
Abstract
Purpose
To explore transcriptome and immunological features of patients with Ewing sarcoma (ES) using all publicly available microarray data.
Methods
Data of 479 ES tissues were integrated and normalized. Gene expression, immune infiltration, and cancer-specific pathways were analyzed. Genes of interest were knocked down, followed by cell proliferation and colony formation assays.
Results
Consistent with the previous reports of differential expressed genes (DEGs) in ES, our analysis identified CCND1, HMCN1, and NKX2-2 were among the most highly expressed, while TWNC1, MYBPC1, and CKM were among the lowest expressed genes. GO, KEGG, and GSEA enrichment analysis identified that the DEGs related to bone and muscle functioning, those that contributed to crucial cellular, and metabolism pathways such as actin binding, apoptosis, TCA cycle, and cell cycle were also significantly enriched. Immune infiltration analysis discovered that many T cell subsets including CD4T, CD8 T, and Gamma delta T cells were highly infiltrated, while monocytes and B cells were less infiltrated in tumors. A total of 138 genes were both significantly up-regulated in tumors and associated with decreased survival, while 38 significantly down-regulated genes were associated with increased survival, many of which were previously reported as oncogenes and tumor suppressors in ES and other cancers. Silencing of four newly identified top ranked up-regulated genes with decreased survivals in ES inhibited proliferation and colony formation of ES cells.
Conclusion
This study may provide a clear representative transcriptome profile of ES, providing diagnostic biomarkers, pathways, and immune infiltrative characteristics targets for ES.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ewing sarcoma (ES) is a highly malignant bone and soft tissue sarcoma that occurs mostly in children and adolescents (Grünewald et al. 2018). The 5-year overall survival rate has risen to 70–75% in children and adolescents (Whelan et al. 2018; Womer et al. 2012), while adult survival is a little lower than in them (Ahmed et al. 2013). 20–25% of patients had metastases at the time of diagnosis, and they are likely to be resistant to intensive therapy and have shown a very poor cure rate (Gaspar et al. 2015; Khanna et al. 2017).
The disease is characteristic of fusion mutations of the ES gene to an E26 transformation-specific transcription factor, most commonly FLI1 (Riggi et al. 2021). Moreover, many genes such as LSD1 and some tyrosine kinases have been reported to be overexpressed and considered targets in ES (Balamuth and Womer 2010; Sankar et al. 2014). However, the underlying mechanism of how these driver mutations transform tumorigenesis is still not clear. Thus, a comprehensive understanding of genetic profiles would allow us to uncover crucial differential genes, biomarkers, pathways, and potential therapeutical options for ES patients. However, due to the rare occurrence, resources of genetic studies of ES, especially for RNA-seq data, is limited. Compared with RNA-seq, small cohorts of microarray studies have been reported during the last decades (Agelopoulos 2015; Postel-Vinay et al. 2012; Savola et al. 2011; Scotlandi et al. 2009; Specht et al. 2014; Surdez et al. 2021; Volchenboum et al. 2015), which could be unified and summarized in a representative way. In this study, we performed integrated analyses of all publicly available microarray datasets of ES patients to our best knowledge and uncovered a genetic profile for a better understanding of the characteristics and optional therapeutic targets for ES patients.
Materials and methods
Datasets
A total of ten datasets, GSE12102, GSE17679, GSE34620, GSE37371, GSE45544, GSE63157, GSE73166, GSE68776, and GSE142162 (For detailed information, see Table1), attributed to GPL6244, GPL5175, and GPL570 platforms, were obtained from the NCBI database (www.ncbi.nlm.nih.gov/geo/). A total of 20 samples from the GSE37371 dataset, which were tested on the GPL96 platform, were excluded from the study. In total, microarray data of 479 tumor samples and 22 normal muscle samples were applied for subsequent analyses.
Data procession
The Affy package (version 1.68.0) of R software (version 4.0.3) was used to normalize the raw data of datasets (CEL files). According to the microarray annotation file, the expression value of the probe was correlated with the gene expression. We then calculated the average expression level for genes corresponding to multiple probes, sorted and selected the highest value, then combined all the data into one dataset based on overlapping genes.
Differential gene analysis
Limma package (version 3.46.0) was applied to remove batch effects from the dataset obtained above, and the tumor and normal tissues were compared to identify differentially expressed genes. Genes with adjusted p < 0.05 and |logFC|> 1 were deemed to have significantly changed expression.
Survival analysis
The differentially expressed genes are likely to play important roles in the development of ES. Clinical data for GSE17618 (which is part of GSE17679) matched our hypotheses. Overall survival analysis was performed using the Survival (version 3.2.7) package and Survminer (version 0.4.9) package. The samples (total 44 samples) were split into two groups (high/low) based on the expression of significantly different genes, with a median gene expression level as the cutoff. The Kaplan–Meier survival curves were drawn using the ggplot2 package (version 3.3.3) and log-rank testing for significance. Statistical significance was defined as p < 0.05.
