
Abstract
Purpose
CD19 is a cell surface protein that is found on both healthy and malignant B cells. Accordingly, it has become an important target for novel treatments for non-Hodgkin lymphomas and B-cell leukaemia. Three anti-CD19 monoclonal antibodies with distinct mechanisms of action have been developed for the treatment of B-cell malignancies.
Methods
We reviewed the preclinical and clinical data on the development of the newly approved anti-CD19 monoclonal antibodies blinatumomab, tafasitamab and loncastuximab tesirine, and consider their place in the treatment of relapsed or refractory B-cell malignancies.
Results
Blinatumomab is a bispecific T-cell engager that binds to both CD19 on B cells and CD3 on T cells, facilitating antibody-dependent cytotoxicity. Blinatumomab significantly prolongs overall survival in patients with relapsed or refractory B-cell acute lymphoblastic leukaemia, although cytokine release syndrome and severe neurotoxicity may necessitate discontinuation. Tafasitamab, which has modified anti-CD19 Fab and Fc regions, has significantly enhanced affinity for both CD19 and effector cell receptors compared with unmodified anti-CD19. In L-MIND, tafasitamab plus lenalidomide provided an overall response rate (ORR) of 57.5% in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) in patients non-transplant eligible. Loncastuximab tesirine is an antibody–drug conjugate that has been studied as monotherapy and in combination with ibrutinib in 3L + relapsed or refractory DLBCL. The ORR was 48.3% in a phase II trial of loncastuximab tesirine. The optimal place of anti-CD19 monoclonal antibodies in therapy has yet to be determined, but the prospect of improved outcomes for at least some patients with treatment-resistant B-cell malignancies appears likely, particularly in those with limited therapeutic options and poor prognosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The lymphomas are a heterogeneous group of malignant diseases caused by the clonal proliferation of lymphocytes. Histologically, they are classified as Hodgkin or non-Hodgkin lymphomas (NHL), with the latter accounting for 90% of cases (Jamil et al. 2021). NHL is largely a disease of later life, with the median age at diagnosis being 67 years (Sapkota et al. 2020). Diffuse large B-cell lymphoma (DLBCL) is the most common type of NHL, accounting for 25–30% of cases (Jamil et al. 2021). An aggressive lymphoma with a high mortality rate (Camicia et al. 2015), DLBCL is characterized by large lymphoid cells that carry B-cell surface antigens such as CD19 and CD20 (Jamil et al. 2021).
The monoclonal antibody rituximab is directed against CD20. With US Food and Drug Administration (FDA) approval in 1997, it became the first biological to be licensed for the treatment of a B-cell malignancy (Horst et al. 2020). Since then, the addition of rituximab to the CHOP regimen (cyclophosphamide, doxorubicin, vincristine and prednisone) has significantly improved response rates and survival in patients with newly diagnosed DLBCL (Tilly et al. 2015; Coiffier et al. 2002). Approximately 60% of patients achieve remission with R-CHOP; a further 30–40% experience a disease relapse, while 10% have refractory disease (Gisselbrecht and Neste 2018). In patients with relapsed or refractory disease, the standard second-line treatment is salvage chemotherapy followed by autologous stem cell transplantation (ASCT); however, 50% of patients who receive salvage chemotherapy will have an inadequate response and thus be ineligible for ASCT, while a further 25% will relapse following ASCT (Gisselbrecht and Neste 2018). Thus, only around 25% of these patients will derive long-term benefit. Prognosis in this patient population is poor (Crump et al. 2017), and the relative lack of effective subsequent options continues to be one of the main unmet needs in the field of haemato-oncology (Harris et al. 2020).
