of ZNF667-AS1, PVT1, RMST, LINC00955, and TCL6 and achieves AUC 0.812, sensitivity 85.71%, and specificity 69.41%.
Conclusion
In our work, we identified several lncRNAs as potential biomarkers and developed models for diagnosis and prognostication in relation to stage and early relapse after nephrectomy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite the great efforts and progress in imaging techniques and therapeutical options in the last decades, renal cell carcinoma (RCC) still remains one of the deadliest urogenital cancers worldwide, being the ninth most common neoplasm in the United States and accounting for 2% of global cancer cases (Padala et al. 2020). The incidence of RCC rises especially in higher income regions where accessibility of primary health care and imaging techniques enables early identification of RCC cases (Capitanio et al. 2019). However, more than 50% of cases are discovered incidentally (Escudier et al. 2019); as only a small fraction of patients present with typical symptoms of RCC, and no systemic screening programe has been developed so far (Decastro and McKiernan 2008; Padala et al. 2020). And still, even the growing accessibility of imaging does not cover the diagnostic need, as the locally advanced disease is already present in a notable proportion of patients, with almost 17% harboring distant metastasis (Capitanio et al. 2019). Moreover, a significant portion of patients after successful nephrectomy experience relapse relatively early, up to 2 years after the surgery. Currently recognized prognostic models, namely stage, size, grade, and necrosis (SSIGN) score, the University of California Los Angeles Integrated Staging System (UISS), and molecular models such as ClearCode34 can provide only limited prognostic value. No preference is placed on any of them by official oncology guidelines (Escudier et al. 2019). Assessment of prognosis is currently based mainly on histological and clinical factors, and the pressing clinical need to predict early relapse after nephrectomy stays unmet as no reliable biomarker has been discovered and implemented yet.
Long non-coding RNAs are such potential biomarkers, as their expression is dysregulated in association with many human diseases not only as a secondary effect but also as a causative event (Chi et al. 2019). As potent regulators of gene expression on all levels from chromatin organization all the way to the protein folding and beyond, lncRNAs have a vital impact on cell functioning (Quinn and Chang 2016; Chi et al. 2019). Dysregulation of lncRNA levels affects other cellular components and contributes to development of pathologic conditions but can also serve as a unique snapshot of current situation in any given cell (Chandra Gupta and Nandan Tripathi 2017; Bohosova et al. 2021). In the recent decade, there was a plethora of publications focused on dysregulation of lncRNA in many human diseases, although, predominantly in cancer, including renal cell carcinoma (Chandra Gupta and Nandan Tripathi 2017; Bhan et al. 2017).
In a present study, we aimed to identify diagnostic and prognostic lncRNAs, specifically dysregulated in patients with early relapse after nephrectomy and thus provide potential new tool for diagnosis and prognostication of patients in higher risk of disease progression.
Materials and methods
Samples and patients
Patients diagnosed with renal cell carcinoma were enrolled during their treatments at the Masaryk Memorial Cancer Institute, Brno, the Czech Republic, between 2014 and 2018. Enrollment, sample, and clinical data management were handled according to the Declaration of Helsinki. Signed informed consent has been collected from all the patients before the study enrollment. The study design was approved by the Ethics Committee of the Masaryk Memorial Cancer Institute and Ethics Committee of Masaryk University. From all 259 patients, tumor renal parenchyma and adjacent non-tumor tissue have been taken during the nephrectomy. All tissue was first submerged in RNA-later and then stored at -80 °C until further processing. In Table 1 relevant clinical characteristics for the select cohorts of patients used in sequencing and validation part of the study are shown.
Out of the 259 patients enrolled in the study, several (N = 13) patients had to be excluded due to extremely short overall survival after nephrectomy, and death unrelated to the disease relapse (up to ten months from surgery), or invalid clinical data. The remaining 246 patients were stratified into 2 independent cohorts for the sequencing and validation part of the study. The sequencing cohort of 48 patients consisted of two groups which were later compared with each other—early relapse and late-relapse, both groups contained 24 patients (together 96 samples, as we processed both the tumor and non-tumor tissue from each patient). Early-relapse patients were defined as relapses up to 25 months after nephrectomy (median 10,58 months, range 2.41–21.97). Late-relapse group consisted of patients without relapse events for the period of more than 50 months after the nephrectomy. For validation, we used all the remaining patients a divided them again into two groups—early and late-relapse patients. Early-relapse group consisted of all the remaining patients (N = 15) who experienced relapse before the 25-month mark. In the late-relapse group were all the remaining patients (N = 183) who relapsed after 25 months or had not relapsed at all during the 5-year follow-up.