The profile of differentially expressed genes (DEGs)
The heatmap, which displays the expression level of the significant DEGs in samples of the dataset, was created using the pheatmap package (version 1.0.12). The DEGs were displayed using a volcano plot created with the ggplot2 package (version 3.3.3).
Pathway analysis
The GO database contains three domains: biological process (BP), cellular component (CC), and molecular function (MF). To analyze the function of DEGs, the clusterProfiler package (version 3.18.1) and the KEGG.db package (version 3.2.4) were used to perform GO analysis and KEGG pathway analysis, respectively. The parameter of GO analysis is set as minGSSize = 1, pvalueCutoff = 0.01, qvalueCutoff = 0.01. Gene set enrichment analysis (GSEA) was performed with h.all.v7.4.symbols.gmt as a reference gene set. The clusterProfiler Package (version 3.18.1) and enrichplot (version 1.10.2) were used to perform GSEA analysis and visualize the results.
Immune infiltration analysis
Microarray datasets were uploaded and analyzed in the ImmuCellAI website (http://bioinfo.life.hust.edu.cn/ImmuCellAI#!/analysis) (Miao et al. 2020). A significance test was further performed on the basis of the results of immune infiltration.
Cell culture
The ES cell line A-673 (RRID:CVCL_0080) was obtained from Procell Life Science Ltd (Wuhan, China). The cell line was maintained in DMEM supplemented with 10% of FBS and 1% of penicillin–streptomycin. Cells were incubated at 37 °C in a humidified atmosphere containing 5% of CO2. We declare that the cell line is free of mycoplasma infection and has been identified within 3 years. The testing method is as follows: DNA was extracted by a commercial kit from CORNING (AP-EMN-BL-GDNA-250G). Twenty short tandem repeat (STR) loci plus the gender-determining locus, amelogenin, were amplified by six multiplex PCR and separated on ABI 3730XL Genetic Analyzer. The signals were then analyzed by the software GeneMapper.
Small interfering RNA transfection
A-673 cells were seeded in six-well culture plates and transfected with siRNA and GP-transfect-Mate (GenePharma, Suzhou, China) according to the manufacturer’s instructions. The culture medium was replaced after 5 h and gene expressions were determined by the reverse transcription‑quantitative polymerase chain reaction (RT‑qPCR) after 2 days. The sequences of all siRNAs tested in the present study are listed in Table S1.
RT‑qPCR analysis
Total RNA was isolated from ES cell line A-673 using a Tiangen ®TRNzol Universal Reagent (TIANGEN Biotech, Beijing, China) as per the manufacturer's protocol. cDNA was reverse transcribed from the total RNA using the HiScript® III RT SuperMix with gDNA wiper for RT‑qPCR (Vazyme Biotech, Nanjing, China) according to the manufacturer’s protocol. RT‑qPCR reactions were performed on an ABI 7900 real‑time PCR system (Thermo Fisher Scientific, Waltham, USA) using the following conditions: 95 ˚C for 30 s, followed by 40 cycles of 95 ˚C for 10 s and 60 ˚C for 30 s. The relative expressions of genes were determined by the 2−ΔΔCt method with GAPDH as an internal reference. The primer sequences applied in the present study are listed in Table S2.
Cell proliferation assay
A-673 cells were seeded in the six-well culture plates and transfected with siRNAs and GP-transfect-Mate mixtures (GenePharma, Suzhou, China) according to the manufacturer’s instructions. After 48 h, We counted cells directly under the microscope as the starting cell count. Then we counted the number of cells at 72 h, 96 h and 120 h, respectively. Finally, the proliferation rate relative to the number of starting cells was calculated.
Colony-forming cell assay
A-673 cells were seeded in the six-well culture plates and transfected with siRNAs and GP-transfect-Mate mixtures (GenePharma, Suzhou, China) according to the manufacturer’s instructions. The culture medium was replaced after 5 h. cells were then harvested after 1 day, and 500 of them were reseeded on 6-cm plates with DMEM complete medium for approximately 14 days of incubation. Colonies were finally fixed with methanol, stained with crystal violet dye, and counted using ImageJ software.
Prognosis analysis of single-sample geneset group
The samples were clustered using the R package mclust (version 5.4.10) based on the most significant gene set from GSEABase (http://www.gsea-msigdb.org/gsea/msigdb/human/geneset/HALLMARK_E2F_TARGETS.html), and variations in the gene expression of different clusters were presented using heatmaps. Based on these differences, the samples (total 44 samples) were classified into two groups. The R package survival (version 3.2.7) and Survminer (version 0.4.9) were used to perform univariate COX regression analysis on different groups to detect the degree of prognostic impacts.
