
Abstract
Purpose
Breast cancer, the most prevalent cancer worldwide, consists of 4 main subtypes, namely, Luminal A, Luminal B, HER2-positive, and Triple-negative breast cancer (TNBC). Triple-negative breast tumors, which do not express estrogen, progesterone, and HER2 receptors, account for approximately 15-20% of breast cancer cases. The lack of traditional receptor targets contributes to the heterogenous, aggressive, and refractory nature of these tumors, resulting in limited therapeutic strategies.
Methods
Chemotherapeutics such as taxanes and anthracyclines have been the traditional go to treatment regimens for TNBC patients. Paclitaxel, docetaxel, doxorubicin, and epirubicin have been longstanding, Food and Drug Administration (FDA)-approved therapies against TNBC. Additionally, the FDA approved PARP inhibitors such as olaparib and atezolizumab to be used in combination with chemotherapies, primarily to improve their efficiency and reduce adverse patient outcomes. The immunotherapeutic Keytruda was the latest addition to the FDA-approved list of drugs used to treat TNBC.
Results
The following review aims to elucidate current FDA-approved therapeutics and their mechanisms of action, shedding a light on the various strategies currently used to circumvent the treatment-resistant nature of TNBC cases.
Conclusion
The recent approval and use of therapies such as Trodelvy, olaparib and Keytruda has its roots in the development of an understanding of signaling pathways that drive tumour growth. In the future, the emergence of novel drug delivery methods may help increase the efficiency of these therapies whiel also reducing adverse side effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer (BC) is the most frequently diagnosed cancer worldwide with an estimated 2.3 million new cases in 2020, representing 11.7% of all cancer cases. BC accounted for 684,996 deaths in 2020 (Sung et al. 2020). In Canada, for example, 27,700 cases were diagnosed in 2021 with an estimated 5400 deaths as a result (Canadian Cancer Statistics 2021). In the United States of America (USA) in 2021, a total of 290,560 new cases of BC (287,850 females and 2710 males) and 44,130 deaths (43,250 females and 530 males) were reported (Siegel et al. 2022). While breast cancer is the most diagnosed type of cancer in females worldwide, in developed countries such as the USA, Canada, Australia, and England, lung cancer causes more deaths (Bray et al. 2018; Lukong 2017). Since the 2000s, the reduction in mortality due to breast cancer can be attributed to an increase in screening, awareness, and development of effective therapies made available in these regions (Bray, et al. 2018; Lukong 2017). Breast cancer develops from the abnormal proliferation of mammary epithelial cells to form carcinoma in situ which can then become invasive and metastatic (Makki 2015; Place et al. 2011). These tumors can be classified using either histological or molecular features.
Molecular subtype classification involves the assessment of the presence or absence of ER, PR, and HER2 (Perou et al. 2000; Sørlie et al. 2001). These subtypes are luminal A, luminal B, HER2-positive, and Triple-negative breast cancers (TNBCs) (Lukong 2017; Fragomeni et al. 2018). Luminal A breast cancers constitute ~ 30–40% of all invasive cases (Fragomeni et al. 2018). These tumors are ER-positive, PR-positive but HER2-negative and are generally low grade (Fragomeni et al. 2018). Luminal B tumors consist of 20–30% of all cases and are typically ER-positive, PR-negative/positive, and HER2-negative/positive (Fragomeni et al. 2018). They are also higher grade, with a poorer prognosis than luminal A cases (Fragomeni et al. 2018). HER2-positive breast cancers have two subcategories: HER2-enriched (ER and PR-negative but HER2-positive) and luminal HER2 (ER and PR-positive and HER2-positive) (Kneubil et al. 2013; Vici et al. 2015). Overall, 12–20% of breast cancer cases are classified as HER2-positive (Fragomeni et al. 2018). Triple negative breast cancer (TNBC) is diagnosed in 15–20% of all breast cancer patients and is defined by the absence of ER, PR, and HER2 expression (Fragomeni et al. 2018; Foulkes et al. 2010).
Patients with TNBCs are more predisposed to adverse outcomes, recurrence, and metastasis than patients with other breast cancer subtypes. Patients with TNBC were significantly more likely to die within 10 years of diagnosis than patients with other breast cancer subtypes (Dent et al. 2007). In an analysis of 1025 breast cancer-specific deaths, patients with the TNBC subtype were found to have significantly reduced survival probability compared to patients with hormone receptor-positive or negative tumors (Lin et al. 2012). This difference was observed even after adjustment for risk factors such as age, race, and tumor size (Lin et al. 2012). Dent and colleagues found that 33.9% of TNBC patients experienced recurrence and metastasis compared to 20.4% of patients with other breast tumors (Dent et al. 2007). They reported that these metastases were more likely to be locoregional at first before spreading to distant tissue (Dent et al. 2007). Furthermore, TNBC metastases were more likely to occur in visceral organs and soft tissue, as reported by Liedtke et al. (Liedtke et al. 2008). An analysis by Lin and scientists found that first recurrences were most likely to be located in the brain, lung, or locoregional sites (Lin et al. 2012). Of these sites, the central nervous system appeared to be the most prevalent, with first and subsequent recurrences of TNBC occurring in the TNBC in 174 out of 480 TNBC patients analyzed (Lin et al. 2012). These studies also reported that TNBC tumors were less likely to recur in the bone than hormone receptor-positive breast tumors (Dent et al. 2007; Lin et al. 2012; Liedtke et al. 2008).
The development of TNBCs can be attributed to several risk factors including genetic predisposition, race, age, obesity, BRCA1 and BRCA2 mutations are considered risk factors for the development of TNBC tumors (Schneider et al. 2008). A meta-analysis of 46.870 patients, including 868 carriers of BRCA1 mutations found that patients carrying the tumors were significantly more likely to develop TNBC than both non-carriers as well as patients carrying BRCA2 mutations (Chen et al. 2018). Additionally, African American women are more likely to develop TNBC and have adverse outcomes than European–Americans (Lin et al. 2012; Prakash et al. 2020). Out of 190 TNBC patients, it was found that a significant proportion were either African American or Asian women compared to patients without TNBC (Yeh et al. 2017). The analysis also found that a prior history of breast cancer was a strong risk factor associated with TNBC development in these patients (Yeh et al. 2017). The subtype of breast cancer also develops more frequently in women under the age of 50 and is associated with obesity though studies on the latter risk factor have shown contrasting results (Lin et al. 2012; Dolle et al. 2009). While there was a higher prevalence of TNBC in obese premenopausal women compared to their normal-weight counterparts, Body Mass Index (BMI) had no apparent effect on the risk of TNBC development in postmenopausal women (Lin et al. 2012).
The treatment of breast cancer is dictated by the molecular subtype of the tumor. Luminal A and B carcinoma overexpress ER and are therefore treated by targeting the estrogen receptor pathway using endocrine therapy (Hanker et al. 2020). Endocrine therapies include Selective Estrogen Receptor Modulators (SERMs) such as Tamoxifen, Selective Estrogen Receptor Downregulators (SERDs) such as Fulvestrant, and Aromatase Inhibitors (AIs) like Letrozole (Hanker et al. 2020). Generally administered after surgery, these drugs have drastically reduced mortality and remission rates in patients since their introduction (Hanker et al. 2020). HER2-positive cases are treated with HER2-targeting agents trastuzumab and lapatinib (Arteaga et al. 2011). To improve the efficiency of both endocrine and HER2-targeting therapies, combinatorial approaches using either of these agents along with PI3K inhibitors, CDK 4/6 inhibitors, or PD1 inhibitors are being tested (Hanker et al. 2020; Arteaga et al. 2011).
On the other hand, TNBC tumors present a significant challenge to the development and use of targeted therapies due to the lack of ER, PR, and HER2 expression (Pareja et al. 2016). The term TNBC was first used to describe cases that only responded to chemotherapy (Brenton et al. 2005). Despite the absence of these biomarkers, TNBCs are heterogenous, with various subtypes, each distinguished by the up or downregulation of different protein pathways (Bianchini et al. 2016). TNBC cases are classified into basal-like 1 (BL1), basal-like 2 (BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem-like (MSL), and luminal androgen receptor (LAR) subtypes (Pareja et al. 2016; Lehmann et al. 2016).
Regardless of the subtype, TNBC patients have primarily been treated with cytotoxic chemotherapy either before (neoadjuvant) or after surgery (adjuvant) (Bianchini et al. 2016). TNBC cases often benefit from chemotherapy to a greater degree than other breast cancer subtypes (Carey et al. 2007). For example, a study by von Minckwitz and colleagues showed that neoadjuvant chemotherapy using anthracyclines and taxanes benefited 30–40% of patients with early-stage TNBC (Minckwitz et al. 2012). Apart from anthracyclines such as doxorubicin (Adriamycin) and taxanes such as paclitaxel (Taxol), the FDA has approved PARP inhibitors such as olaparib (Lynparza) and immunotherapy such as atezolizumab (Tecentriq) to be used in combination with the chemotherapy agents (Pareja et al. 2016; Collignon et al. 2016; Schmid et al. 2018). More recently, the immune-targeted therapy Keytruda was fast-tracked for approval to treat metastatic triple negative breast cancer (Raedler and Keytruda (Pembrolizumab) 2015).
Due to the lack of biomarkers for targeted therapies and an aggressive, often invasive, disease progression, there are few FDA-approved drug treatments for triple-negative breast cancer. This review describes the mechanisms of action of these therapies as well as the limited scope of treatments available for TNBC to open avenues for future drug development.
TNBC subtypes
BRCA (BReast CAncer) genes (BRCA 1 and 2 or 1/2) are tumor-suppressor genes that play a role in DNA damage repair and mutations of these genes that results in defective DNA repair mechanisms may increase the risk of developing breast cancer (Venkitaraman 2014). TNBCs with germline BRCA1/2 can be referred to as “BRCAness” tumors (Summa et al. 2013). The prevalence of BRCA1/2 among women with TNBC varies significantly by ethnicity/race and age (Hahnen et al. 2017; Lukong et al. 2017). This means that not all TNBCs are equal.
Gene expression studies have revealed that TNBC is a heterogeneous subgroup of tumors that is also distinguishable by the up or downregulation of different cellular pathways (Bianchini et al. 2016). A landmark study by Lehmann and colleagues led to the classification of TNBC into seven distinct subtypes based on gene expression profiling (Lehmann et al. 2016). These include six stable subtypes: (i) basal-like 1 (BL1), (ii) basal-like 2 (BL2), (iii) immunomodulatory (IM), (iv) mesenchymal (M), (v) mesenchymal stem-like (MSL) and (vi) luminal androgen receptor (LAR) subtypes, and an unstable (UNS) subtype (vii), together referred to sometimes as Lehmann TNBC subtypes (Pareja et al. 2016; Lehmann et al. 2016). BL1 breast cancers overexpress genes in the DNA damage response pathway and are stained by Ki67 while BL2 tumors are associated with the upregulation of growth factor signaling, TP53, and myoepithelial markers (Fragomeni et al. 2018; Pareja et al. 2016). IM tumors are defined by hyperactive immune signaling cascades and infiltration of lymphocytes into the tumor (Fragomeni et al. 2018; Pareja et al. 2016). Mesenchymal and mesenchymal stem-like TNBCs share several similarities (enrichment of epithelial-to-mesenchymal transition genes) but are differentiated by the increase in expression of mesenchymal stem cell genes in the latter subtype (Pareja et al. 2016). The LAR subtype, as the name suggests, presents similarly to luminal breast cancers due to androgen receptor activation (Pareja et al. 2016). It is important to note that Lehmann et al. later revised their previous sub-classification to include only four subtypes (TNBCtype-4): BL1, BL2, M, and LAR because the immunomodulatory signature for instance occurred in all the other TNBC molecular subtypes, making up about 20% of all TNBCs (Lehmann et al. 2016). Studies by Burstein et al. also led to the classification of four TNBC subtypes that comprised LAR, mesenchymal (MES), basal-like immunosuppressed (BLIS) and basal-like immune-activated (BLIA) (Burstein et al. 2015). No surprisingly, BLIA showed a better prognosis compared with BLIS based on its more favourable immunological background (Burstein et al. 2015).