Isolation and quality control
Frozen tissue samples were thawed at the processing site and from each specimen, a 0.5 × 0.5 cm piece was cut and further processed. Total RNA enriched for small RNAs was extracted using mirVana™ miRNA isolation kit (Invitrogen, Waltham, MA, USA) with a minor alteration of the manufacturer’s protocol. Specimens were first homogenized using ceramic beads (Qiagen, Hilden, Germany) along with the Lysis/Binding buffer from the mirVana™ miRNA isolation kit (Invitrogen, Waltham, MA, USA). We then proceeded to the RNA extraction as suggested by the manufacturer. The concentration of the extracted RNA was measured using NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Extracted RNA was stored at − 80 °C until further processing.
Library preparation and transcriptome sequencing
Prior to the library preparation, the concentration of the samples chosen for the sequencing analysis was measured again for better precision of input amounts of RNA using Qubit™ 2.0 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) fluorometer. The integrity of RNA was determined using Agilent 2200 TapeStation system and RNA ScreenTape (Agilent, Santa Clara, CA, USA). As some samples still contained some residual genomic DNA which would interfere with the sequencing of the desired fragments, we removed it using the DNA-free™ DNA Removal Kit (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol. The concentration of the pure RNA was measured again using Qubit™ 2.0 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) fluorometer and then we proceeded to the ribosomal RNA depletion. First, the samples were diluted in 26 ul of nuclease-free water (Qiagen, Hilden, Germany) to achieve 500 ng of total RNA input. For the RNA depletion, we used the RiboCop rRNA Depletion Kit V1.2 (Lexogen, Vienna, Austria) according to the manufacturer's protocol in order to eliminate the ribosomal RNA which would overwhelm the sequencing capacity. The concentration of the purified RNA was measured again using Qubit™ 2.0 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) fluorometer.
We then proceeded to the sequencing library preparation using Ultra™ II Directional RNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, MA, USA), AM-Pure® XP Beads (Beckman Coulter, Brea, CA, USA), and NEBNext® Multiplex Oligos for Illumina® (Dual Index Primers Set 1) (New England Biolabs, Ipswich, MA, USA). Minor adjustments have been made to the manufacturer's protocol: RNA has been fragmented for a longer time (8 min despite higher RNA quality); incubation with the USER Enzyme has been carried out as a first step in the PCR Enrichment of Adaptor Ligated DNA reaction; this reaction has been also run in the Biometra Optical Thermocycler® (Analytik Jena, Jena, Germany) which allows following the amplification curve real-time and stopping the reaction when the desired amplification signal is achieved. To visualize the amplification, we added 2 ul of EvaGreen® Dye, 20X in Water (Biotium, Fremont, CA, USA) into the PCR reaction mix, and individual tubes were taken out of the machine when the fluorescence reached 5000. Purifying beads volume had to be adjusted as the volume of the mixture was lower before the PCR and higher after the PCR. Final DNA libraries were stored at − 20 °C until further processing.
Quality and quantity of the libraries was measured using the Qubit™ 2.0 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) fluorometer and Agilent 2200 TapeStation system and High Sensitivity D1000 ScreenTape (Agilent, Santa Clara, CA, USA) according to the manufacturer's protocol. All 96 samples have been divided into 8 groups, so 12 patients have been sequenced in each run. Also, we stratified samples semi-randomly, as to have tumor and non-tumor tissue and early as well as late-relapse patients in each run to minimize batch effect. Samples have been pooled equimolar at the 4 nM concentration, which has been rechecked using the Qubit™ 2.0 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) fluorometer. The size of each pool was checked using Agilent 2200 TapeStation system and High Sensitivity D1000 ScreenTape (Agilent, Santa Clara, CA, USA) according to the manufacturer's protocol. Pools containing fragments of undesirable length have been purified using Agencourt AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA).
Following the Illumina Denature and Dilute protocol, the polls have been denatured and diluted to 1,8 pM final concentration and loaded onto a sequencing cassette from NextSeq 500/550 High Output v2 kit, 75 cycles (Illumina, San Diego, CA, USA) and run according to the manufacturer's protocol.