The success of rituximab has helped establish immunotherapy, and monoclonal antibodies in particular, as an integral part of anticancer therapy (Horst et al. 2020). Monoclonal antibodies exert their anticancer effects by activating cytotoxic pathways, including antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC) (Horst et al. 2020; Abramson et al. 2020). Direct effects are also possible: for example, receptor blockade may cause cell death by diminishing downstream growth signals. However, as mentioned above, a substantial proportion of patients with DLBCL experience early relapse following rituximab-based therapy, or are refractory to it. This has prompted the development of monoclonal antibodies targeting B-cell antigens other than CD20. In addition, considerable effort has been devoted to the development of optimised monoclonal antibodies with enhanced cytotoxic effector functions (Katz and Herishanu 2014).
In addition, CD19 has attracted considerable interest as a potential target to treat B-cell malignancies (Katz and Herishanu 2014; Zalevsky et al. 2009). Several treatments that target cancer cells via CD19 have either entered the market or are undergoing regulatory evaluation, including modified monoclonal antibodies against CD19 (e.g. tafasitamab) and chimeric antigen receptor (CAR) T-cell therapies (e.g. tisagenlecleucel). Compared with CAR T-cell therapies, which at present are resource-intensive in terms of implementation, manufacturing time, and costs (Harris et al. 2020; Abramson et al. 2020; Patriarca and Gaidano 2021), anti-CD19 monoclonal antibodies will likely be accessible to a greater proportion of patients with B-cell malignancies.
The aim of this review is to present and summarize our current knowledge on the newly approved anti-CD19 monoclonal antibodies blinatumomab, tafasitamab and loncastuximab tesirine, and to consider their place in the treatment of relapsed or refractory B-cell malignancies.
Rationale for targeting CD19 in B-cell malignancies
CD19 is a transmembrane receptor that is specific for the B-cell lineage (Katz and Herishanu 2014). Unlike CD20, CD19 is expressed throughout the entire B-cell maturation process (Katz and Herishanu 2014), and studies in mice lacking CD19 have demonstrated that it has an important role in B-cell maturation and activation (Zalevsky et al. 2009; Poe et al. 2012). CD19 has thus become an interesting target for the immunotherapy of NHLs and B-cell leukaemias (Anderson et al. 1984; Ginaldi et al. 1998; Olejniczak et al. 2006; Narkhede and Tafasitamab 2020). Although levels of expression are lower for CD19 than CD20, CD19 is expressed by a broader spectrum of lymphoid malignancies due to its presence from the early stages of B-cell development (Zalevsky et al. 2009; Anderson et al. 1984; Horton et al. 2008).
Development and optimisation of anti-CD19 antibodies
Over the past three decades, several anti-CD19 monoclonal antibodies have been evaluated for the treatment of B-cell malignancies (Katz and Herishanu 2014; Zalevsky et al. 2009; Narkhede and Tafasitamab 2020; Roßkopf et al. 2020). Initial results with unmodified or drug-conjugated anti-CD19 antibodies were modest, and research efforts subsequently focused on antibody optimisation and strategies to potentiate immune effector cell activity (Horst et al. 2020; Katz and Herishanu 2014; Narkhede and Tafasitamab 2020). Currently available drugs contain humanized anti-CD19 that has undergone modification to potentiate its anticancer activity (Katz and Herishanu 2014; Narkhede and Tafasitamab 2020). Three different modification strategies have been implemented, leading to three different types of anti-CD19 antibodies: bispecific T-cell engagers (BiTEs; e.g. blinatumomab), fragment crystallizable (Fc)–engineered and Fab affinity-matured antibodies (e.g. tafasitamab [MOR208]), and new-generation antibody–drug conjugates (e.g. loncastuximab tesirine [ADCT-402]) (Katz and Herishanu 2014; Narkhede and Tafasitamab 2020).
Bispecific T-cell engagers
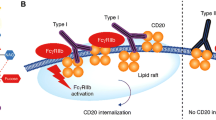
BiTEs are able to bind to two antigens simultaneously: typically, one antigen on a malignant cell, and the other on a T cell. This brings malignant cells and effector cells into close proximity to each other, facilitating ADCC (Fig. 1) (Abramson et al. 2020; Katz and Herishanu 2014). Blinatumomab was the first BiTE antibody to be approved; it binds to CD19 on malignant B cells, and to CD3 on T cells (Abramson et al. 2020). The two binding sites are connected by a linker (Abramson et al. 2020).