Results
Study overview and dataset characteristics
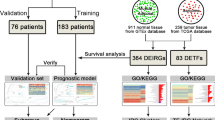
As shown in Fig. 1, step-by-step analyses were carried out using the integrated microarray dataset derived from nine independent microarray studies, covering 479 ES samples and 22 muscle samples. Prognostic impact (Kaplan–Meier) analysis of DEGs was done on a sub-cohort (GSE17618) where the survival data is available.

Study workflow shows overall steps carried out in the integrated microarray analyses
Identification of DEGs in ES
After a proper normalization of batch effects among different arrays (Fig. S1), we observed a total of 2383 DEGs (Table S3), including 1601 up-regulated and 782 down-regulated genes, using the P value < 0.05 and |logFC|> 1 criterion in ES samples compared to normal muscle samples, with green and red representing low and high expression levels, respectively (Fig. S4A). Similarly, a volcano plot of DEGs was depicted and the top ten logFC genes were listed (Fig. 2A). To our interest, several genes on our list have been reported to be involved in the progression of ES, such as NKX2-2, HMCN1, HOXD10, and PRKCB. NRX2-2 participates in the formation of the core regulatory circuitry, activates EWS–FLI1 transcription, and significantly promotes the proliferation of ES cells both in vitro and in vivo (Sarver et al. 2015; Shi et al. 2020; Surdez et al. 2012; Svoboda et al. 2014). To our interest, the DEGs were overwhelmingly attributed to the development of muscle and bone, indicating that the disease interrupts muscle and bone growth, which is also evidenced by plain radiographs exhibiting permeative and infiltrative destruction of the affected bone (mainly in the diaphysis of the long bone) (Ozaki 2015). Similarly, intramyofiber invasion, a particular phenomenon in which malignant cells exhibit intracellular spread into skeletal muscle cells, usually occurs in ES (Mahjoub et al. 2006).

Identification of differentially expressed genes (DEGs) and enriched pathways in ES. (A) A volcano plot of DEGs in tumor samples compared to normal samples. Red indicates the gene expression was up-regulated in tumor samples compared to normal samples (P < 0.05 and logFC > 1); green indicates the gene expression was down-regulated in tumor samples compared with normal samples (P < 0.05 and logFC < -1); Black indicates the |logFC|< 1. (B–D) GO analysis. The size of the dots represents the gene ratio. E KEGG pathway enrichment analysis. The size of the dots represents the number of DEGs in the pathway. F GSEA analysis. Red indicates NES > 0; green indicates NES < 0. The size of the dots represents the number of DEGs in the pathway. (G–H) The GSEA plot of the two pathways: G2M checkpoint and E2F Targets
Identification of enriched pathways in ES
The GO database categorizes gene and protein functions into three categories: biological process (BP), cellular component (CC), and molecular function (MF). The top 20 significant categories in BP, CC, and MF are shown Fig. 2B–D. The DEGs were primarily involved in the muscle-related processes in the BP category such as muscle system, muscle contraction, and muscle tissue development. Contractile fibers, myofibrils, sarcomere-related genes, and those associated with the growth of muscles showed considerably higher expression in CC. Furthermore, the MF analysis revealed an abundance of actin, actin filament, and calmodulin binding-related genes. The genes related to the bone and muscle functioning also showed higher expression. The DEGs were largely associated with the development of muscle and bone, suggesting that the development of Ewing sarcoma impacts muscle and bone growth. In addition, KEGG pathway enrichment analysis discovered that the DEGs were found to be engaged in several crucial cellular pathways associated with cancer development and cell activity, such as the p53 signaling pathway and the cell cycle (Fig. 2E). We also found that several non-cancer related pathways including citrate cycle, dilated cardiomyopathy, cardiac muscle contraction and focal adhesion were enriched in KEGG analysis. Interestingly, many of these pathways such as dilated cardiomyopathy and cardiac muscle contraction, have also been discovered to be related to ES progress in a recent study (Ding et al. 2021). Finally, GSEA analysis showed 25 significantly activated and 7 suppressed pathways (Fig. 2F). The top two activated pathways (NES > 0) were E2F targets and G2M checkpoint, both of which were related to the cell cycle, indicating that DEGs have possible effects on the cell cycle (Figs. 2G–H) and many other altered gene sets involved in such as KRAS, apoptosis, and DNA repair pathways (Figs. S2-3). In addition, seven suppressed pathways including oxidative phosphorylation were enriched by GSEA analysis. Indeed, ES is a highly aggressive and metabolically active malignant tumor and its metabolic activity can broadly be characterized by features of glycolytic activity and oxidative phosphorylation (Atreyi Dasgupta and Shuck 2017). A previous study has also shown that strong oxidative phosphorylation metabolism is associated with a well-defined range of EWSR1-FLI1 activity (Aynaud et al. 2020).