Data analysis
Raw data from the Illumina NextSeq 500 was converted to fastq using bcl2fastq2 Con-version software (version 2.20.0), and read quality was checked using FastQC (version 0.11.9) (Andrews 2010). Adapter sequences were identified using the Kraken system (version 16-098) (Davis et al. 2013), and poor read ends were removed using Trimmomatic (version 0.39). The 3 'ends with a threshold value less than five and reads shorter than 35 bp have been considered poor and were removed. The modified libraries were mapped with the STAR tool (version 2.7.0d) (Dobin et al. 2013) to the human genome, the sequence of which was downloaded from the GENCODE (version 37) database. During mapping, each reading was allowed to map to up to 20 different locations. Genes were quantified using RSEM software (version 1.3.1) (Li and Dewey 2011), and differentially expressed lncRNAs were identified using the DESeq2 tool (version 1.18.1) (Love et al. 2014) and edgeR package (version 3.30.3). Six samples have failed library preparation resulting in almost no reads, and thus were removed from any subsequent analysis. Another 24 samples were removed due to low number of gene-aligned reads (less than 10 million). Further, 14 samples were removed due to failed DESeq2 normalization and 4 samples were removed due to more issues. Remaining 56 samples were analyzed and the results are shown at the Results section.
PCR validation of the results
Based on the results of sequencing, 20 candidate lncRNAs were selected. Ten candidates were chosen based on the comparison of early and late-relapse patients (prognostic lncRNAs), other ten were chosen based on the comparison of tumor and non-tumor tissue (diagnostic lncRNAs). In diagnostic lncRNAs, we simply took first ten lncRNAs with assays available in our supplier, Thermo Fisher. In prognostic lncRNAs, we took lncRNAs with base mean > 20 (except TSPEAR due to its low padj value we decided to test it as well). When choosing specific assays from our supplier, we decided to take only assays which spanned exons, had best coverage and possibly the highest RefSeq number.
For the validation of the sequencing results, we used the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) for reverse transcription. TaqMan™ Gene Expression Master Mix (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) was used in qPCR run on QuantStudio 12 K Flex Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol. TaqMan® Gene Expression Assays used for qPCR along with the catalogue number of product (Assay ID) (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) are listed in Table 2 below. The expression of PPIA was used as a normalization standard and reference gene based on literature search and our previous experience with the expression of PPIA in RCC tissue (Bohosova et al. 2022).
Statistical analysis
For the statistical analysis of the validation phase results, we used normalized expression data which were evaluated using Mann–Whitney U Test, Wilcoxon test, ROC analysis, and Kaplan–Meier analysis (GraphPad Prism 8, GraphPad Software, La Jolla, CA, USA). P values lower than 0,05 were considered statistically significant. Combined ROC curves were prepared using both GraphPad Prism 8 and JMP software (SAS-JMP software, SAS Institute, Cary, North Carolina, USA). All statistically significant lncRNAs in stage and relapse comparison and all diagnostic lncRNAs with AUC > 0.9200 were analyzed in different combinations of the lncRNAs in the panels. Based on the results of combined ROC analysis for several lncRNAs, a diagnostic Dx score has been developed:
Results
lncRNA expression profiles in RCC patients
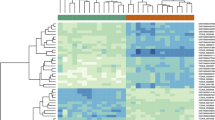
Next-generation sequencing results were analyzed using the DESeq2 tool. Comparing the tumor and paired non-tumor tissue, we identified 2800 dysregulated lncRNAs (Fig. 1a, b). Twenty most dysregulated lncRNAs are listed in Table 3. However, when comparing early and late responders, we found only three significantly dysregulated lncRNAs in patients with early relapse of the disease (Table 4). In Tables 5 and 6 are listed all the lncRNAs chosen for validation along with their specific performance in sequencing.