Targeting TNBC subtypes
Classification of TNBC subtypes can help guide therapy selection. BL1 and BL2 tumors, for example, are particularly susceptible to PARP inhibitors due to the dysregulation of DNA damage repair that characterizes these cancers (Yin et al. 2020). Additionally, the increased activity of growth factor receptors, which differentiates BL2 from BL1 tumors, makes anti-growth factor drugs such as Lapatinib, Gefitinib, and Cetuximab ideal candidates to treat BL2 TNBCs (Yin et al. 2020). Both BL1 and BL2 cancers respond to platinum-based chemotherapy (Maqbool et al. 2022). The M and MSL subtypes can be targeted using the mTOR inhibitor Rapamycin with M-type TNBCs being susceptible to growth factor inhibition and the MSL-type being susceptible to the Src inhibitor dasatinib (Maqbool et al. 2022). Immunomodulatory TNBCs highly express immune system-associated genes which makes them vulnerable to anti-immune checkpoint therapeutics such as PD1 and PDL1 inhibitors (Atezolizumab and Pembrolizumab, for example) (Yin et al. 2020). Finally, LAR subtype TNBCs can be targeted using anti-androgen receptor therapies such as Bicalutamide and Enzalutamide (Barton et al. 2016). A summary of the various molecular subtypes of TNBC and their corresponding therapies is shown in Table 1.
FDA-approved TNBC therapies
PARP inhibitors
In their landmark study in 2005, farmer and colleagues showed that embryonic stem cells deficient in BRCA1 and/or BRCA2 were particularly susceptible to cell death when subjected to small-molecule PARP inhibitors (Farmer et al. 2005). Due to the prevalence of deleterious, germline BRCA mutations in TNBC, the study opened possibilities to target PARP and treat these cancers specifically, leading to the development of Lynparza and Talzenna (Collignon et al. 2016; Farmer et al. 2005). In 2018, the FDA approved Lynparza (manufactured by AstraZeneca) and Talzenna (manufactured by Pfizer) for the treatment of germline BRCA-mutated, metastatic triple negative breast cancer (U.S. 2018a).
Initially developed and approved for the treatment of germline BRCA-mutated ovarian cancer in 2014, Lynparza (generic name olaparib) was then tested on patients with HER2-negative metastatic breast cancer in a randomized, open-label, phase 3 clinical trial (named OlympiAD) in 2017 (Kim et al. 2022; Robson et al. 2017). Patients who received olaparib showed significantly longer progression-free survival versus patients who were given traditional chemotherapy (Robson et al. 2017). The trial indicated that olaparib monotherapy reduced the risk of death or disease progression by 42% (Robson et al. 2017). Similarly, Talzenna (generic name talazoparib) was tested on patients with advanced, HER2-negative breast cancer in the EMBRACA phase 3 clinical trial in 2017 (Litton et al. 2018). The trial showed that talazoparib treatment increased progression-free survival by 3 months compared to traditional chemotherapy in these patients (Litton et al. 2018). Lynparza and Talzenna have different chemical structures but share the same mechanism of action in that they target and inhibit the role of PARP in the DNA damage response pathway (Shen et al. 2015).
Poly (ADP-ribose) polymerases (PARPs)
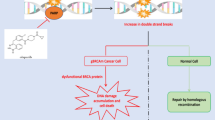
Poly (ADP-ribose) polymerases (PARPs) are a family of related enzymes that covalently add poly (ADP-ribose) chains onto their targets, in a process called PARylation (Schreiber et al. 2006). PARPs play key roles in various cellular processes that include transcription, replication, recombination, and notably, DNA repair. Among PARP family members, PARP1 is the primary DNA damage sensor and generates about 90% of poly (ADP-ribose) chains following DNA damage. PARP1 contains six functional domains, which include three zinc finger-related domains (DNA binding domains), one BRCA1 C-terminus domain (auto-modification domain), a tryptophan-/glycine-/arginine-rich domain (WGR domain), and one catalytic domain (Krishnakumar and Kraus 2010). Further, the catalytic domain of PARP1 is made up of two subdomains: a helical domain (HD) and an ADP-ribosyltransferase catalytic domain (ART) (Krishnakumar and Kraus 2010). The (ADP-ribose) polymerase activity of PARP1 is strongly regulated by interaction with single-stranded DNA breaks. PARP1 recognizes and interacts with DNA single-strand breaks via its zinc finger-related domains. In its inactive, non-DNA binding status, the HD of PARP1 inhibits the binding between PARP1 and its cofactor β-nicotinamide adenine dinucleotide (β-NAD) via the ART subdomain. PARP1 binds to SSBs, thus abrogating the auto-inhibitory function of HD and resulting in the activation of ART (Rouleau et al. 2010). Once on the SSBs, PARP1 recruits scaffolding proteins such as DNA ligase III and DNA polymerase β (Lee et al. 2014). PARP1 also interacts with PARG (poly ADP-ribose glycohydrolase) to attach ADP-ribose moieties to histones, facilitate chromatin remodeling, and recruit DNA damage repair proteins (Livraghi and Garber 2015). Once the chromatin is unwound, the assembled repair complex uses the undamaged template strand to fix the break (Livraghi and Garber 2015). Auto-PARylation of PARP1 releases PARP1 from the repaired DNA, thus reinstituting the auto-inhibitory status (Rouleau et al. 2010). In the presence of PARP inhibitors, the PARP-dependent DNA repair system cannot be activated leading to the development of double-strand breaks and susceptibility of BRCA1/2-mutant breast cancer cells for instance to synthetic lethality (Farmer et al. 2005; Bryant et al. 2005). Additionally, studies have shown that PARP1 recognition of DNA damage also facilitates the homologous recombination repair (HR) pathway, though the enzyme does not play an active role in HR beyond binding to the double-stranded break site (Hay et al. 2009; Schultz et al. 2003). In the presence of PARP inhibitors (PARPi), the PARP-dependent DNA repair system cannot be activated. Disruption of both HR and the PARP-mediated base excision repair (BER) pathways through PARP inhibition is often lethal to cells (Farmer et al. 2005). BRCA1/2-mutant breast cancer cells for instance are susceptible to PARPi-induced synthetic lethality (Farmer et al. 2005; Bryant et al. 2005).
Mechanism of action of PARP inhibitors: lynparza and talzenna
The mechanism of action of PARP inhibitors in tumors is inextricably linked with BRCA1/2 (Farmer et al. 2005; Bryant et al. 2005). PARP-deficient mice do not develop tumors and are otherwise healthy and fertile but the inhibition of PARP in BRCA1/2 deficient cancer cells has been shown to cause cell death (Farmer et al. 2005; Bryant et al. 2005; Conde et al. 2001). Therefore, it is important to understand the function of BRCA1 and BRCA2 (in DNA damage response) to appreciate why germline BRCA mutated breast cancers are susceptible to PARP inhibition (D'Andrea 2018).
The BRCA proteins play a role in repairing double-stranded DNA breaks through the process of homologous recombination repair (HR) (Byrum et al. 2019). BRCA1 and BRCA2 migrate to genomic lesions and interact with proteins such as BARD1, RAD51, MRN to regulate chromatin remodeling, exchange of information from the undamaged template, and strand resection after repair (Caestecker and Walle 2013). Both BRCA1 and BRCA2 are also critical to the protection of replication forks during the S phase (D'Andrea 2018). Cells lacking BRCA1/2 show reduced proliferation, increased chromosomal aberrations, and increased susceptibility to cancer development (Venkitaraman 2014). Chromosome instability due to HR repair pathway defects (caused by BRCA1/2 mutations) normally activates apoptosis to prevent tumorigenesis (Lee et al. 2014). However, p53 mutations and selective pressure favor uncontrolled cell proliferation, resulting in the formation of tumors despite checkpoint controls to account for DNA damage (Lee et al. 2014). In these cancer cells, HR repair dysfunction is compensated for by the activation of single-stranded DNA break repair pathways, chief among which is the BER pathway (Farmer et al. 2005). PARP activation, a result of DNA damage, is a major driver in BER (Livraghi and Garber 2015).
PARP inhibitors like Lynparza and Talzenna function through two mechanisms (Fig. 1) (D'Andrea 2018). Firstly, the inhibition of PARP catalytic activity prevents the recruitment of DNA damage repair machinery, blocking the base excision repair process, leading to replication fork stalling during the S phase and the conversion of the single-strand nick into a double-stranded break (Fig. 1) (D'Andrea 2018). The double-stranded break is recognized by HR pathway proteins but due to defects in the pathway caused by BRCA1/2 mutations, the repair is unsuccessful, leading to apoptosis (Fig. 1) (D'Andrea 2018). A second proposed mechanism involves the prevention of PARP detachment from the DNA damage site (single and/or double-stranded) (Fig. 1) (Shen et al. 2015). The trapping of PARP at the site requires the recruitment of HR pathway machinery, which is defective in BRCA1/2-mutant breast cancer tumors (Fig. 1) (D'Andrea 2018). Interestingly, studies have indicated that Talzenna is a more effective “trapping agent” than Lynparza though both molecules have similar PARP inhibition activity (Shen et al. 2015). There are additional mechanisms that govern PARP inhibitor sensitivity in human breast tumors deficient in BRCA1/2 such as the PARP/POLQ pathway (D'Andrea 2018). However, these mechanisms are still under investigation (D'Andrea 2018).