Anti-cancer mechanism of action of antibodies directed against CD19 on the surface of malignant B cells (Hartley 2020; Duell et al. 2019; Cheson et al. 2021). Blinatumomab (top left) binds simultaneously to CD19 and to CD3 receptors on T-cells, which brings the effector and malignant cells into close proximity to each other, and facilitates antibody-dependent cellular cytotoxicity (ADCC). Tafasitamab (top right) binds with high affinity to both CD19 and Fc gamma receptors (FcγR) on effector cells. Binding to FcγRIII on natural killer cells facilitates ADCC, while binding to FcγR on macrophages facilitates antibody-dependent cellular phagocytosis (ADCP). Tafasitamab also has direct cytotoxic effects. Loncastuximab tesirine (bottom right) is an anti-CD19 antibody–drug conjugate that contains a cytotoxic pyrrolobenzodiazepine dimer (PBD). Antibody binding to cell surface CD19 leads to internalization of the complex and intracellular release of the PBD payload. The PBD then binds to the minor groove of DNA, and forms inter-strand cross-links that are not recognized by DNA repair mechanisms, thereby leading to cell death
Fc-engineered and Fab affinity-matured antibodies
The cytotoxic effects of monoclonal antibodies are mediated by their Fc regions, which are therefore critical for therapeutic efficacy (Horst et al. 2020). Engineering of the Fc domain, via modification of the Fc glycosylation profile (glyco-engineering) or site-directed mutagenesis of the Fc domain (protein engineering), is a novel approach to antibody modification (Horst et al. 2020; Narkhede and Tafasitamab 2020; Roßkopf et al. 2020; Kellner et al. 2017). ADCC and ADCP responses are induced when the Fc domain binds to Fc gamma receptors (FcγR) that are expressed on a variety of effector cells, including natural killer (NK) cells and macrophages (Horst et al. 2020). NK cells, which express only the FcγRIIIa member of the FcγR family,Footnote 1 are the most potent inducers of ADCC (Horst et al. 2020), while macrophages mediate ADCP.
Tafasitamab, the first Fc-engineered monoclonal antibody to be approved for the treatment of DLBCL, has two amino acid mutations in the Fc domain (S239D and I332E) that, remarkably, result in a ≥ 40-fold increase in its affinity for FcγR, including FcγRIIIa (Horst et al. 2020; Narkhede and Tafasitamab 2020; Horton et al. 2008). At the same time, humanization and affinity maturation of the variable region (Fab) nearly doubles the affinity of tafasitamab for CD19 compared with its immunoglobulin G precursors (Horton et al. 2008). Augmented affinity for both effector and target cells bestows functional advantages upon tafasitamab that are reminiscent of a bispecific antibody; as a result of these modifications, tafasitamab is associated with a 10- to 1000-fold increase in ADCC compared with an unmodified analogue.
Tafasitamab was optimized using a proprietary technology that uses a novel method of variable fragment (Fv) humanization to maximize the human sequence content and enhance affinity for antigen and for effector cell FcγRs. As a result of this process, tafasitamab can be considered an enhanced next-generation anti-CD19 monoclonal antibody. Its mechanism of action is summarized in Fig. 1.