Analysis of association between DEGs and survival
The up-regulated and down-regulated genes were screened using the logFC > 1 or logFC < -1 criteria. Then GSE17618 datasets were used to determine the survival. A total of 138 overlapping DEGs were identified in the two datasets which satisfied the criteria P value < 0.05, HR > 1, and logFC > 1. Similarly, a total of 38 overlapping DEGs were identified in the two datasets, which satisfied the criteria P value < 0.05, HR < 1 and logFC < -1 (Fig. 3A), and a heat map of the of all the overlapping DEGs (Fig. S4B) and the top 10 up-regulated and down-regulated overlapping DEGs and their prognostic impacts are shown (Fig. 3B). In addition, we selected the most significant pathway gene sets in the GSEA analysis to validate the association of pathways with prognosis. As shown in Fig. 3C, enrichment of E2F-Targets pathway-based gene set grouping showed decreased overall survival in our dataset. Many previously identified DEGs including CCNB2, RACGAP1, and CKS2 also participated in this pathway (Fig. 3D). Together, these results demonstrated that E2F-Targets pathway may play an important role in the progression of ES.

Analysis of association between DEGs and survival. (A) Screening of DEGs. A total of 138 overlapping DEGs were identified in the two datasets which satisfied the criteria HR > 1 and logFC > 1. A total of 38 overlapping DEGs were identified in the two datasets which satisfied the criteria HR < 1 and logFC < -1. B Heatmap of the top ten overlapping DEGs (10 + 10) mentioned above. C The samples were grouped based on the expression level of the E2F-Targets geneset, and survival analysis was performed using GSE17618 survival information. The red and blue lines represent two different groups. D Gene expression heatmap of E2F-Targets geneset for grouped samples
Immune infiltration analysis
We performed the immune infiltration analysis between tumor and normal samples. We calculated the proportion of each immune infiltrated cell type (Fig. S5) and observed that a number of T cell subgroups, such as CD4 T, CD8 T, Gamma delta T, Tcm, and Tex, were highly infiltrated in tumor samples than those in normal samples. On the contrary, larger amounts of monocytes and B cells were infiltrated in normal samples than in tumor samples (Fig. 4A, B), while other lymphocytes were not significantly altered (Fig. S6).

Immune infiltration analysis results. Significant highly (A) and lowly (B) immune infiltrated lymphocytes in tumor than those in normal samples are shown
In vitro verification of four up-regulated DEGs with decreased survival
To further confirm and evaluate potential therapeutic values from the novel identified up-regulated DEGs with the impacts of decreased survival in the current study, we chose to validate genes that were the top ten up-regulated DEGs with decreased survival and not shown to be associated with tumorigenesis. Among them, studies have proved that CCNB1, RACGAP1 and NUSAP1 promote the progression of multiple cancers (Aljohani et al. 2022; Gu et al. 2022; Guo et al. 2020), and NCAPG and CCNB2 have been found to be associated with a poor prognosis of ES (Kakar et al. 2022; Zhao et al. 2022), while ASPM, CENPF, CKS2, and DTL have not been studied in ES to the best of our knowledge. Interestingly, these four genes were also found in the GO analysis and GSEA results (Fig. 2B–H). As shown in Fig. 5A, B, increased expressions of these four genes are significantly linked to worse overall survival. We then transiently transfected siRNAs targeting ASPM, CENPF, CKS2, and DTL, and the confirmed high knock-down efficiencies by qRT-PCR in A-673 cells (Fig. 5C). Direct cell counting under microscope after transfection revealed that all knock-down experimental groups had significantly fewer cells on days 2 and 3 compared to the siRNA control group (Fig. 5D). Furthermore, clonogenic assay demonstrated that silencing of the four genes severely reduced the ability of A673 cells to form colonies (Fig. 5E, F).