Mechanism of action of PARP inhibitors. PARP recognizes and binds to single-stranded breaks in DNA and initiates the recruitment of base excision repair (BER) machinery to repair the break. When inhibited, PARP becomes trapped at the site of the SSB, causing a double stranded break. In the absence of BRCA1/2 and homologous repair mechanisms, this break remains resulting in apoptosis downstream
Clinical application
Lynparza (olaparib)
In January 2018, Lynparza became the first treatment approved by the FDA for HER2-negative metastatic breast cancer patients with BRCA1/2 mutations (U.S. 2018a; Caulfield et al. 2019). To be eligible, patients are required to have undergone chemotherapy and/or hormone therapy if their tumor is hormone-positive (Caulfield et al. 2019). The National Comprehensive Cancer Network (NCCN) guidelines made Lynparza a Category 1 recommendation (Caulfield et al. 2019). The drug, manufactured by AstraZeneca, is available in tablet form and is taken in 300 mg doses twice daily until disease remission (Zimmer et al. 2018). In the phase 3 clinical trial OlympiAD, 97% of patients experienced side effects such as anemia, nausea, diarrhea, and fatigue amongst others (Robson et al. 2017). However, discontinuation due to these effects only occurred in 5% of the cohort so, overall, the drug was well tolerated (Robson et al. 2017). Lynparza’s effectiveness as a monotherapy in BRCA1/2-deficient primary TNBC tumors was demonstrated by a recent clinical trial conducted by Eikesdal et al. (2021). Patients, who had not received prior chemotherapy, with primary TNBC received Lynparza for 10 weeks (Eikesdal et al. 2021). Out of 32 patients, 18 responded to the treatment (Eikesdal et al. 2021). While Lynparza is primarily being used as a monotherapy today, clinical trials are being conducted to test its efficacy in combination with other established therapies (Zimmer et al. 2018). For example, the phase I/II trial by Dent and colleagues is currently testing the use of Lynparza with Paclitaxel in patients with metastatic triple negative breast cancer (Dent et al. 2013).
Talzenna (talazoparib)
In October 2018, the FDA approved an alternative PARP inhibitor for the treatment of HER2-negative metastatic breast cancers with germline BRCA mutations (U.S. 2018b). Developed and manufactured by Pfizer, clinical studies showed that Talzenna performed similarly to Lynparza (McCann 2019). Patients with BRCA-mutated triple negative breast cancer as well as those with hormone receptor-positive, HER2-negative breast cancer are eligible for treatment (McCann 2019). Talzenna is administered orally in 1-mg doses, taken once daily until disease progression is halted (McCann 2019). Its most adverse side effects include anemia, thrombocytopenia, and fatigue (Litton et al. 2018). While 98.6% of patients experienced adverse side effects, only 5.9% of this subset of patients in the EMBRACA trial discontinued treatment as a result (Litton et al. 2018). Like Lynparza, Talzenna is currently being tested in combination with cytotoxic chemotherapy in clinical trials, though results have yet to be reported (McCann 2019).
Anthracycline-based chemotherapy
Anthracyclines are a class of drugs that act as DNA intercalating agents, thereby interfering with the activity of Topoisomerase II (Top2) in eukaryotic cells (Marinello et al. 2018). Daunomycin, the predecessor to doxorubicin, was isolated from S. peucetius bacteria and thereby dubbed the “antitumor antibiotic” (Arcamone et al. 1969). Doxorubicin was subsequently isolated from S. peucetius caesius, a variant strain developed through mutagenesis of S. peucetius (Arcamone et al. 1969). At the time, the development of doxorubicin allowed for the administration of lower chemotherapy doses due to its higher potency compared to daunomycin, despite the fewer side effects caused by the latter (Bonadonna et al. 1969). Shortly thereafter, in 1974, the drug was approved by the FDA for the treatment of metastatic breast cancer (Cortazar et al. 2012). In the following decades since its approval, several clinical trials, using triple negative breast cancer patients amongst others, have shown the efficacy of doxorubicin in increasing survival by 3–6 months when compared to the regimen without the drug (A'Hern et al. 1993; Paridaens et al. 2000). More recent trials are testing the efficiency of doxorubicin in combination with taxanes such as paclitaxel as well as cyclophosphamide (Bergin and Loi 2019). While the drug is widely prescribed for various cancers today, it is severely cardiotoxic in cumulative doses and is therefore administered periodically (Findlay et al. 2007). Currently, doxorubicin is manufactured by Bedford Laboratories under the brand name Adriamycin (Khasraw et al. 2012).
Epirubicin, manufactured by Pfizer under the name Ellence, was approved for the adjuvant treatment of breast cancer in 1999 (Khasraw et al. 2012). It is an epimer of doxorubicin and therefore has a similar therapeutic profile (Findlay et al. 2007). However, epirubicin has a more favorable toxicity profile due to reduced cardiac and hematologic toxicity (Khasraw et al. 2012). Therefore, epirubicin can be administered at higher doses before causing adverse cardiovascular events which may result in improved response rates (Findlay et al. 2007; Khasraw et al. 2012). A clinical trial in 2010 showed that when epirubicin was added to an adjuvant chemotherapy regimen, it resulted in a 90% recurrence-free survival rate (RFS), higher than a doxorubicin-based regimen (Burnell et al. 2010).
Overall, decades of clinical evidence and patient data have led to both doxorubicin and epirubicin becoming a major component of both early and advanced breast cancers today (Collignon et al. 2016). As epimers, they share a similar mechanism of action in their interference in topoisomerase 2 functioning and intercalation of DNA (Beretta et al. 2008).
Mechanism of action of anthracyclines
Anthracyclines primarily target DNA by inserting between base pairs and remaining intercalated through ionic and steric bonding (Fig. 2) (Marinello et al. 2018). However, the ability of these drugs to bind to DNA is not central to their antitumor activity (Beretta et al. 2008). Evidence suggests that these intercalating agents are cytotoxic as a result of their interference with topoisomerase II (Top2), an enzyme that regulates the supercoiling and unwinding of DNA during transcription, replication, and recombination (Fig. 2) (Nitiss 2009). Both doxorubicin and epirubicin are well documented as Top2 “poisons (Marinello et al. 2018).

The mechanism of action of anthracyclines. Transcription and replication, the vital relieving of stress due to DNA super coiling, is conducted through the introduction of double stranded breaks by topoisomerase 1/2. These breaks are then sealed by DNA repair machinery. Doxorubicin inserts itself between DNA base pairs, thereby trapping topoisomerase 1/2 in place after it has catalyzed the double stranded break. This halts replication, leading to cell death
Top2 enzymes create double-strand breaks in DNA, causing DNA relaxation and coiling where necessary (Fig. 2) (Nitiss 2009). They act as a homodimer of two isozymes (Top2α and Top2β) and use ATP for catalytic activity (Marinello et al. 2018). The isozymes do not differ in their activity but are differentially expressed, with Top2α highly expressed in proliferating cells and Top2β expressed at equal levels in all cells (Beretta et al. 2008). Top2β knockout in mice was found to cause prenatal death as a result of severe developmental defects affecting neuronal cells (Lyu et al. 2006). Top2α was shown to interact with and regulate the transcription of ribosomal RNA genes in highly proliferating cells, a crucial process in cell growth (Ray et al. 2013). These studies provide a context for the importance of Top2 and why interference of its activity is crucial to the cytotoxic and therapeutic effects of DNA intercalating agents such as anthracyclines.
Top2 begins its DNA cleaving activity by binding specific sites (called the G-segment), such as promoter regions or other sequences along actively transcribed genes (Marinello et al. 2018). The G-segment is nicked on each strand, resulting in a double-strand break with a 5’ overhang on the cleaved strands (Marinello et al. 2018). The active tyrosine site in the enzyme (Y782) then binds to and stabilizes the DNA termini at the break site (Beretta et al. 2008). As ATP binds to Top2, it undergoes a conformational change and passages a second, unbroken strand (T-segment) through the double-stranded break site (Beretta et al. 2008). The passage step is vital to the uncoiling of DNA as it allows for the separation of two coiled segments (Marinello et al. 2018). Finally, Top2 hydrolyzes ATP, seals the G-segment break, and resets the system, allowing for the process to repeat at a different site (Beretta et al. 2008).
Intercalating agents such as doxorubicin and epirubicin insert themselves between adjacent base pairs in a DNA sequence (Fig. 2) (Marinello et al. 2018). The drug-DNA complex effectively “traps” Top2 when the enzyme binds to the sequence and attempts to perform its function (Fig. 2) (Pommier et al. 2016). Specifically, while the planar moieties of doxorubicin and epirubicin form stacking interactions with DNA base pairs, their side chains recognize and bind to Top2 (Marinello et al. 2018). Once trapped at the binding site, the Top2-drug-DNA complex acts as an impediment to replication and transcription (Pommier et al. 2016). For example, as the replication fork approaches the Top2-drug-DNA complex, the enzymes involved in replication collide with the immovable complex, resulting in incomplete replication products (termed replication run-off) (Pommier et al. 2016). Top2 poisons such as doxorubicin and epirubicin are, therefore, particularly potent during the S phase of the cell cycle, when DNA replication and transcription are highly active (Marinello et al. 2018). The DNA fragments are, effectively, permanent double-strand breaks in DNA and are recognized as such by the DNA damage repair machinery (Pommier et al. 2016).
The DNA damage response (DDR) pathway, mediated primarily by ataxia–telangiectasia mutated (ATM) and ataxia–telangiectasia and Rad3 related (ATR) kinases, involves the activation of p53, ERK and checkpoint kinase 2 (CHEK2) (Kumari et al. 2017; Yang et al. 2016). The pathway arrests the cell cycle and activates apoptosis if DNA repair cannot occur, as is the case in most tumors with mutations in the repair pathway (Pommier et al. 2016). While there is extensive evidence for the involvement of the well-documented, canonical activated p53 pathway in doxorubicin-induced cell death, recent evidence has suggested that pERK can cause apoptosis in breast cancer cells treated with doxorubicin, regardless of the expression or functioning of p53 (Kumari et al. 2017). Direct interaction of Bim with Bcl-xl is capable of inducing cell death in prostate cancer cells treated with doxorubicin (Yang et al. 2016). Overall, the treatment of breast cancers with doxorubicin and epirubicin results in permanent DNA damage which in turn activates p53-mediated and p53-independent apoptosis pathways, resulting in cytotoxicity (Pommier et al. 2016).
Clinical application
Adriamycin (doxorubicin)
Current clinical practice in the United States, as described by the National Comprehensive Cancer Network guidelines, recommends the use of doxorubicin and liposomal doxorubicin as single agents in the treatment of triple negative tumors and tumors with germline BRCA1/2 mutations (Gradishar et al. 2020). Doxorubicin, in a single chemotherapy agent system, is administered in 60–75 mg/m2 doses every 3 weeks in combination with the recommended cardio-protective drugs to account for the high risk of cardiac toxicity (26%) (Gradishar et al. 2020). Liposomal doxorubicin, a variant with a different delivery system, has a less frequent schedule (50 mg/m2 every 4 weeks) and a lower risk of cardiac toxicity (7%) (Gradishar et al. 2020; Rayner and Cutts 2014). Liposomal doxorubicin has been found to reduce the risk of other doxorubicin-associated side effects such as nausea, vomiting, alopecia, and neutropenia (Gradishar et al. 2020). Doxorubicin has also been recommended for combinatorial chemotherapy with cyclophosphamide or docetaxel (Gradishar et al. 2020). Two clinical trials have investigated the combinatorial efficacy of doxorubicin with paclitaxel/docetaxel and cyclophosphamide (Biganzoli et al. 2002; Nabholtz et al. 2003). In combination therapies, doxorubicin doses are reduced to 40–60 mg/m2 every 3–4 weeks with a maximum recommended cumulative dose of 450–500 mg/m2 (Gradishar et al. 2020; Rayner and Cutts 2014). Currently, the use of Adriamycin in combination with carboplatin and cyclophosphamide is being investigated in a neoadjuvant setting (McAndrew and DeMichele 2018).