a Clastrogram and the heatmap showing 2800 significantly dysregulated lncRNAs in 28 samples of tumor tissue (pink) and s28 non-tumor tissue samples (blue). In the heatmap, red shows higher expression, and blue shows lower expression. lncRNAs with foldchange > 2 and p < 0.05 were considered significant. b Clastrogram and the heatmap showing showing 20 most significantly dysregulated lncRNAs in 28 samples of tumor tissue (pink) and 28 non-tumor tissue samples (blue). In the heatmap, red shows higher expression, and blue shows lower expression. lncRNAs with foldchange > 2 and p < 0.05 were considered significant
qPCR validation of sequencing results
Twenty selected candidate lncRNAs were analyzed using qPCR. Their expression levels have been measured in tumor and paired non-tumor tissue of all patients in the validation cohort and Cq values were normalized to the expression of PPIA. Fourteen lncRNAs have been successfully validated as significantly dysregulated in tumor tissue compared to the paired non-tumor tissue. Four lncRNAs (ZNF667-AS1, RMST, COLCA1, and SNHG3) were significantly dysregulated also in patients with early relapse compared to the late-relapsing patients. We also compared patients in lower stages (stage 1 and 2) with patients in higher stages (stage 3 and 4). Six lncRNAs also shown association with stage of the patients. Results of all lncRNAs with statistically significant dysregulation of expression (in any comparison) are shown in Fig. 2. In all validated lncRNAs we also did ROC analysis (Figs. 3 and 4). Understandably, lncRNAs differed in the ability to distinguish a tumor and non-tumor tissue, however, ten of them showed satisfactory AUC above 0.75. All results are summarized in Table 7.

Significantly dysregulated lncRNAs, each graph shows all comparisons analyzed for each lncRNA (tumor vs. non-tumor, stage I + II vs. stage III + IV, early relapse vs. late relapse)

Graph showing ROC curves of candidate lncRNAs dysregulated in tumor tissue compared to non-tumor tissue of RCC patients

Graph showing ROC curves of candidate lncRNAs dysregulated in tumor tissue of patients with or without early relapse (labeled Relapse) of with different stage of the disease (labeled Stage)
In order to gain better discriminatory results, we combined statistically significantly dysregulated lncRNAs into a diagnostic, and prognostic models. Logistic regression analysis showed that linear combination of AZGP1P1, CDKN2B-AS1, COL18A1, and RMST provided best diagnostic discrimination (AUC 0.9808, sensitivity 95.96%, specificity 90.4%) outperforming other combinations of lncRNAs (Table 8). In stage comparison, the combination of ZNF667-AS1, PVT1, RMST, LINC00955, and TCL6 yielded best results with AUC 0,812, sensitivity 85.71% and specificity 69,41%. And although even if the combination of COLCA1, RMST, SNHG3, and ZNF667-AS1 in relapse comparison did bring higher AUC than RMST, SNHG3, and ZNF667-AS1 alone, it did not outperformed AUC of COLCA1 alone, which makes this lncRNA a superior prognostic biomarker (Fig. 5).

Graph showing combined ROC curves of the most significantly dysregulated lncRNAs diagnostic of prognostic setting
Discussion
In this study, we provide valuable results showing set of exceptional diagnostic and prognostic biomarkers feasible in RCC therapy. Our main aim was to sequence transcriptome of a large set of patients and provide lncRNA expression profiles of a novel and unique patient cohort. We identified more than 2800 significantly dysregulated lncRNAs, out of which we chose twenty for further validation on an independent cohort. Moreover, we aimed to validate diagnostic and prognostic value of selected candidate lncRNAs and their association with early relapse and/or stage, which was successful as 14 out of 20 lncRNAs did perform statistically significantly (p < 0.0001 in diagnostic setting, p < 0.05 in prognostic setting). To achieve better discriminatory results, we tested several combinations of lncRNAs and identified panels with the best diagnostic and prognostic value. However, in case of early relapse prediction, even the combination achieving the highest AUC did not outperform lncRNA COLCA1. To our best knowledge, we are the first to show that this lncRNA holds such a prognostic power and is significant not only regarding the early relapse after nephrectomy but in RCC in general, as there is only one other work mentioning COLCA1 in relation to RCC as one of nine redox-related lncRNA associated with overall survival of RCC patients (Qi-Dong et al. 2020).
There are however, several other lncRNAs in our panel, which are not frequent in similar RCC-focused studies. For example AZGP1P1 has been associated only with prognosis in breast cancer (Liu et al. 2019), LINC00462, which has been associated with ferroptosis according to one recent work (Wu et al. 2022b), LINC00955 without any other evidence, RMST, which is known to be involved in the development of other tumors, but not RCC (Chen et al. 2022) and similarly ZNF667-AS1, also known as MORT (Vrba and Futscher 2018).