In vitro verification of four up-regulated DEGs with decreased survival. (A) Comparison of expression of four DEGs (ASPM, CENPF, CKS2, and DTL) between tumor and normal samples. Normal samples (N): blue, tumor samples (T): red. B The Kaplan–Meier survival curves of the selected four genes. C RT‑qPCR verification of silencing efficiencis of siRNAs targeting the respective four genes. (D) Cell proliferation assay tesing knock-down effects of the four genes. A-673 cells were cultured in six-well plates and transfected with siRNAs, respectively. Cell counts were performed on day 1, day 2 and day 3. Data are shown as mean ± SD, n = 3 independent experiments. (E–F) Clonogenic assay testing knock-down effects of the four genes. 500 cells with transfection of respective siRNAs were transferred to six-well plates and the cell culture was maintained for 14 days for colony formation observation. E Representative images of cell colonies. F Quantitative results of colony-forming assay. Data (Fig. 5A) are shown as mean ± SD, n = 22 (normal) v.s. n = 479 (tumor). Data (Fig. 5C, D, F) are shown as mean ± SD, n = 3 independent experiments. (ns: p > 0.05, *: p < 0.05, **: p < 0.01, ***: p < 0.001)
Discussion
Due to its rare occurrence, ES lacks a thorough transcriptome study in contrast to those cancer types that have been extensively sequenced and profiled. Here, we undertook an integrative analysis of publicly accessible microarray datasets of ES with the intention of creating a comprehensive transcriptome profile of ES for a better understanding of its features and identifying specific therapy alternatives. Consistent with previous microarray studies (Fagone et al. 2015), we showed cyclin D1 to be the highest expressed gene in ES through the current integrated microarray database analysis (Magro et al. 2015). Cyclin D1 has been shown to be frequently up-regulated in many different cancer types and its up-regulation triggers tumorigenesis through induction of transcription, translation, and protein stability (Kim and Diehl 2009). We analyzed the role of cyclin D1 in ES through pathway enrichment analysis, and found that E2F targets and G2/M checkpoints were the two most highly enriched pathways. This is consistent with previously discovered functions of cyclin D1, although its precise role in ES is still unclear. Our results suggest that the overexpression of E2F-cyclin D1 co-stimulating regulatory loops can enhance cell cycle progression, potentially leading to uncontrolled tumor cell growth (Alao 2007; Schulze et al. 1994). Our analysis has also shown a positive prognostic role of the E2F-Targets pathway. Therefore, Cyclin D1 may also be employed as a molecular diagnostic biomarker in ES. Interestingly, another ES variant, the BCOR–CCNB3 fusion-positive sarcoma, has also been shown to up-regulate CCND1 (Kao et al. 2018). On the basis of the successes of CDK4/6 inhibitors in dedifferentiated liposarcoma (Lin et al. 2020), as well as the current clinical data supporting the use of CDK4/6 inhibitors in subsets of sarcoma primarily driven by CDK4/6 deregulation (Hsu et al. 2022), available CDK4/6 small molecule inhibitors targeting the E2F pathway may be promising for the treatment of variants of ES including the BCOR–CCNB3 fusion subset.
NKX2-2 is another among the highest expressed genes in our analysis. To our interest, NKX2-2 has been exclusively studied in ES and is recognized as a specific biomarker of this malignancy. NKX2-2 is a major target gene and a core co-regulatory member of the driver mutation fusion gene, EWS–FLI1 in ES (Shi et al. 2020; Smith et al. 2006). Our study again confirms NKX2-2 as a valuable biomarker for ES.
Up-regulation of the ASPM gene has recently been shown and recognized as a poor outcome factor in glioma (Chen et al. 2020) and liver malignancies (Lin et al. 2008). CKS2 is recognized as a tumor promoter that functions as a regulator of the gene translation of numerous validated targets including p53, CDK1,cyclin A, caspase-3, and Bax (You et al. 2015). CENPF is a microtubule-binding protein that has been found to be associated with poor prognosis in several types of cancer (Sun et al. 2019). However, the mechanism behind this correlation remains unclear. DTL, an E3 ubiquitin-protein ligase, has also been shown a poor prognostic role to promote cancer progression in several cancer types (Cui et al. 2019; Luo et al. 2022). On the other hand, the present study identified NDRG2 and TOB1 as down-regulated DEGs with prolonged survival in ES. NDRG2 has been shown to inhibit the occurrence and metastasis of tumors and increase the sensitivity of anti-cancer drugs. Additionally, the functions of NDRG2 as a tumor suppressor contribute to tumor growth inhibition and anti-metastasis in various tumors (Lee et al. 2022). Like many other tumor suppressor proteins, down-regulated TOB1 expression has been reported in various cancers, mostly in breast, pancreas, thyroid, and stomach. TOB1 expression levels are inversely associated with the tumorigenicity and metastatic ability of breast cancer cell lines as well as with tumor progression in patients with breast cancers (Lee et al. 2015). To the best of our knowledge, we present here for the first time evidence of up-regulation and poor prognostic outcomes associated with four genes in ES. These findings underscore the importance of conducting further in-depth studies of these genes in the future.