Ellence (epirubicin)
Epirubicin was approved by the FDA for the treatment of breast cancer in 1999 and has since been tested for its efficacy both as a monotherapy and a combination therapy with taxanes (Rayner and Cutts 2014). While it shares the same mechanism of action and clinical efficacy as doxorubicin, several clinical trials comparing equimolar doses of the two drugs have found that patients treated with epirubicin reported less cardiac toxicity, nausea, alopecia, and myelosuppression (Khasraw et al. 2012). When used as a monotherapy, epirubicin can be used in higher doses than doxorubicin (Rayner and Cutts 2014). The drug is administered in 100–120 mg/m2 every 3–4 weeks with a cumulative dose limit of 900 mg/m2 (Rayner and Cutts 2014). Like doxorubicin, there are clinical trials currently testing the efficacy of epirubicin with carboplatin (McAndrew and DeMichele 2018). Additionally, a recent study is testing the combination of epirubicin and doxorubicin with the newly approved immunotherapy agent atezolizumab (U.S. 2018c).
Immunotherapy (atezolizumab and pembrolizumab)
The lack of ER, HER2, and PR expression in triple negative breast tumors presents a significant challenge in designing targeted therapies (Katz and Alsharedi 2017). To address this, several studies have looked into specific processes that can be targeted in TNBC without a wider systemic effect (Katz and Alsharedi 2017). Gene expression and clinical data analyses of vital signaling processes in triple-negative breast cancer revealed that higher immune response levels were associated with a significantly better clinical outcome (Desmedt et al. 2008; Lehmann et al. 2011). The infiltration of CD8+ lymphocytes was reported to predict increased patient survival specifically in TNBC versus other subtypes of breast cancer (Liu et al. 2012). Additionally, the treatment of triple negative breast tumors with anthracycline chemotherapy has been shown to induce the immune system by activating CD8+ T cells which kill cancer cells (Stagg and Allard 2013). Immune response modulation, therefore, is a promising, targeted approach to the treatment of triple-negative breast tumors (Katz and Alsharedi 2017).
Atezolizumab: Atezolizumab (brand name: Tecentriq) was developed as an IgG1 monoclonal antibody targeting the protein PD-L1 (programmed cell death-ligand 1) to prevent its interaction with its receptor PD-1, resulting in the reversal of T-cell suppression (Schmid et al. 2018). The antibody was developed to specifically inhibit the PD-L1 to PD-1 interaction while still allowing for the alternative ligand PD-L2 to bind PD-1, thereby reducing autoimmune side effects (Herbst et al. 2014). In a randomized, phase 3 clinical trial, atezolizumab was tested on 451 patients in combination with nanoparticle albumin-bound (nab) paclitaxel (Schmid et al. 2018). Compared to patients who were treated with placebo and chemotherapy, patients treated with atezolizumab survived longer without disease progression with an objective response rate of 56% versus 45.9% (atezolizumab treatment group being higher) (Schmid et al. 2018). The success of the trial led to the approval of Tecentriq, by the FDA, for the treatment of triple negative breast cancer in May 2020 (U.S. 2020a).
Pembrolizumab: Pembrolizumab (brand name: Keytruda) was first developed and approved as a treatment for unresectable or metastatic melanoma (Raedler and Keytruda (Pembrolizumab) 2015). Unlike Atezolizumab, Pembrolizumab is a humanized IgG4κ antibody that targets the receptor PD-1 rather than its ligand (Kwok et al. 2016). Through the KEYNOTE-522 clinical trial, the effects of pembrolizumab treatment followed by chemotherapy in patients with early-stage TNBC were compared with placebo plus chemotherapy treatment (Schmid et al. 2020). It was found that a significant number of patients (64.8% versus 51.2%) benefited from the PD-1 antibody followed by adjuvant chemotherapy (Schmid et al. 2020). Following these results, the FDA approved Keytruda for the treatment of high-risk, early-stage triple-negative breast cancer in July 2021.
Mechanisms of action of atezolizumab and pembrolizumab
Immunotherapeutics, such as atezolizumab, primarily function by activating a patient’s immune system to recognize and kill cancer cells (Sun et al. 2018). However, cancer progression occurs through the evasion of the immune system (Chen et al. 2016). One of the major pathways of immune suppression involves the regulation of immune checkpoints, a series of ligand-receptor interactions which determine whether the T-cell response is activated or inhibited (Sun et al. 2018).
Both preclinical and clinical data have shown that CTLA-4 and PD-L1 are key proteins in the regulation of immune checkpoints in cancer cells and their upregulation was shown to negatively affect T-cell response to cancer progression (Ribas 2012). As atezolizumab targets the PD-L1-PD-1 axis, the focus of this section will be on PD-1 functioning and its effect on the immune system (Schmid et al. 2018).
PD-1 is a receptor, expressed on stimulated T-cells, which recognizes and binds its ligand PD-L1, thereby initiating an inhibitory signal (Ishida et al. 1992). It was initially characterized as an important regulator of programmed cell death in lymphoid cell lines that were induced to die (Ishida et al. 1992). The receptor is expressed on the surface of memory T-cells that have been previously induced with an antigen as well as on dendritic cells and natural killer cells (Akinleye and Rasool 2019). It recognizes and binds to its ligands PD-L1 and PD-L2, though evidence has shown that PD-L1 is the dominant ligand in the interaction (Sun et al. 2018). PD-L1 is primarily expressed on antigen-presenting cells, healthy tissue as well as tumor cells, and stroma (Sun et al. 2018). In a proinflammatory setting, multiple cell types tend to increase PD-L1 production in response to IFN-γ and IL4 through STAT1 activation (Sun et al. 2018; Akinleye and Rasool 2019). Indeed, this may explain the production of PD-L1 in tumor cells in response to cytokines in the tumor microenvironment (Akinleye and Rasool 2019).
Previous studies have demonstrated that PD-L1 binding to PD-1 inhibits T-cell proliferation and survival, among other effects (Butte et al. 2007; Keir et al. 2006). Specifically, the PD-1/PD-L1 axis, when induced, affects the production of proinflammatory cytokines IFN-γ, TNF-α, and IL-2 (Keir et al. 2006). In healthy tissue, PD-1/PD-L1 signaling serves to prevent autoimmunity, also known as peripheral T-cell tolerance (Keir et al. 2006). Indeed, PD-1 expression on T-cells has to be induced by antigen-presenting cells carrying antigen-MHC complexes which results in the inhibition of future immune activation events that are normally regulated through CD28 or IL-2 (Akinleye and Rasool 2019). In effect, PD-1/PD-L1 helps “check” the immune system from an overactive response to infection and/or inflammation through the inhibition of effector cell functioning through its control of cytokine production in T-cells, resulting in a signaling cascade that affects T-cell proliferation and survival (Akinleye and Rasool 2019).
To escape antitumor immune responses, cancer cells (including TNBC cells) take advantage of the PD-1/PD-L1 axis (Sun et al. 2018). PD-1 expression was found to be increased in tumor-infiltrating lymphocytes in patients with TNBC, implying the inhibited state of the immune response to cancer (Mittendorf et al. 2014). Furthermore, several cancer subtypes, including TNBC, have been shown to express higher levels of PD-L1 either through the upregulation of adaptive or innate immune resistance pathways (Akinleye and Rasool 2019). In the innate immune resistance pathway, PD-L1 expression is increased through the activation of PI3K/AKT in some cancer subtypes, regardless of the cytokines present in the tumor microenvironment (Akinleye and Rasool 2019). The inhibition of AKT was found to reduce the expression of PD-L1 in TNBC cells, implying that TNBC cells overexpress PD-L1 through this mechanism (Mittendorf et al. 2014). PD-L1 expression can also be induced through IFN-γ production in the tumor microenvironment (adaptive immune resistance) (Akinleye and Rasool 2019). The propagation of this inhibitory effect on T-cells results in dysfunction and exhaustion of the immune response to tumors (Akinleye and Rasool 2019).
As an immune checkpoint inhibitor, atezolizumab binds to PD-L1 and suppresses the functioning of the PD-1/PD-L1 axis (Fig. 3) (Schmid et al. 2018). By binding to PD-L1, atezolizumab not only prevents PD-1 activation but also allows for the B7-CD28 mediated activation of CD8+ T-cells in response to tumor antigens (Ribas 2012). Furthermore, PD-L1 blockade reverses CD8+ T-cell exhaustion, hence restoring their cancer-killing function (Pauken and Wherry 2015). Once PD-L1/PD-1 signaling is inhibited, the T-cells can respond to the inflammatory cytokines described previously to perform their anti-tumor function (Fig. 3) (Akinleye and Rasool 2019). Particularly, evidence indicates IFN-γ signaling is vital to the regulation of CD8+ T-cells after PD-L1 blockade (Sun et al. 2018). Further signaling changes caused by PD-L1 inhibition are currently being investigated along (Akinleye and Rasool 2019). While pembrolizumab binds to a different target, namely, the PD-1 receptor, its effect on the suppression of the PD-1/PD-L1 axis is like atezolizumab (Schmid et al. 2018). The efficacy of the two drugs in the treatment of advanced squamous non-small-cell lung cancer, in combination with chemotherapy, was compared and it was found that pembrolizumab plus chemotherapy resulted in superior overall survival and disease-free progression rates (Zhang et al. 2018). A similar comparison has not yet been made in TNBC patients.

Mechanism of action of the anti-cancer activity of immunotherapeutics. The interaction of the receptor PD-1 with its ligand PD-L1 normally reduces inflammatory response. Tumour cells use this interaction to suppress anti-cancer T-cell response. Inhibition of either the receptor PD-1 (using pembrolizumab) or the ligand PD-L1 (using atezolizumab) results in the activation of the immune response against the tumour cells
Clinical application
In March 2020, Atezolizumab became the first PD-1 inhibitor to be approved by the FDA for the treatment of advanced and/or metastatic triple negative breast cancer (U.S. 2020a). The drug is now used alongside anthracycline or taxane-based chemotherapy and is administered in 840-mg doses every 2 weeks until the patient is progression-free or adverse side effects occur (Schmid et al. 2018). Patients treated with Atezolizumab and chemotherapy were shown to have increased nausea (46% versus 38%), neutropenia (20.8% versus 15.3%), and hypothyroidism (13.7% versus 3.4%) versus patients who were only treated with chemotherapy (Schmid et al. 2018). However, only 15.9% of patients in the phase 3 clinical trial withdrew from the atezolizumab treatment group versus 8.2% withdrawals in the chemotherapy-only group (Schmid et al. 2018). Currently, clinical trials are underway to test the effectiveness of combining atezolizumab with carboplatin and cyclophosphamide as well as HDAC inhibitors for the treatment of TNBC patients (U.S. 2020b, 2020c).
Pembrolizumab was approved for the treatment of high-risk, early-stage TNBC cases in July 2021 (Administration 2021). Treatments are administered at doses of 200 mg every 3 weeks or 400 mg every 6 weeks for a total of 24 weeks alongside chemotherapy (before surgery) (Schmid et al. 2020). Subsequently, Pembrolizumab treatment is continued, without accompanying chemotherapy, for 27 weeks (Schmid et al. 2020). The most common side effects observed in the clinical trial were febrile neutropenia, anemia, and pyrexia (Schmid et al. 2020).