Antibody–drug conjugates
Antibody–drug conjugates combine selectivity and cytotoxic potency because they facilitate the targeted delivery of cytotoxic agents to cancer cells via antibodies directed against tumour-associated cell surface antigens, such as CD19 (Kahl et al. 2019; Diamantis and Banerji 2016). They, therefore, have the potential to optimize efficacy while reducing systemic toxicity (Jain et al. 2020). Loncastuximab tesirine, an anti-CD19 antibody–drug conjugate, was approved by the FDA in April 2021 (US Food and Drug Administration. 2021a), and is undergoing evaluation by the European Medicines Agency (EMA). In loncastuximab tesirine, a humanized anti-CD19 antibody is stochastically conjugated to a cytotoxic pyrrolobenzodiazepine (PBD) dimer (Kahl et al. 2019). Following antibody-CD19 complex cellular internalization and sequestration in lysosomes, PBD is released and acts as a DNA minor-groove cross-linking agent that is able to escape DNA repair mechanisms; as a result, it exhibits enhanced biological activity and intracellular persistence compared with conventional DNA cross-linking agents (Fig. 1) (Kahl et al. 2019; Hartley 2020).
Preclinical and clinical studies of anti-CD19 antibodies
Blinatumomab
Blinatumomab was initially investigated as a treatment for relapsed or refractory B-cell acute lymphoblastic leukaemia (ALL), see Table 1 (Topp et al. 2014, 2015; Kantarjian et al. 2017), and has been approved for this use in both adults and children by the FDA and EMA (US Food and Drug Administration 2021b; European Medical Agency 2021). However, the association of blinatumomab with cytokine release syndrome and neurotoxicity has led to the inclusion of a ‘black box’ warning in the US prescribing information (US Food and Drug Administration 2021b). Blinatumomab has also been formally studied as a treatment for relapsed or refractory NHL (Viardot et al. 2016, 2020; Goebeler et al. 2016).
Non-Hodgkin lymphoma
In a phase I dose-escalation study in 76 heavily pre-treated patients with relapsed or refractory NHL (NCT00274742), the maximum tolerated dose of blinatumomab (administered via continuous intravenous infusion [IVI]) was determined to be 60 μg/m2/day (Goebeler et al. 2016). In patients who received this dosage, the overall response rate (ORR) was 69% across NHL subtypes and 55% in patients with DLBCL. However, neurological events were dose-limiting.
In a later dose-finding study in 25 heavily pre-treated patients with DLBCL (NCT01741792), the ORR after one cycle of blinatumomab was 43% (9 of 21 evaluable patients), which included CR in 4 patients. Grade 3 neurological AEs reported in more than one patient were encephalopathy and aphasia (Viardot et al. 2016).
A pooled analysis has been conducted of data from 17 patients from these two trials and from the phase II portion of a third trial (Coyle et al. 2020), all of whom had relapsed or refractory DLBCL and a CR to treatment with blinatumomab (Viardot et al. 2020). Neither the median duration of CR nor median overall survival (OS) were reached after median follow-up times of 15.6 months and 16.4 months, respectively, and the probability of OS at 2 years was estimated to be 77.9%. Fourteen patients had grade ≥ 3 treatment-emergent AEs, and eight patients had serious AEs.
Tafasitamab
Preclinical studies
In vitro/ex vivo and in vivo studies in mice and primates demonstrated the potential of tafasitamab as a therapeutic agent. In an in vitro study of patient-derived B-cell lymphoma and leukaemia lines, tafasitamab substantially increased ADCC relative to an unmodified anti-CD19 antibody, and was also associated with increased ADCP and apoptosis (Horton et al. 2008). In a mouse lymphoma xenograft model, tafasitamab was found to significantly inhibit lymphoma growth, and demonstrated greater antitumour activity than unmodified anti-CD19 (Horton et al. 2008). Tafasitamab also produced rapid, dose-dependent B-cell depletion in blood and lymphoid tissues in cynomolgus monkeys, which have similar immune systems to humans (Zalevsky et al. 2009); a concomitant reduction in NK cells suggested their involvement in the observed B-cell clearance. In contrast, neither B-cell depletion nor reductions in NK cell count were observed with unmodified anti-CD19 antibodies.