Most sarcomas including ES are considered “cold” cancers based on their low tumor mutation burden (TMB) and low immunogenicity (Rytlewski et al. 2021). Although short of experimental proof, our immune infiltration results indicate ES may still be able to stimulate an immune response, evidenced by increased immune infiltration of many types of lymphocytes including memory and effector T cells, NKT cells, and DC cells. Thus, CAR- or immune checkpoint inhibitors-based immunotherapy may still be promising therapeutic options for ES patients.
Conclusion
Our findings provided a clearer representative transcriptome profile of ES. These uncovered prognostics-associated genes, pathways, and immune infiltrative characteristics might be valuable for future study.
Abbreviations
- ES:
-
Ewing sarcoma
- GEO:
-
Gene Expression Omnibus
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- CC:
-
Cellular component
- MF:
-
Molecular function
- BP:
-
Biological process
- GSEA:
-
Gene set enrichment analysis
- NC:
-
Negative control
References
Agelopoulos K, Richter GHS, Schmidt E, Dirksen U, von Heyking K, Moser B, Muller-Tidow C (2015) Deep sequencing in conjunction with expression and functional analyses reveals activation of FGFR1 in ewing sarcoma. Clinical Cancer Res 21:4935–4946
Ahmed, S.K., Robinson, S.I., Okuno, S.H., Rose, P.S., and Laack, N.N.I. (2013). Adult ewing sarcoma: survival and local control outcomes in 102 patients with localized disease. Sarcoma.
Alao JP (2007) The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer 6:24
Aljohani, A.I., Toss, M.S., Green, A.R., and Rakha, E.A. (2022) The clinical significance of cyclin B1 (CCNB1) in invasive breast cancer with emphasis on its contribution to lymphovascular invasion development. Breast Cancer Res Treat.
Atreyi Dasgupta, M.T., Nino Rainusso, Ronald J. Bernardi, Ryan , and Shuck, L.K., David M. Loeb and Jason T. Yustein (2017). Metabolic modulation of Ewing sarcoma cells inhibits tumor.
Aynaud MM, Mirabeau O, Gruel N, Grossetete S, Boeva V, Durand S, Surdez D, Saulnier O, Zaidi S, Gribkova S et al (2020) Transcriptional programs define intratumoral heterogeneity of ewing sarcoma at single-cell resolution. Cell Rep 30(1767–1779):e1766
Balamuth NJ, Womer RB (2010) Ewing’s sarcoma. Lancet Oncol 11:184–192
Chen X, Huang L, Yang Y, Chen S, Sun J, Ma C, Xie J, Song Y, Yang J (2020) ASPM promotes glioblastoma growth by regulating G1 restriction point progression and Wnt-beta-catenin signaling. Aging (albany NY) 12:224–241
Cui H, Wang Q, Lei Z, Feng M, Zhao Z, Wang Y, Wei G (2019) DTL promotes cancer progression by PDCD4 ubiquitin-dependent degradation. J Exp Clin Cancer Res 38:350
Ding K, Qiu W, Yu D, Ma H, Xie K, Luo F, Li S, Li Z, Wei J (2021) Bioinformatics analysis of ZBTB16 as a prognostic marker for Ewing’s sarcoma. Biomed Res Int 2021:1989917
Fagone P, Nicoletti F, Salvatorelli L, Musumeci G, Magro G (2015) Cyclin D1 and Ewing’s sarcoma/PNET: a microarray analysis. Acta Histochem 117:824–828
Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley M-C, Kovar H, Grimer R, Whelan J, Claude L (2015) Ewing sarcoma: current management and future approaches through collaboration. J Clin Oncol 33:3036–3046
Grünewald TG, Cidre-Aranaz F, Surdez D, Tomazou EM, de Álava E, Kovar H, Sorensen PH, Delattre O, Dirksen U (2018) Ewing Sarcoma. Nat Rev Disease Primers 4:1–22
Gu Y, Chen B, Guo D, Pan L, Luo X, Tang J, Yang W, Zhang Y, Zhang L, Huang J et al (2022) Up-regulation of RACGAP1 promotes progressions of hepatocellular carcinoma regulated by GABPA via PI3K/AKT pathway. Oxid Med Cell Longev 2022:3034150
Guo H, Zou J, Zhou L, Zhong M, He Y, Huang S, Chen J, Li J, Xiong J, Fang Z et al (2020) NUSAP1 Promotes gastric cancer tumorigenesis and progression by stabilizing the YAP1 protein. Front Oncol 10:591698
Hsu JY, Seligson ND, Hays JL, Miles WO, Chen JL (2022) Clinical utility of CDK4/6 Inhibitors in sarcoma: successes and future challenges. JCO Precis Oncol 6:e2100211
Kakar MU, Mehboob MZ, Akram M, Shah M, Shakir Y, Ijaz HW, Aziz U, Ullah Z, Ahmad S, Ali S et al (2022) Identification of differentially expressed genes associated with the prognosis and diagnosis of hepatocellular carcinoma by integrated bioinformatics analysis. Biomed Res Int 2022:4237633
Kao YC, Owosho AA, Sung YS, Zhang L, Fujisawa Y, Lee JC, Wexler L, Argani P, Swanson D, Dickson BC et al (2018) BCOR-CCNB3 fusion positive sarcomas: a clinicopathologic and molecular analysis of 36 cases with comparison to morphologic spectrum and clinical behavior of other round cell sarcomas. Am J Surg Pathol 42:604–615
Khanna N, Pandey A, Bajpai J (2017) Metastatic Ewing’s sarcoma: revisiting the “evidence on the fence.” In J Med Paediatric Oncol: off JIn Soc Med Paed Oncol 38:173
Kim JK, Diehl JA (2009) Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol 220:292–296
Lee HS, Kundu J, Kim RN, Shin YK (2015) Transducer of ERBB2.1 (TOB1) as a tumor suppressor: a mechanistic perspective. Int J Mol Sci 16:29815–29828
Lee, K.W., Lim, S., Kim, K.D. (2022) The Function of N-Myc Downstream-Regulated Gene 2 (NDRG2) as a Negative Regulator in Tumor Cell Metastasis. Int J Mol Sci 23.