Taxane-based chemotherapy
In the modern clinical setting, the taxane family of chemotherapeutic drugs is one of the most effective antitumor therapies in general and particularly for triple-negative breast cancers due to the lack of expression of traditional targets (Nabholtz and Gligorov 2005). In 1971, Wani and colleagues derived paclitaxel (brand name: Taxol) from the bark extract of Taxus brevifolia, an evergreen yew from the Pacific Northwest, and described its antitumor and specifically, antileukemic properties (Wani, et al. 1971). Several years later, Schiff et al. discovered that Taxol reduced HeLa cell division significantly through its stabilization of microtubule assembly (Schiff et al. 1979). The limited availability of the drug led to the development and isolation of docetaxel, a semi-synthetic analog of paclitaxel from bark extracts of Taxus baccata (Ringel and Horwitz 1991). While structurally similar to paclitaxel barring minor chemical modifications, docetaxel was found to have a greater affinity to beta-tubulin, reduced efflux rate, and no cardiotoxic effects compared to paclitaxel (Nabholtz and Gligorov 2005).
The mechanism of action of taxanes was considered unique and therefore sparked interest in the development of paclitaxel and docetaxel for cancer therapy (Rowinsky et al. 1990). Several phase I/II clinical trials were then conducted to determine the dosage, toxicity, and efficacy of paclitaxel in treating doxorubicin/mitoxantrone-resistant metastatic breast tumors (Holmes et al. 1991; Nabholtz et al. 1996; Wilson et al. 1994; Seidman et al. 1998). Based on these and other clinical trials, the US FDA approved paclitaxel for the treatment of metastatic breast cancers which progressed despite prior anthracycline treatment (Cortazar et al. 2012). A later phase III clinical trial showed the increased effectiveness of paclitaxel in treating hormone-receptor negative (triple negative) metastatic breast cancers as well as anthracycline-resistant tumors (Henderson et al. 2003). Between 1992 and 1993, several phase I/II studies of docetaxel as a first and second-line therapy against metastatic breast cancer showed increased tumor response to the drug, especially in cases of anthracycline resistance (Nabholtz and Gligorov 2005; Oosterom 1995; Trudeau 1995). Based on a response rate of 37.9% in anthracycline-refractory tumors, the FDA granted accelerated approval for docetaxel in the treatment of metastatic breast cancer in 1996 (Cortazar et al. 2012).
The poor solubility, retention, and side effects associated with paclitaxel led to the development of a better delivery system and formulation for the drug in the form of albumin-bound paclitaxel (brand name: Abraxane) (Ibrahim et al. 2005). The formulation significantly reduced hypersensitivity, neuropathy, erythrocyte aggregation, and severe anaphylaxis while increasing drug transport efficacy by delivering paclitaxel in an albumin suspension (Ibrahim et al. 2005). A multicenter phase II clinical trial showed a 48% response rate in patients with metastatic breast cancer, no severe hypersensitivity reactions, and significant antitumor activity when used as a first-line treatment (Ibrahim et al. 2005).
Mechanism of action of taxanes
Unlike anthracyclines, PARP inhibitors, and immunotherapies, taxanes function through their binding to and stabilization of microtubules, preventing their disassembly into beta-tubulin components (Fig. 4) (Gallego-Jara et al. 2020). In fast-dividing cancer cells, the prevention of disassembly significantly reduces the microtubule dynamics required for cell–cell division, signaling, and migration, amongst other cellular processes (Dumontet and Jordan 2010). Therefore, there are several proposed mechanisms of action of taxol in its mediation of cell death (Gallego-Jara, et al. 2020).

Schematic of the mechanism of action of Taxanes. By binding to microtubules, taxanes prevent their disassembly, thereby disrupting the dynamic instability of microtubules, leading to centrosomal impairment and suppression of spindle dynamics during mitosis
Microtubules are a vital component of the cellular cytoskeleton, acting as filaments driving key processes such as intracellular transport, cell division, and polarity (Brouhard and Rice 2018). Each microtubule consists of 13 protofilaments arranged in parallel, assembled around a hollow cylindrical core (Fig. 4) (Lasser et al. 2018). These filaments are highly dynamic polymers of αβ-tubulin monomeric units and are regulated by the GTPase activity of tubulin (Fig. 4) (Akhmanova and Steinmetz 2015). While GTP binds both α and β-tubulin, primarily, GTP-bound β-tubulin is hydrolyzed during the polymerization of microtubules (Fig. 4) (Akhmanova and Steinmetz 2015). GDP-bound β-tubulin has reduced affinity to surrounding tubulin units, favoring depolymerization and leading to the dynamic behavior of microtubules where GDP-tubulin is constantly lost at one end while being replaced by GTP-tubulin at the other end (Fig. 4) (Akhmanova and Steinmetz 2015). If the rate of GTP-tubulin addition exceeds the rate of GDP-tubulin dissociation, the microtubules obtain a GTP cap (plus end) and a slow-growing minus end, thereby gaining polarity, a vital aspect of mitotic spindle formation during the cell cycle (Akhmanova and Steinmetz 2015). This dynamic instability of microtubules is vital to the cytoskeletal remodeling that occurs during mitosis as well as intracellular transportation (Brouhard and Rice 2018; Mitchison and Kirschner 1984). In rapidly dividing tumor cells, the microtubules that constitute the mitotic spindles are highly sensitive to therapeutic disruption due to their importance in sister chromatid separation (Brouhard and Rice 2018).
Taxol performs its function by stabilizing the microtubule through its binding to β-tubulin (Fig. 4) (Kellogg et al. 2017; Nogales 2000). In particular, the Taxol molecule binds to the M-loop in β-tubulin and stabilizes its association with adjacent β-tubulin molecules, thereby strengthening protofilament-protofilament interaction and reducing the rate of depolymerization caused by calcium and cold temperatures (Fig. 4) (Nogales 2000; Weaver 2014). Functionally, this causes cell cycle arrest in the G2/M-phase due to the inhibition of chromatin separation which results in the mitotic checkpoint activation and cell death thereafter (Weaver 2014; Ganguly et al. 2010; Milas et al. 1995). Induction of apoptosis through cell cycle arrest is widely regarded as the primary mechanism of action of taxanes. Paclitaxel has also been found to induce apoptosis by mediating an increase in Reactive Oxidative Species (ROS), downregulation of Bcl-2, and inhibition of the AKT/MAPK pathway to reduce cell proliferation in ovarian, canine mammary, and osteosarcoma cell lines (Strobel et al. 1996; Ren et al. 2018; Li et al. 2020). Heightened ROS levels were also found to coincide with increased expression of endoplasmic reticulum-stress proteins such as GRP78 and IRE1α in osteosarcoma cells (Li et al. 2020). The induction of endoplasmic reticulum stress then releases free Ca2+ and increases ROS production from mitochondria damaged by the calcium ion overload (Csordás and Hajnóczky 2009). Together, these effects initiate cytochrome C release as well as caspase 3 cleavage, both of which are mechanisms of mitochondria-mediated apoptosis (Suh et al. 2013). Additional research has shown that the induction of autophagy may be an alternative mechanism for taxol functioning. In gastric cancer cells, paclitaxel treatment demonstrated the inhibition of proliferation and induction of autophagy through p62 protein degradation (Yu et al. 2017). In non-small-cell lung cancer cells, the promotion of autophagy through esomeprazole treatment reversed taxol resistance, specifically through the reduction of intracellular pH and inhibition V-ATPase (Bai et al. 2021). However, these results are contradicted by others who have shown that autophagy inhibition reverses taxol resistance and induces caspase-dependent apoptosis (Peng et al. 2014; Kim et al. 2013; Zamora et al. 2019; Song et al. 2017).
Clinical application
Paclitaxel (taxol)
Paclitaxel is currently used to treat HR-negative, HER2-negative breast tumors as well as tumors with BRCA 1/2 germline mutations (Gradishar et al. 2020). Current guidelines recommend the use of single chemotherapy agents to mitigate the adverse side effects that affect patients. Paclitaxel, in particular, is effective in either weekly doses at 80 mg/m2 or every 3 weeks at 175 mg/m2 (Gradishar et al. 2020). Clinical trials have shown that the weekly regimen appears to improve overall survival while preserving the same response rate as the 3-weekly approach (Mauri et al. 2010).
Nab-paclitaxel, an alternative form of paclitaxel, consists of albumin-bound paclitaxel nanoparticles that have a mean diameter of 130 nm (Schettini et al. 2016). It was developed primarily to reduce the adverse side effects otherwise caused by the paclitaxel solvent (Gallego-Jara et al. 2020). The conjugation of paclitaxel to albumin allows for the rapid delivery of the drug through the gp60/caveolin-1 receptor pathway in tumor cells, resulting in greater drug penetration as well as higher maximum tolerable doses in patients (Schettini et al. 2016). Indeed, phase 3 clinical trials have shown that weekly nab-paclitaxel doses of 125 mg/m2 have improved patient survival rates and reduced adverse side effects such as hypertension and neutropenia (Untch et al. 2016; Gradishar et al. 2005). Currently, this regimen is the recommended scheme for the treatment of triple-negative breast tumors and can be used instead of paclitaxel or docetaxel regimens (Gradishar et al. 2020).
Docetaxel (taxotere)
Approved by the FDA in 1996 for the treatment of metastatic and triple negative breast tumors, docetaxel has since been used either as a monotherapy or in combination with anthracyclines in the treatment of triple negative breast cancers (Rayner and Cutts 2014; Nabholtz and Gligorov 2005). Indeed, the greater efficacy of docetaxel compared to paclitaxel has made it a safer alternative for patients as it performs the same function as paclitaxel at a lower effective dose (Nabholtz and Gligorov 2005). Currently, docetaxel is administered in 60–100 mg/m2 doses every 3 weeks (there appears to be no discernable difference between weekly and 3-weekly treatment cycles) in both neoadjuvant and adjuvant settings (Mauri et al. 2010). An ongoing clinical trial is testing the effectiveness of combining 75 mg/m2 docetaxel doses with carboplatin, a platinum-based chemotherapeutic, every 3 weeks (Ademuyiwa et al. 2021).
Antibody-drug conjugate therapy
The attachment of antibodies and drugs was developed to efficiently deliver small molecule inhibitor molecules to cancer cells specifically (Nagayama et al. 2020). The monoclonal antibody component of the complex ensures the specificity of drug delivery and therefore increases the potency of the treatment while also reducing toxicity to healthy tissue (Nagayama et al. 2020). By recognizing and binding to an antigen-specific to cancer cells, the monoclonal antibody can then drive changes in tumor cell signaling or induce an immune response against the tumor (Chau et al. 2019). Currently, there are approximately 30 FDA-approved monoclonal antibody therapies against cancer in the market (Carter and Lazar 2018). These antibodies can be conjugated with effector molecules, such as small molecule inhibitors, cytotoxins, and radioactive isotopes, using a linker region that is cleaved at the site of the tumor thereby releasing the drug (Chau et al. 2019). The drug is then absorbed by the tumor cells, where it induces cell death (Chau et al. 2019).