In an ex vivo study using cell lines from patients with B-cell chronic lymphocytic leukaemia (CLL), tafasitamab was found to be a significantly more potent inducer of ADCC than either unmodified anti-CD19 antibodies or rituximab (Awan et al. 2010). Additionally, tafasitamab was a moderate inducer of ADCP and had direct cytotoxic effects, but did not activate CDC. This study confirmed that tafasitamab-induced ADCC is mediated by NK cells. Importantly, tafasitamab-induced ADCC was found to be potentiated by the addition of lenalidomide, which stimulates the proliferation and activation of NK cells (Gribben et al. 2015), prompting subsequent clinical investigation into the efficacy of this combination.
The effects of tafasitamab have also been studied in vitro and ex vivo in B-cell ALL (Rafiq et al. 2012; Kellner et al. 2013). In vitro, tafasitamab was found to have potent ADCC-inducing effects and modest direct cytotoxicity, but was superior in these respects to either rituximab or alemtuzumab (anti-CD52) (Rafiq et al. 2012). An ex vivo study, using cell lines from paediatric and adult patients with B-lineage ALL, has also been undertaken to elucidate the Fc-mediated effects of tafasitamab (Kellner et al. 2013). The study confirmed that tafasitamab induces NK cell-mediated ADCC; rituximab and unmodified anti-CD19 antibodies (which were used as controls) were, respectively, ineffective and less potent than tafasitamab.
Clinical studies
The first clinical trial of tafasitamab was a phase I dose-escalation study in patients with relapsed or refractory CLL (n = 27; Table 2) (Woyach et al. 2014). No maximum tolerated dose was reached, and treatment was generally well tolerated. Preliminary evidence of efficacy was obtained, with 18 patients (66.7%) achieving a response defined using physical examination criteria and laboratory data, and 8 patients (29.6%) achieving a response defined using radiological (i.e. computed tomography) criteria.
A phase IIa trial in patients with relapsed or refractory NHL was initiated following the successful long-term use of salvage/maintenance treatment with tafasitamab monotherapy in a patient with DLBCL who had early relapse following standard first- and second-line treatment (Jurczak et al. 2016, 2018, 2019). In this open-label, multicentre, single-arm study, 92 patients received up to three 28-day cycles of tafasitamab as monotherapy (12 mg/kg per week by IVI) (Jurczak et al. 2019). The primary efficacy endpoint was investigator-assessed ORR, which was achieved in 23.9% (95% CI 15.6–33.9). The median duration of response was 24.0 months (95% CI 11.1–not reached [NR]) overall, and 20.1 months (95% CI 1.1–NR) in the 35 patients with DLBCL; treatment was well tolerated, with most treatment-emergent AEs being of mild intensity. Interestingly, and consistent with the role of tafasitamab–NK cells interactions, median progression-free survival (PFS) was at least twice as long in patients with NK cell counts > 100 cells/μL at baseline compared with those with counts < 100 cells/μL: 4.2 versus 2.1 months, respectively, in patients with DLBCL, and 8.8 versus 3.2 months in patients with follicular lymphoma.
The first results of the phase II L-MIND trial, an open-label, single-arm study of tafasitamab in combination with lenalidomide in adults (n = 81) with relapsed or refractory DLBCL who were ineligible for high-dose chemotherapy and subsequent ASCT, were reported in 2020 (Salles et al. 2020). Tafasitamab (12 mg/kg) was administered intravenously over 2 h every week (cycles 1–3) or every 2 weeks (cycle 4 onwards), and oral lenalidomide (starting dose 25 mg) was given daily on days 1–21 of each 28-day cycle. Treatment was continued for up to 12 cycles, and was followed by tafasitamab monotherapy until disease progression occurred. Recently, the results of the 3-year follow-up of the L-MIND clinical trial have been published (Duell et al. 2021). After ≥ 35 months’ follow-up, ORR was 57.5%, including a complete response in 40.0% of patients and a partial response in 17.5% of patients. Median duration of response (DoR) was 43.9 months, median OS was 33.5 months, and median PFS was 11.6 months (Duell et al. 2021). The combination of tafasitamab and lenalidomide was well tolerated, with most non-haematological AEs being of grade 1 or 2 intensity.