Lin SY, Pan HW, Liu SH, Jeng YM, Hu FC, Peng SY, Lai PL, Hsu HC (2008) ASPM is a novel marker for vascular invasion, early recurrence, and poor prognosis of hepatocellular carcinoma. Clin Cancer Res 14:4814–4820
Lin DI, Hemmerich A, Edgerly C, Duncan D, Severson EA, Huang RSP, Ramkissoon SH, Connor YD, Shea M, Hecht JL et al (2020) Genomic profiling of BCOR-rearranged uterine sarcomas reveals novel gene fusion partners, frequent CDK4 amplification and CDKN2A loss. Gynecol Oncol 157:357–366
Luo Y, He Z, Liu W, Zhou F, Liu T, Wang G (2022) DTL Is a Prognostic biomarker and promotes bladder cancer progression through regulating the AKT/mTOR axis. Oxid Med Cell Longev 2022:3369858
Magro G, Brancato F, Musumeci G, Alaggio R, Parenti R, Salvatorelli L (2015) Cyclin D1 is a useful marker for soft tissue Ewing’s sarcoma/peripheral primitive neuroectodermal tumor in children and adolescents: a comparative immunohistochemical study with rhabdomyosarcoma. Acta Histochem 117:460–467
Mahjoub WK, Chretien F, Gherardi R, Ortonne N (2006) Intracellular spread of Ewing’s sarcoma within skeletal muscle fibres: is there a role for cytotoxic tumour-infiltrating lymphocytes? Histopathology 48:623–626
Miao YR, Zhang Q, Lei Q, Luo M, Xie GY, Wang H, Guo AY (2020) ImmuCellAI: a unique method for comprehensive T-cell subsets abundance prediction and its application in cancer immunotherapy. Adv Sci (weinh) 7:1902880
Ozaki T (2015) Diagnosis and treatment of Ewing sarcoma of the bone: a review article. J Orthop Sci 20:250–263
Postel-Vinay S, Veron AS, Tirode F, Pierron G, Reynaud S, Kovar H, Oberlin O, Lapouble E, Ballet S, Lucchesi C et al (2012) Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet 44:323–327
Riggi N, Suva ML, Stamenkovic I (2021) Ewing’s Sarcoma. N Engl J Med 384:154–164
Rytlewski J, Milhem MM, Monga V (2021) Turning ‘Cold’ tumors ‘Hot’: immunotherapies in sarcoma. Ann Transl Med 9:1039
Sankar S, Theisen ER, Bearss J, Mulvihill T, Hoffman LM, Sorna V, Beckerle MC, Sharma S, Lessnick SL (2014) Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin Cancer Res 20:4584–4597
Sarver AE, Sarver AL, Thayanithy V, Subramanian S (2015) Identification, by systematic RNA sequencing, of novel candidate biomarkers and therapeutic targets in human soft tissue tumors. Lab Invest 95:1077–1088
Savola S, Klami A, Myllykangas S, Manara C, Scotlandi K, Picci P, Knuutila S, Vakkila J (2011) High Expression of complement component 5 (C5) at tumor site associates with superior survival in Ewing’s sarcoma family of tumour patients. ISRN Oncol 2011:168712
Schulze A, Zerfass K, Spitkovsky D, Henglein B, Jansen-Durr P (1994) Activation of the E2F transcription factor by cyclin D1 is blocked by p16INK4, the product of the putative tumor suppressor gene MTS1. Oncogene 9:3475–3482
Scotlandi K, Remondini D, Castellani G, Manara MC, Nardi F, Cantiani L, Francesconi M, Mercuri M, Caccuri AM, Serra M et al (2009) Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol 27:2209–2216
Shi X, Zheng Y, Jiang L, Zhou B, Yang W, Li L, Ding L, Huang M, Gery S, Lin DC et al (2020) EWS–FLI1 regulates and cooperates with core regulatory circuitry in Ewing sarcoma. Nucleic Acids Res 48:11434–11451
Smith R, Owen LA, Trem DJ, Wong JS, Whangbo JS, Golub TR, Lessnick SL (2006) Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 9:405–416
Specht K, Sung YS, Zhang L, Richter GH, Fletcher CD, Antonescu CR (2014) Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer 53:622–633