One of the first antibody–drug conjugates (ADCs), gemtuzumab ozagamicin, was approved by the FDA for the treatment of patients with acute myeloid leukemia in 2000 (Nagayama et al. 2020; Sievers et al. 2001; Bross et al. 2001). Unfortunately, as the therapy did not significantly improve patient survival and caused increased off-target toxicity, it was removed from the market in 2010 and a lower dose of the drug was approved for use in 2017 (Nagayama et al. 2020). This showed that while ADCs theoretically seem straightforward to develop, there are significant challenges that limit the potency and specificity of these therapies (Chau et al. 2019). Current ADCs use a variety of cytotoxic “payload” molecules such as calicheamicins or SN38 (DNA-damaging agents), maytansines, or auristatins (anti-tubulin agents) or antitumor antibiotics (Nagayama et al. 2020; Chau et al. 2019). The next generation of ADCs included brentuximab vedotin, used to treat Hodgkin’s lymphoma and anaplastic large-cell lymphoma, and trastuzumab emtansine, used for HER2-positive breast cancer treatment (Nagayama et al. 2020). Unlike first-generation ADCs, these therapies were effective in reducing toxicity while also improving overall patient survival (Nagayama et al. 2020).
The development of ADCs for the treatment of patients with TNBC has been summarized by Nagayama and colleagues (Nagayama et al. 2020). Of the ADCs that have been tested, sacituzumab govitecan-hziy (brand name: Trodelvy) was approved by the FDA, in 2021, for the treatment of metastatic TNBC patients who had received at least two treatments previously (Bardia et al. 2021). In a phase 3 clinical trial, Sacituzumab govitecan treatment was compared to chemotherapy in patients with metastatic, treatment-refractory, TNBC (Bardia et al. 2021). Overall survival among patients treated with the ADC was 12.1 months versus 6.7 months for those treated with chemotherapy, displaying an objective response rate of 35% versus 5% respectively (Bardia et al. 2021). These significant improvements made by the administration of Trodelvy led to its approval by the FDA.
Mechanism of action of sacituzumab govitecan
In general antibody–drug conjugates consist of 3 components: an antibody against a target specific to cancer cells, an anti-cancer cytotoxic drug, and a linker region to conjugate the two (Fig. 5a) (Chau et al. 2019). Sacituzumab govitecan is an ADC consisting of an anti-Trop2 monoclonal antibody conjugated with the cytotoxic drug SN-38 (a topoisomerase I inhibitor) through a proprietary, pH-sensitive, cleavable linker (Fig. 5a) (Bardia et al. 2021; Moon et al. 2008). While the therapy was designed to target tumors expressing high levels of Trop2, the Phase III clinical trial by Bardia and colleagues showed that sacituzumab govitecan was significantly beneficial to patients with metastatic TNBC, regardless of Trop2 expression, though a greater benefit was found in patients with high Trop2-expression tumors (Bardia et al. 2021). In tumors expressing Trop2, the antibody component of sacituzumab govitecan would be able to recognize the surface protein and bind to it, resulting in the internalization of the ADC through the formation of an endosome (Fig. 5b) (Bravaccini and Maltoni 2021). The subsequent acidification of the endosome results in the cleavage of the linker, thereby releasing the drug, SN-38, into the cytoplasm upon the fusion of the endosome with the lysosome (Fig. 5b) (Nagayama et al. 2020). As a topoisomerase I inhibitor, SN-38 prevents the repair of single-strand breaks in DNA, resulting in DNA damage and cell death thereafter (Fig. 5b) (Bravaccini and Maltoni 2021). Therefore, the mechanism of action of an ADC is dependent on the selection of an antigen that is highly expressed in cancer tissue while having reduced expression in the surrounding normal tissue (Nagayama et al. 2020). Sacituzumab-govitecan targets the antigen Trop2, a glycoprotein that is highly expressed in TNBC tissue (Bardia et al. 2021).

Schematic of sacituzumab govitecan and its mechanism of action. a Representation of the 3 main components of sacituzumab govitecan, the antibody, the cytotoxic payload and the linker between them. b The mechanism of action of Trodelvy in a triple negative breast cancer cell. The antibody recognizes and binds to Trop2, is then internalized into the cell. This process induces the cleavage of the linker, releasing SN-38 into the cytoplasm after which it binds to and inhibits TOP1B, causing double stranded DNA breaks and cell death thereafter. Created with Biorender.com
Tumor-associated calcium signal transducer 2 (Trop2) is a transmembrane glycoprotein that plays a role in calcium signaling and interacts with signaling regulators such as insulin-like growth factor 1 (IGF1), protein kinase C, and cyclin D1 (Goldenberg et al. 2018). It is primarily associated with cell migration, proliferation, and anchorage-independent growth in cancer cells (Nagayama et al. 2020). Since its discovery 40 years ago, it has been known by multiple names including trophoblast cell-surface antigen 2, membrane component chromosome 1 surface marker 1, gastrointestinal antigen 733–1, and epithelial glycoprotein-1 (Goldenberg et al. 2018). In a study conducted in mice, the Goldenberg group identified mouse monoclonal antibodies that bound to Trop2 in cancerous lung, breast, colon, kidney, and ovarian tissues, thereby indicating that the glycoprotein was widely expressed by multiple cancer types (Stein et al. 1990). In patients with these cancers, including TNBC, increased Trop2 expression correlates with worse prognoses (Stepan et al. 2011). Notably, Trop2 overexpression has been observed in over 80% of TNBC cases while surrounding non-cancerous breast tissue expresses lower levels of the glycoprotein (Son et al. 2018). Thus, the differential expression of Trop2 could be used to target cancer cells with antibodies and deliver drugs specifically while reducing off-target effects (Goldenberg et al. 2018). To this end, the humanized monoclonal antibody (known as hRS7) against Trop2 was developed to specifically recognize and bind to TNBC cancer cells that expressed Trop2 (Bardia et al. 2021).
The payload of Trodelvy is SN-38, the active ingredient in irinotecan, which itself is a well-known inhibitor of topoisomerase I and causes DNA damage (Goldenberg and Sharkey 2019). While irinotecan is highly potent against various human cancer cell lines, with its IC50 in the nanomolar range, its low bioavailability presented challenges to its therapeutic application (Goldenberg and Sharkey 2019; Sharkey et al. 2015). Particularly, the conversion of irinotecan to its active form SN-38 within a patient’s liver, intestine or plasma was highly inefficient (Sharkey et al. 2015). Therefore, the active metabolite SN-38 was directly conjugated to the antibody at a ratio of 7.6 molecules of SN-38 for every 1 molecule of the hRS7 antibody (Goldenberg and Sharkey 2019). Upon internalization and release into the cell, SN38 binds to and stabilizes topoisomerase IB, forming an SN-38-TOPIB-DNA complex (Peters and Chapter 2020). This prevents TOPIB-induced single-stranded DNA breaks from repairing and when the DNA replication fork (during S-phase) encounters the complex, irreversible double-stranded breaks are formed, leading to cell death as a result (Fig. 5b) (Peters and Chapter, 2020).
It should be noted that while this mechanism of action is similar to that of anthracyclines, SN-38 targets topoisomerase I, which catalyzes single-stranded DNA breaks while anthracyclines target topoisomerase II, which catalyzes double-stranded DNA breaks. Interestingly, however, p53-mediated apoptosis appears to be a common mechanism through which both SN38 and anthracyclines mediate their cytotoxic function (Takeba et al. 2007; Derenzini et al. 2009).
Clinical application
Currently, Trodelvy has been indicated for the treatment of metastatic TNBC patients who have previously received at least 2 therapies, one of which for the metastasis itself (Bardia et al. 2021). A dose of 10 mg per kg is injected intravenously once a week in 21-day treatment cycles until disease progression or intolerable toxicity occurs (Bardia et al. 2021). The most common adverse side effects include neutropenia, diarrhea, nausea, fatigue, and anemia (Bardia et al. 2021).
Conclusion and perspectives
The emergence of therapies such as Trodelvy, olaparib, and Keytruda, an antibody–drug conjugate, a PARP inhibitor, and an immunotherapeutic respectively, can be traced back to an increase in understanding, not only of the tumor microenvironment but of the signaling pathways that affect tumor growth. These drugs represent newer, more targeted tools that clinicians can now use to combat an otherwise treatment-refractory disease in TNBC in a manner that reduces patient risk while not compromising on reducing tumor recurrence. The approval of these agents by the FDA, either as mono or combinatorial therapies, allows for a more nuanced approach to the treatment of TNBC.
While chemotherapy remains the standard, not only in TNBC treatment but also in most cancers, the emergence of new drug delivery methods, as shown by nab-paclitaxel, will help reduce the adverse side effects normally associated with this class of anti-cancer agents. The emerging use of nano-delivery systems may be the key to increasing the pharmacokinetic efficiency of existing, approved therapies thereby reducing systemic toxicity and circumventing drug resistance, a problem that has plagued chemotherapy use for decades (Yao et al. 2020).
References
Ademuyiwa FO et al (2021) Immunogenomic profiling and pathological response results from a clinical trial of docetaxel and carboplatin in triple-negative breast cancer. Breast Cancer Res Treat 189(1):187–202
Administration, U.S.F.D. FDA approves pembrolizumab for high-risk early-stage triple-negative breast cancer. 2021 [cited 2022 11/1/2022]; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pembrolizumab-high-risk-early-stage-triple-negative-breast-cancer.