On the other hand, we also identified several notoriously known lncRNAs typically associated with development of RCC. There is CDKN2B-AS1, known also as ANRIL, is up-regulated in RCC tissue which corresponds with our results (Angenard et al. 2019; Dasgupta et al. 2020; Xie et al. 2021) and there are even some single nucleotide polymorphisms identified which are typically associated with RCC (Li et al. 2014). COL18A1 is downregulated as in our study also in other works (Yang et al. 2018; Angenard et al. 2019; Liu et al. 2022) and its expression seems to be specific for chromophobe RCC (He et al. 2016). Dysregulation of LINC00472 along with PVT1 and LUCAT1 has been described in our previous work (Bohosova et al. 2022). LINC00472 has been mentioned also in other works as downregulated in RCC and associated with progression of the disease. Even some mechanisms of LINC00472 functioning have been elucidated (Gao and Wang 2021; Xiang et al. 2021; Wang et al. 2022). PVT1 and LUCAT1 are very well-described lncRNAs in RCC as their impact on overall survival has been shown among several other works also in meta-analysis from Wang et al. (Wang et al. 2019). Involvement of PVT1 into the development of RCC has been thoroughly reviewed also in Bohosova et al. (2021). Similarly, TCL6 has also been shown to be involved in development of RCC as a tumor suppressor (Rysz et al. 2022) and interestingly, artificial increase of TCL6 in cancer cells sensitizes them to paclitaxel, which could pose a novel therapeutical opportunity (Chen et al. 2020).
Although most results correspond with other works, there were some discrepancies in the direction of change in expression, namely in SNHG3 and LINC00887. While SNHG3, an autophagy-related lncRNA (Xuan et al. 2021), was downregulated in our study, other works show its upregulation in RCC (Zhang et al. 2019; Yang et al. 2020; Xu et al. 2021). Similarly, LINC00887 was downregulated in our study and up-regulated in study of Wu et al. (Wu et al. 2022a). However, the main drawback of current state of knowledge regarding the lncRNA dysregulation in RCC lies in the sequencing data, as in majority of works the same set of TCGA sequencing data was analyzed. Therefore, high degree of inter-rater reliability is expected, while some discrepancies between TCGA and our independent cohort are inevitable.
In conclusion, we identified distinct expression profiles of lncRNA in renal tumor tissue and based on our exploratory data, we successfully validated several highly reliable diagnostic and prognostic tissue biomarkers. Moreover, combination of lncRNAs with the best statistical values provided even better discriminatory values. Our study shows that besides diagnosis and overall survival prognostication, lncRNAs can discern also between patients with different length of relapse-free survival and thus should be considered for further independent validation in larger cohorts.
Data availability
Raw sequencing data were generated at CEITEC Genomics Core Facility and are publicly available at the Sequence Read Archive under accession number PRJNA929051. Derived data supporting the findings of this study are available from the corresponding author on request.
References
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data
Angenard G, Merdrignac A, Louis C et al (2019) Expression of long non-coding RNA ANRIL predicts a poor prognosis in intrahepatic cholangiocarcinoma. Dig Liver Dis off J Ital Soc Gastroenterol Ital Assoc Study Liver 51:1337–1343. https://doi.org/10.1016/j.dld.2019.03.019
Bhan A, Soleimani M, Mandal SS (2017) Long non-coding RNA (LncRNA) and cancer: a new paradigm. Cancer Res 77:3965–3981. https://doi.org/10.1158/0008-5472.CAN-16-2634
Bohosova J, Kubickova A, Slaby O (2021) lncRNA PVT1 in the pathogenesis and clinical management of renal cell carcinoma. Biomolecules 11:664. https://doi.org/10.3390/biom11050664
Bohosova J, Kasik M, Kubickova A et al (2022) LncRNA PVT1 is increased in renal cell carcinoma and affects viability and migration in vitro. J Clin Lab Anal 36:e24442. https://doi.org/10.1002/jcla.24442