Sun J, Huang J, Lan J, Zhou K, Gao Y, Song Z, Deng Y, Liu L, Dong Y, Liu X (2019) Overexpression of CENPF correlates with poor prognosis and tumor bone metastasis in breast cancer. Cancer Cell Int 19:264
Surdez D, Benetkiewicz M, Perrin V, Han ZY, Pierron G, Ballet S, Lamoureux F, Redini F, Decouvelaere AV, Daudigeos-Dubus E et al (2012) Targeting the EWSR1-FLI1 oncogene-induced protein kinase PKC-beta abolishes ewing sarcoma growth. Cancer Res 72:4494–4503
Surdez D, Zaidi S, Grossetete S, Laud-Duval K, Ferre AS, Mous L, Vourc’h T, Tirode F, Pierron G, Raynal V et al (2021) STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer Cell 39(810–826):e819
Svoboda LK, Harris A, Bailey NJ, Schwentner R, Tomazou E, von Levetzow C, Magnuson B, Ljungman M, Kovar H, Lawlor ER (2014) Overexpression of HOX genes is prevalent in Ewing sarcoma and is associated with altered epigenetic regulation of developmental transcription programs. Epigenetics 9:1613–1625
Volchenboum SL, Andrade J, Huang L, Barkauskas DA, Krailo M, Womer RB, Ranft A, Potratz J, Dirksen U, Triche TJ et al (2015) Gene expression profiling of ewing sarcoma tumors reveals the prognostic importance of tumor-stromal interactions: a report from the children’s oncology group. J Pathol Clin Res 1:83–94
Whelan J, Le Deley M-C, Dirksen U, Le Teuff G, Brennan B, Gaspar N, Hawkins DS, Amler S, Bauer S, Bielack S (2018) High-dose chemotherapy and blood autologous stem-cell rescue compared with standard chemotherapy in localized high-risk Ewing sarcoma: results of Euro-EWING 99 and Ewing-2008. J Clin Oncol 36:3110
Womer RB, West DC, Krailo MD, Dickman PS, Pawel BR, Grier HE, Marcus K, Sailer S, Healey JH, Dormans JP (2012) Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s oncology group. J Clin Oncol 30:4148
You H, Lin H, Zhang Z (2015) CKS2 in human cancers: Clinical roles and current perspectives (Review). Mol Clin Oncol 3:459–463
Zhao R, Xiong C, Zhang C, Wang L, Liang H, Luo X (2022) Construction of a prognosis-related gene signature by weighted gene coexpression network analysis in ewing sarcoma. Comput Math Methods Med 2022:8798624
Acknowledgements
This study was funded in part by the Startup Foundation for Advanced Talents and Science and Technology Innovation Foundation at Yangzhou University (H.B.S), the Innovative Training Grant of College Students in Jiangsu Province (202111117097Y, Y.G, and H.B.S) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX21_1655).
Author information
Authors and Affiliations
Contributions
BJC and HJS collected the data, drew the figures, and wrote the partial manuscript. HBS proposed the idea and modified, supervised, wrote, and approved the final version of the manuscript. ZHF performed in vitro experiment. MK helped to edit the manuscript. LLC, SYY, ZYL, and CLB helped to provide scientific views and research suggestions. NW helped to manage associated paperwork. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the current research work has no commercial or financial relationships that could be construed as a potential conflict of interest.
Data availability
A total of ten datasets, GSE12102, GSE17679, GSE34620, GSE37371, GSE45544, GSE63157, GSE73166, GSE68776, and GSE142162, were obtained from the NCBI database (www.ncbi.nlm.nih.gov/geo/).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, B., Sun, H., Fan, Z. et al. Integrative analyses of bulk microarray data to discover genes, pathways, and immune infiltration characteristics associated with targeting of Ewing sarcoma. J Cancer Res Clin Oncol 149, 6967–6977 (2023). https://doi.org/10.1007/s00432-023-04642-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-023-04642-0