A’Hern RP, Smith IE, Ebbs SR (1993) Chemotherapy and survival in advanced breast cancer: the inclusion of doxorubicin in Cooper type regimens. Br J Cancer 67(4):801–805
Akhmanova A, Steinmetz MO (2015) Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol 16(12):711–726
Akinleye A, Rasool Z (2019) Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J Hematol Oncol 12(1):92
Arcamone F et al (1969) Adriamycin, 14-hydroxydaimomycin, a new antitumor antibiotic from S. Peucetius var. Caesius. Biotechnol Bioeng 11(6):1101–1110
Arteaga CL et al (2011) Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol 9(1):16–32
Bai Z et al (2021) Esomeprazole overcomes paclitaxel-resistance and enhances anticancer effects of paclitaxel by inducing autophagy in A549/Taxol cells. Cell Biol Int 45(1):177–187
Bardia A et al (2021) Sacituzumab govitecan in metastatic triple-negative breast cancer. N Engl J Med 384(16):1529–1541
Barton VN et al (2016) Anti-androgen therapy in triple-negative breast cancer. Thera Adv Med Oncol 8(4):305–308
Beretta GL, Zunino F (2008) Molecular mechanisms of anthracycline activity. In: Krohn K (ed) Anthracycline chemistry and biology II: mode of action, clinical aspects and new drugs. Springer, Berlin, pp 1–19
Bergin ART, Loi S (2019) Triple-negative breast cancer: recent treatment advances. F1000Res 8:1342
Bianchini G et al (2016) Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol 13(11):674–690
Biganzoli L et al (2002) Doxorubicin and paclitaxel versus doxorubicin and cyclophosphamide as first-line chemotherapy in metastatic breast cancer: The European Organization for Research and Treatment of Cancer 10961 Multicenter Phase III Trial. J Clin Oncol 20(14):3114–3121
Bonadonna G et al (1969) Clinical evaluation of adriamycin, a new antitumor antibiotic. Br Med J 3(5669):503–506
Bravaccini S, Maltoni R (2021) Trop-2 therapy in metastatic triple-negative breast cancer in Italy: clinical opportunity and regulatory pitfalls. J Pers Med 11(11):1211
Bray F et al (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J Clin 68(6):394–424
Brenton JD et al (2005) Molecular classification and molecular forecasting of breast cancer: ready for clinical application? J Clin Oncol 23(29):7350–7360
Bross PF et al (2001) Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res 7(6):1490–1496
Brouhard GJ, Rice LM (2018) Microtubule dynamics: an interplay of biochemistry and mechanics. Nat Rev Mol Cell Biol 19(7):451–463
Bryant HE et al (2005) Specific killing of BRCA2-deficient tumors with inhibitors of poly(ADP-ribose) polymerase. Nature 434(7035):913–917
Burnell M et al (2010) Cyclophosphamide, epirubicin, and Fluorouracil versus dose-dense epirubicin and cyclophosphamide followed by Paclitaxel versus Doxorubicin and cyclophosphamide followed by Paclitaxel in node-positive or high-risk node-negative breast cancer. J Clin Oncol 28(1):77–82
Burstein MD et al (2015) Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res 21(7):1688–1698
Butte MJ et al (2007) Programmed death-1 ligand 1 interacts specifically with the B7–1 costimulatory molecule to inhibit T cell responses. Immunity 27(1):111–122
Byrum AK, Vindigni A, Mosammaparast N (2019) Defining and modulating “BRCAness.” Trends Cell Biol 29(9):740–751
Caestecker KW, Van de Walle GR (2013) The role of BRCA1 in DNA double-strand repair: past and present. Exp Cell Res 319(5):575–587
Carey LA et al (2007) The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res 13(8):2329
Carter PJ, Lazar GA (2018) Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov 17(3):197–223
Caulfield SE, Davis C, Byers KF (2019) Olaparib: a novel therapy for metastatic breast cancer in patients with a BRCA1/2 mutation. J Adv Practition Oncol 10(2)
Chau CH, Steeg PS, Figg WD (2019) Antibody–drug conjugates for cancer. Lancet 394(10200):793–804
Chen J et al (2016) Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol 27(3):409–416
Chen H et al (2018) Association between BRCA status and triple-negative breast cancer: a meta-analysis. Front Pharmacol 9:909–909
Collignon J et al (2016) Triple-negative breast cancer: treatment challenges and solutions. Breast Cancer (dove Medical Press) 8:93–107
Conde C et al (2001) Loss of poly(ADP-ribose) polymerase-1 causes increased tumor latency in p53-deficient mice. EMBO J 20(13):3535–3543
Cortazar P et al (2012) US Food and Drug Administration approval overview in metastatic breast cancer. J Clin Oncol 30(14):1705–1711
Csordás G, Hajnóczky G (2009) SR/ER-mitochondrial local communication: calcium and ROS. Biochim Biophys Acta 1787(11):1352–1362
D’Andrea AD (2018) Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (amst) 71:172–176
De Summa S et al (2013) BRCAness: a deeper insight into basal-like breast tumors. Ann Oncol 24(Suppl 8):viii13-iii21
Dent R et al (2007) Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 13(15 Pt 1):4429–4434
Dent RA et al (2013) Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res 15(5):R88
Derenzini M et al (2009) The p53-mediated sensitivity of cancer cells to chemotherapeutic agents is conditioned by the status of the retinoblastoma protein. J Pathol 219(3):373–382
Desmedt C et al (2008) Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res 14(16):5158
Dolle JM et al (2009) Risk factors for triple-negative breast cancer in women under the age of 45 years. Cancer Epidemiol Biomark Prevent 18(4):1157–1166
Dumontet C, Jordan MA (2010) Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 9(10):790–803
Eikesdal HP et al (2021) Olaparib monotherapy as primary treatment in unselected triple negative breast cancer☆. Ann Oncol 32(2):240–249
Farmer H et al (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434(7035):917–921
Findlay B et al (2007) A dose escalation trial of adjuvant cyclophosphamide and epirubicin in combination with 5-fluorouracil using G-CSF support for premenopausal women with breast cancer involving four or more positive nodes. Ann Oncol 18(10):1646–1651
Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363(20):1938–1948
Fragomeni SM, Sciallis A, Jeruss JS (2018) Molecular subtypes and local-regional control of breast cancer. Surg Oncol Clin N Am 27(1):95–120
Gallego-Jara J et al (2020) A compressive review about taxol((R)): history and future challenges. Molecules 25(24):5986
Ganguly A, Yang H, Cabral F (2010) Paclitaxel-dependent cell lines reveal a novel drug activity. Mol Cancer Ther 9(11):2914–2923
Goldenberg DM, Sharkey RM (2019) Antibody-drug conjugates targeting TROP-2 and incorporating SN-38: a case study of anti-TROP-2 sacituzumab govitecan. Mabs 11(6):987–995
Goldenberg DM, Stein R, Sharkey RM (2018) The emergence of trophoblast cell-surface antigen 2 (TROP-2) as a novel cancer target. Oncotarget 9(48):28989–29006
Gradishar WJ et al (2005) Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol 23(31):7794–7803
Gradishar WJ et al (2020) Breast Cancer, Version 3.2020. NCCN Clin Pract Guidelines Oncol. 18(4):452
Hahnen E et al (2017) Germline mutations in triple-negative breast cancer. Breast Care (basel) 12(1):15–19
Hanker AB, Sudhan DR, Arteaga CL (2020) Overcoming endocrine resistance in breast cancer. Cancer Cell 37(4):496–513
Hay T et al (2009) Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res 69(9):3850–3855
Henderson IC et al (2003) Improved outcomes from adding sequential Paclitaxel but not from escalating Doxorubicin dose in an adjuvant chemotherapy regimen for patients with node-positive primary breast cancer. J Clin Oncol 21(6):976–983
Herbst RS et al (2014) Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515(7528):563–567
Holmes FA et al (1991) Phase II trial of taxol, an active drug in the treatment of metastatic breast cancer. J Natl Cancer Inst 83(24):1797–1805
Ibrahim NK et al (2005) Multicenter phase II trial of ABI-007, an albumin-bound paclitaxel, in women with metastatic breast cancer. J Clin Oncol 23(25):6019–6026
C.C.S.A.C. and c.w.t.C.C. Society, Canadian Cancer Statistics 2021. 2021, Canadian Cancer Society: Toronto, ON
Ishida Y et al (1992) Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo j 11(11):3887–3895
Katz H, Alsharedi M (2017) Immunotherapy in triple-negative breast cancer. Med Oncol 35(1):13
Keir ME et al (2006) Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med 203(4):883–895
Kellogg EH et al (2017) Insights into the distinct mechanisms of action of taxane and non-taxane microtubule stabilizers from Cryo-EM structures. J Mol Biol 429(5):633–646
Khasraw M, Bell R, Dang C (2012) Epirubicin: is it like doxorubicin in breast cancer? A clinical review. Breast 21(2):142–149
Kim HJ et al (2013) Cytoprotective role of autophagy during paclitaxel-induced apoptosis in Saos-2 osteosarcoma cells. Int J Oncol 42(6):1985–1992
Kim G et al. (2022) FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. (1078-0432 (Print))
Kneubil MC et al (2013) Breast cancer subtype approximations and loco-regional recurrence after immediate breast reconstruction. Eur J Surg Oncol 39(3):260–265
Krishnakumar R, Kraus WL (2010) The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 39(1):8–24
Kumari R et al (2017) Constitutively activated ERK sensitizes cancer cells to doxorubicin: involvement of p53-EGFR-ERK pathway. J Biosci 42(1):31–41
Kwok G et al (2016) Pembrolizumab (Keytruda). Hum Vaccin Immunother 12(11):2777–2789
Lasser M, Tiber J, Lowery LA (2018) The role of the microtubule cytoskeleton in neurodevelopmental disorders. Front Cell Neurosci 12(165)
Lee JM, Ledermann JA, Kohn EC (2014) PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol 25(1):32–40
Lehmann BD et al (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121(7):2750–2767
Lehmann BD et al (2016) Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS ONE 11(6):e0157368
Li M et al (2020) Paclitaxel inhibits proliferation and promotes apoptosis through regulation ROS and endoplasmic reticulum stress in osteosarcoma cell. Mol Cell Toxicol 16(4):377–384
Liedtke C et al (2008) Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol 26(8):1275–1281
Lin NU et al (2012) Clinicopathologic features, patterns of recurrence, and survival among women with triple-negative breast cancer in the National Comprehensive Cancer Network. Cancer 118(22):5463–5472
Litton JK et al (2018) Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med 379(8):753–763
Liu S et al (2012) CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res 14(2):R48
Livraghi L, Garber JE (2015) PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med 13:188
Lukong KE (2017) Understanding breast cancer—the long and winding road. BBA Clin 7:64–77
Lukong KE, Ogunbolude Y, Kamdem JP (2017) Breast cancer in Africa: prevalence, treatment options, herbal medicines, and socioeconomic determinants. Breast Cancer Res Treat 166:351
Lyu YL et al (2006) Role of topoisomerase IIbeta in the expression of developmentally regulated genes. Mol Cell Biol 26(21):7929–7941
Makki J (2015) Diversity of breast carcinoma: histological subtypes and clinical relevance. Clin Med Insights Pathol 8:23–31
Maqbool M, Bekele F, Fekadu G (2022) Treatment strategies against triple-negative breast cancer: an updated review. Breast Cancer (dove Medical Press) 14:15–24
Marinello J, Delcuratolo M, Capranico G (2018) Anthracyclines as topoisomerase II poisons: from early studies to new perspectives. Int J Mol Sci 19(11):3480
Mauri D et al (2010) Overall survival benefit for weekly vs. three-weekly taxanes regimens in advanced breast cancer: a meta-analysis. Cancer Treat Rev 36(1):69–74
McAndrew N, DeMichele A (2018) Neoadjuvant chemotherapy considerations in triple-negative breast cancer. J Target Ther Cancer 7(1):52–69
McCann KE (2019) Advances in the use of PARP inhibitors for BRCA1/2-associated breast cancer: talazoparib. Future Oncol 15(15):1707–1715