Capitanio U, Bensalah K, Bex A et al (2019) Epidemiology of renal cell carcinoma. Eur Urol 75:74–84. https://doi.org/10.1016/j.eururo.2018.08.036
Chandra Gupta S, NandanTripathi Y (2017) Potential of long non-coding RNAs in cancer patients: from biomarkers to therapeutic targets. Int J Cancer 140:1955–1967. https://doi.org/10.1002/ijc.30546
Chen Z, Zhuang Q, Cheng K et al (2020) Long non-coding RNA TCL6 enhances preferential toxicity of paclitaxel to renal cell carcinoma cells. J Cancer 11:1383–1392. https://doi.org/10.7150/jca.32552
Chen X, Liu K, Xu W et al (2022) Tumor-related molecular regulatory mechanisms of long non-coding RNA RMST: recent evidence. Mini Rev Med Chem 22:1374–1379. https://doi.org/10.2174/1389557521666211202150646
Chi Y, Wang D, Wang J et al (2019) Long non-coding RNA in the pathogenesis of cancers. Cells 8:1015. https://doi.org/10.3390/cells8091015
Dasgupta P, Kulkarni P, Majid S et al (2020) LncRNA CDKN2B-AS1/miR-141/cyclin D network regulates tumor progression and metastasis of renal cell carcinoma. Cell Death Dis 11:660. https://doi.org/10.1038/s41419-020-02877-0
Davis MPA, van Dongen S, Abreu-Goodger C et al (2013) Kraken: a set of tools for quality control and analysis of high-throughput sequence data. Methods 63:41–49. https://doi.org/10.1016/j.ymeth.2013.06.027
Decastro GJ, McKiernan JM (2008) Epidemiology, clinical staging, and presentation of renal cell carcinoma. Urol Clin North Am 35(581–592):vi. https://doi.org/10.1016/j.ucl.2008.07.005
Dobin A, Davis CA, Schlesinger F et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinforma Oxf Engl 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Escudier B, Porta C, Schmidinger M et al (2019) Renal cell carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up†. Ann Oncol off J Eur Soc Med Oncol 30:706–720. https://doi.org/10.1093/annonc/mdz056
Gao S, Wang Z (2021) Comprehensive analysis of regulatory network for LINC00472 in clear cell renal cell carcinoma. J Healthc Eng 2021:3533608. https://doi.org/10.1155/2021/3533608
He H-T, Xu M, Kuang Y et al (2016) Biomarker and competing endogenous RNA potential of tumor-specific long noncoding RNA in chromophobe renal cell carcinoma. OncoTargets Ther 9:6399–6406. https://doi.org/10.2147/OTT.S116392
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 12:323. https://doi.org/10.1186/1471-2105-12-323
Li W-Q, Pfeiffer RM, Hyland PL et al (2014) Genetic polymorphisms in the 9p21 region associated with risk of multiple cancers. Carcinogenesis 35:2698–2705. https://doi.org/10.1093/carcin/bgu203
Liu Z, Li M, Hua Q et al (2019) Identification of an eight-lncRNA prognostic model for breast cancer using WGCNA network analysis and a Cox-proportional hazards model based on L1-penalized estimation. Int J Mol Med 44:1333–1343. https://doi.org/10.3892/ijmm.2019.4303
Liu Y, Wang J, Shou Y et al (2022) Restoring the epigenetically silenced lncRNA COL18A1-AS1 represses ccRCC progression by lipid browning via miR-1286/KLF12 axis. Cell Death Dis 13:578. https://doi.org/10.1038/s41419-022-04996-2
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. https://doi.org/10.1186/s13059-014-0550-8
Padala SA, Barsouk A, Thandra KC et al (2020) Epidemiology of renal cell carcinoma. World J Oncol 11:79–87. https://doi.org/10.14740/wjon1279
Qi-Dong X, Yang X, Lu J-L et al (2020) Development and validation of a nine-redox-related long noncoding RNA signature in renal clear cell carcinoma. Oxid Med Cell Longev 2020:6634247. https://doi.org/10.1155/2020/6634247
Quinn JJ, Chang HY (2016) Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17:47–62. https://doi.org/10.1038/nrg.2015.10
Rysz J, Konecki T, Franczyk B et al (2022) The Role of Long Noncoding RNA (lncRNAs) Biomarkers in Renal Cell Carcinoma. Int J Mol Sci 24:643. https://doi.org/10.3390/ijms24010643
Vrba L, Futscher BW (2018) Epigenetic silencing of lncRNA MORT in 16 TCGA cancer types. F1000Research 7:211. https://doi.org/10.12688/f1000research.13944.1
Wang Y, Li Z, Li W et al (2019) Prognostic significance of long non-coding RNAs in clear cell renal cell carcinoma: a meta-analysis. Medicine (baltimore) 98:e17276. https://doi.org/10.1097/MD.0000000000017276
Wang S, Luo C, Li B et al (2022) LINC00472 inhibits cell migration by enhancing intercellular adhesion and regulates H3K27ac level via interacting with P300 in renal clear cell carcinoma. Cell Death Discov 8:454. https://doi.org/10.1038/s41420-022-01243-7
Wu J, Lin R, Zhang L et al (2022a) LINC00887 Fosters development of clear cell renal cell carcinoma via inhibiting CD8+ T Cell immune infiltration. Comput Math Methods Med 2022:2582474. https://doi.org/10.1155/2022/2582474
Wu Z, Huang X, Cai M, Huang P (2022b) Potential biomarkers for predicting the overall survival outcome of kidney renal papillary cell carcinoma: an analysis of ferroptosis-related LNCRNAs. BMC Urol 22:152. https://doi.org/10.1186/s12894-022-01037-0
Xiang Z, Shen E, Li M et al (2021) Potential prognostic biomarkers related to immunity in clear cell renal cell carcinoma using bioinformatic strategy. Bioengineered 12:1773–1790. https://doi.org/10.1080/21655979.2021.1924546
Xie X, Lin J, Fan X et al (2021) LncRNA CDKN2B-AS1 stabilized by IGF2BP3 drives the malignancy of renal clear cell carcinoma through epigenetically activating NUF2 transcription. Cell Death Dis 12:201. https://doi.org/10.1038/s41419-021-03489-y
Xu Z, Ye J, Bao P et al (2021) Long non-coding RNA SNHG3 promotes the progression of clear cell renal cell carcinoma via regulating BIRC5 expression. Transl Cancer Res 10:4502–4513. https://doi.org/10.21037/tcr-21-1802
Xuan Y, Chen W, Liu K et al (2021) A risk signature with autophagy-related long noncoding RNAs for predicting the prognosis of clear cell renal cell carcinoma: based on the TCGA database and bioinformatics. Dis Markers 2021:8849977. https://doi.org/10.1155/2021/8849977
Yang K, Lu X-F, Luo P-C, Zhang J (2018) Identification of six potentially long noncoding RNAs as biomarkers involved competitive endogenous RNA in clear cell renal cell carcinoma. BioMed Res Int 2018:9303486. https://doi.org/10.1155/2018/9303486
Yang W, Zhang K, Li L et al (2020) Discovery and validation of the prognostic value of the lncRNAs encoding snoRNAs in patients with clear cell renal cell carcinoma. Aging 12:4424–4444. https://doi.org/10.18632/aging.102894
Zhang C, Qu Y, Xiao H et al (2019) LncRNA SNHG3 promotes clear cell renal cell carcinoma proliferation and migration by upregulating TOP2A. Exp Cell Res 384:111595. https://doi.org/10.1016/j.yexcr.2019.111595
Acknowledgements
This work was supported by the Ministry of Health, the Czech Republic, grant number NV18-03-00554, conceptual development of research organizations (MOU, 00209805), European Regional Development Fund-Project BBMRI-CZ: Biobank network—a versatile platform for the research of the etiopathogenesis of diseases, No. LM2018125 and the project National Institute for Cancer Research (Programme EXCELES, ID Project No. LX22NPO5102)—Funded by the European Union—Next Generation EU. We acknowledge the CF Genomics of CEITEC supported by the NCMG research infrastructure (LM2018132 funded by MEYS CR) for their support with obtaining scientific data presented in this paper.
Funding
Open access publishing supported by the National Technical Library in Prague.
Author information
Authors and Affiliations
Contributions
Conceptualization, OS, and MS; methodology, OS; validation, DA, ON, and KK; resources, OS; data curation, MS and AP; writing—original draft preparation, JB, writing—review and editing, OS; visualization, KT and DA; supervision, JB; funding acquisition, OS, and AP. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bohosova, J., Kozelkova, K., Al Tukmachi, D. et al. Long non-coding RNAs enable precise diagnosis and prediction of early relapse after nephrectomy in patients with renal cell carcinoma. J Cancer Res Clin Oncol 149, 7587–7600 (2023). https://doi.org/10.1007/s00432-023-04700-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-023-04700-7