Milas L et al (1995) Kinetics of mitotic arrest and apoptosis in murine mammary and ovarian tumors treated with taxol. Cancer Chemother Pharmacol 35(4):297–303
Mitchison T, Kirschner M (1984) Dynamic instability of microtubule growth. Nature 312(5991):237–242
Mittendorf EA et al (2014) PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res 2(4):361
Moon S-J et al (2008) Antibody conjugates of 7-ethyl-10-hydroxycamptothecin (SN-38) for targeted cancer chemotherapy. J Med Chem 51(21):6916–6926
Nabholtz J-M, Gligorov J (2005) The role of taxanes in the treatment of breast cancer. Expert Opin Pharmacother 6(7):1073–1094
Nabholtz JM et al (1996) Multicenter, randomized comparative study of two doses of paclitaxel in patients with metastatic breast cancer. J Clin Oncol 14(6):1858–1867
Nabholtz JM et al (2003) Docetaxel and doxorubicin compared with doxorubicin and cyclophosphamide as first-line chemotherapy for metastatic breast cancer: results of a randomized, multicenter, phase III trial. J Clin Oncol 21(6):968–975
Nagayama A et al (2020) Novel antibody–drug conjugates for triple negative breast cancer. Thera Adv Med Oncol 12:1758835920915980
Nitiss JL (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9(5):338–350
Nogales E (2000) Structural insights into microtubule function. Annu Rev Biochem 69:277–302
Pareja F et al (2016) Triple-negative breast cancer: the importance of molecular and histologic subtyping, and recognition of low-grade variants. NPJ Breast Cancer 2:16036
Paridaens R et al (2000) Paclitaxel versus doxorubicin as first-line single-agent chemotherapy for metastatic breast cancer: a European Organization for Research and Treatment of Cancer Randomized Study with cross-over. J Clin Oncol 18(4):724–733
Pauken KE, Wherry EJ (2015) Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36(4):265–276
Peng X et al (2014) Autophagy promotes paclitaxel resistance of cervical cancer cells: involvement of Warburg effect activated hypoxia-induced factor 1-α-mediated signaling. Cell Death Dis 5(8):e1367–e1367
Perou CM et al (2000) Molecular portraits of human breast tumors. Nature 406(6797):747–752
Peters GJ (2020) Chapter 1-Drug resistance in colorectal cancer: general aspects. In: Cho CH, Hu T (eds) Drug resistance in colorectal cancer: molecular mechanisms and therapeutic strategies. Academic Press, USA, pp 1–33
Place AE, Jin Huh S, Polyak K (2011) The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res 13(6):227
Pommier Y et al (2016) Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 17(11):703–721
Prakash O et al. (2020) Racial disparities in triple negative breast cancer: a review of the role of biologic and non-biologic factors. Front Public Health 8
Raedler LA (2015) Keytruda (Pembrolizumab): first PD-1 inhibitor approved for previously treated unresectable or metastatic melanoma. Am Health Drug Benefits 8(Spec Feature):96–100
Ray S et al (2013) Topoisomerase IIα promotes activation of RNA polymerase I transcription by facilitating pre-initiation complex formation. Nat Commun 4:1598
Rayner DM, Cutts SM (2014) Chapter 45-anthracyclines. In: Ray SD (ed) Side effects of drugs annual. Elsevier, Netherlands, pp 683–694
Ren X et al (2018) Paclitaxel suppresses proliferation and induces apoptosis through regulation of ROS and the AKT/MAPK signaling pathway in canine mammary gland tumor cells. Mol Med Rep 17(6):8289–8299
Ribas A (2012) Tumor immunotherapy directed at PD-1. N Engl J Med 366(26):2517–2519
Ringel I, Horwitz SB (1991) Studies with RP 56976 (taxotere): a semisynthetic analogue of taxol. J Natl Cancer Inst 83(4):288–291
Robson M et al (2017) Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med 377(6):523–533
Rouleau M et al (2010) PARP inhibition: PARP1 and beyond. Nat Rev Cancer 10(4):293–301
Rowinsky EK, Cazenave LA, Donehower RC (1990) Taxol: a novel investigational antimicrotubule agent. JNCI J Nat Cancer Inst 82(15):1247–1259
Schettini F et al (2016) Nab-paclitaxel for the treatment of triple-negative breast cancer: rationale, clinical data and future perspectives. Cancer Treat Rev 50:129–141
Schiff PB, Fant J, Horwitz SB (1979) Promotion of microtubule assembly in vitro by taxol. Nature 277(5698):665–667
Schmid P et al (2018) Atezolizumab and Nab-Paclitaxel in advanced triple-negative breast cancer. N Engl J Med 379(22):2108–2121
Schmid P et al (2020) Pembrolizumab for early triple-negative breast cancer. N Engl J Med 382(9):810–821
Schneider BP et al (2008) Triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res 14(24):8010–8018
Schreiber V et al (2006) Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7(7):517–528
Schultz N et al (2003) Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res 31(17):4959–4964
Seidman AD et al (1998) Dose-dense therapy with weekly 1-hour paclitaxel infusions in the treatment of metastatic breast cancer. J Clin Oncol 16(10):3353–3361
Sharkey RM et al (2015) Enhanced delivery of SN-38 to human tumor xenografts with an anti-Trop-2–SN-38 antibody conjugate (Sacituzumab Govitecan). Clin Cancer Res 21(22):5131–5138
Shen Y, Aoyagi-Scharber M, Wang B (2015) Trapping Poly(ADP-Ribose) polymerase. J Pharmacol Exp Ther 353(3):446–457
Siegel RL et al (2022) Cancer statistics, 2022. CA A Cancer J Clin 72(1):7–33
Sievers EL et al (2001) Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol 19(13):3244–3254
Son S et al (2018) Anti-Trop2 antibody-conjugated bioreducible nanoparticles for targeted triple negative breast cancer therapy. Int J Biol Macromol 110:406–415
Song Y et al (2017) Inhibition of autophagy results in a reversal of taxol resistance in nasopharyngeal carcinoma by enhancing taxol-induced caspase-dependent apoptosis. Am J Transl Res 9(4):1934–1942
Sørlie T et al (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98(19):10869–10874
Stagg J, Allard B (2013) Immunotherapeutic approaches in triple-negative breast cancer: latest research and clinical prospects. Ther Adv Med Oncol 5(3):169–181
Stein R et al (1990) Murine monoclonal antibodies raised against human non-small cell carcinoma of the lung: specificity and tumor targeting. Cancer Res 50(4):1330–1336
Stepan LP et al (2011) Expression of Trop2 cell surface glycoprotein in normal and tumor tissues: potential implications as a cancer therapeutic target. J Histochem Cytochem 59(7):701–710
Strobel T et al (1996) BAX enhances paclitaxel-induced apoptosis through a p53-independent pathway. Proc Natl Acad Sci 93(24):14094–14099
Suh, D.H., et al., Mitochondrial permeability transition pore as a selective target for anti-cancer therapy. Frontiers in Oncology, 2013. 3(41).
Sun C, Mezzadra R, Schumacher TN (2018) Regulation and function of the PD-L1 checkpoint. Immunity 48(3):434–452
Sung H et al (2021) Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J Clin 71(3):209–249
Takeba Y et al (2007) Irinotecan activates p53 with its active metabolite, resulting in human hepatocellular carcinoma apoptosis. J Pharmacol Sci 104(3):232–242
Trudeau ME (1995) Docetaxel (Taxotere): an overview of first-line monotherapy. Semin Oncol 22(6 Suppl 13):17–21
U.S. Food & Drug Administration. FDA approves olaparib for germline BRCA-mutated metastatic breast cancer. 2018a May 5, 2020]; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-germline-brca-mutated-metastatic-breast-cancer.
U.S. Food & Drug Administration. FDA approves talazoparib for gBRCAm HER2-negative locally advanced or metastatic breast cancer. 2018b May 5, 2020]; Available from: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-talazoparib-gbrcam-her2-negative-locally-advanced-or-metastatic-breast-cancer
U.S. Food & Drug Administration. FDA approves atezolizumab for PD-L1 positive unresectable locally advanced or metastatic triple-negative breast cancer. 2020a 8 March, 2020a [cited 2020a 30 May,]; Available from: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-atezolizumab-pd-l1-positive-unresectable-locally-advanced-or-metastatic-triple-negative
U.S. National Library of Medicine. A Study Comparing Atezolizumab (Anti PD-L1 Antibody) In Combination With Adjuvant Anthracycline/Taxane-Based Chemotherapy Versus Chemotherapy Alone In Patients With Operable Triple-Negative Breast Cancer (IMpassion030). 2018c May 13, 2020 [cited 2020 24 May]; Available from: https://clinicaltrials.gov/ct2/show/NCT03498716?term=epirubicin&recrs=abdf&cond=Triple+Negative+Breast+Cancer&cntry=US&draw=2&rank=1.
U.S. National Library of Medicine. Randomized Phase 2 Study of Atezolizumab and Entinostat in Patients With aTN Breast Cancer With Phase 1b Lead In. 2020b [cited 2020b 29 May,]; Available from: https://clinicaltrials.gov/ct2/show/NCT02708680.
U.S. National Library of Medicine. Triple-B Study;Carboplatin-cyclophosphamide Versus Paclitaxel With or Without Atezolizumab as First-line Treatment in Advanced Triple Negative Breast Cancer (Triple-B). 2020c [cited 2020c 29 May]; Available from: https://clinicaltrials.gov/ct2/show/NCT01898117.
Untch M et al (2016) Nab-paclitaxel versus solvent-based paclitaxel in neoadjuvant chemotherapy for early breast cancer (GeparSepto—GBG 69): a randomised, phase 3 trial. Lancet Oncol 17(3):345–356
van Oosterom AT (1995) Docetaxel (Taxotere): an effective agent in the management of second-line breast cancer. Semin Oncol 22(6 Suppl 13):22–28
Venkitaraman AR (2014) Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 343(6178):1470–1475
Vici P et al (2015) Triple positive breast cancer: a distinct subtype? Cancer Treat Rev 41(2):69–76
von Minckwitz G et al (2012) Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J Clin Oncol 30(15):1796–1804
Wani MC et al (1971) Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc 93(9):2325–2327
Weaver BA (2014) How Taxol/paclitaxel kills cancer cells. Mol Biol Cell 25(18):2677–2681
Wilson WH et al (1994) Paclitaxel in doxorubicin-refractory or mitoxantrone-refractory breast cancer: a phase I/II trial of 96-hour infusion. J Clin Oncol 12(8):1621–1629
Yang M-C et al (2016) Bim directly antagonizes Bcl-xl in doxorubicin-induced prostate cancer cell apoptosis independently of p53. Cell Cycle 15(3):394–402
Yao Y et al. (2020) Nanoparticle-based drug delivery in cancer therapy and its role in overcoming drug resistance. Front Mol Biosci 7(193)
Yeh J et al (2017) Clinical characteristics in patients with triple negative breast cancer. Int J Breast Cancer 2017:1796145–1796145
Yin L et al (2020) Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res 22(1):61
Yu Y-F et al (2017) Paclitaxel induces autophagy in gastric cancer BGC823 cells. Ultrastruct Pathol 41(4):284–290
Zamora A et al (2019) Paclitaxel induces lymphatic endothelial cells autophagy to promote metastasis. Cell Death Dis 10(12):956
Zhang Y, Zhou H, Zhang L (2018) Which i s the optimal immunotherapy for advanced squamous non-small-cell lung cancer in combination with chemotherapy: anti-PD-1 or anti-PD-L1? J Immunother Cancer 6(1):135
Zimmer AS et al (2018) Update on PARP inhibitors in breast cancer. Curr Treat Options Oncol 19(5):21
Funding
No funding was received to assist with the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
The main manuscript was written by A.M. and K.L. All figures were prepared by A.M. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors Aditya Mandapati and Kiven Erique report no other conflicts of interest that are relevant to the contents of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mandapati, A., Lukong, K.E. Triple negative breast cancer: approved treatment options and their mechanisms of action. J Cancer Res Clin Oncol 149, 3701–3719 (2023). https://doi.org/10.1007/s00432-022-04189-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-022-04189-6