The proportion of patients with CR was notable because relapsed or refractory DLBCL is difficult to treat and has a poor prognosis. Thus, the L-MIND trial provides clinical evidence that, as suggested by earlier ex vivo experiments, tafasitamab/lenalidomide is a synergistic combination in which the affinity of tafasitamab for both effector and targets cells is magnified by the immunomodulating effects of lenalidomide (such as stimulation of NK cell proliferation, as well as activation and enhancement of NK-mediated ADCC) (Awan et al. 2010; Gribben et al. 2015). In May 2020, the EMA CHMP validated the application for marketing authorization. In July 2020, the US FDA granted accelerated approval. In June 2021, the CHMP recommended the use of tafasitamab and the application is now being reviewed by the European Commission which has the authority to grant marketing authorization in the EU.
To assess the contribution of tafasitamab to the clinical effects of tafasitamab/lenalidomide combination therapy, outcomes in 76 real-world patients treated with lenalidomide monotherapy (the ‘RE-MIND’ cohort) were retrospectively compared with those in 76 propensity score-matched participants from the L-MIND trial (the ‘L-MIND’ cohort) (Nowakowski et al. 2020; Zinzani et al. 2021). Patients in the L-MIND cohort were significantly more likely to respond than those in the RE-MIND cohort (ORR 67.1% vs 34.2%; odds ratio 3.89; 95% CI 1.90–8.14; P < 0.0001 investigator assessment) and had significantly longer OS (median not reached vs 9.3 months; 95% CI 5.0–15.3; P = 0.0008), indicating that tafasitamab and lenalidomide may have synergistic effects in vivo. A second study, RE-MIND2 (NCT04697160), is under way, and will compare outcomes from the L-MIND study with those from matched real-world patients treated with guideline-recommended salvage regimens such as R-GEMOX (rituximab, gemcitabine and oxaliplatin) and BR (bendamustine and rituximab).
A number of clinical trials of tafasitamab are also planned or are currently ongoing (Table 3). The InMIND trial (NCT04680052) is a phase III study of tafasitamab versus placebo, both in combination with lenalidomide and rituximab, in patients with relapsed or refractory follicular lymphoma or marginal zone lymphoma. The phase I First-MIND trial assesses the safety and preliminary efficacy of tafasitamab (with or without lenalidomide) in combination with R-CHOP in newly diagnosed DLCBL (NCT04134936). A large phase III trial in newly diagnosed DLBCL, frontMIND, is recruiting, and will investigate the efficacy and safety of tafasitamab/lenalidomide, added to R-CHOP vs R-CHOP, in high-risk and high-intermediate risk patients (NCT04824092). Lastly, the phase Ib/IIa topMIND trial will evaluate the safety, pharmacokinetics and efficacy of tafasitamab in combination with the phosphatidylinositol 3-kinase δ (PI3Kδ) inhibitor parsaclisib in adults with NHL or CLL (NCT04809467).
Loncastuximab tesirine
In human cell lines expressing CD19, loncastuximab tesirine demonstrated potent, selective cytotoxicity (Zammarchi et al. 2018). Additionally, in models of subcutaneous and disseminated human tumours, loncastuximab tesirine exhibited potent, dose-dependent antitumour activity that was superior to that of antibody–drug conjugates delivering tubulin inhibitors (Zammarchi et al. 2018). Further preclinical studies showed that loncastuximab tesirine was pharmacokinetically stable and had good tolerability in mice and cynomolgus monkeys.
These findings led to the design of a phase I dose-escalation and dose-expansion study of loncastuximab tesirine in adults with relapsed or refractory NHL (NCT02669017; Table 4). In the dose-escalation phase (n = 88), doses ranging from 15 to 200 μg/kg were administered intravenously once every 3 weeks until disease progression or patient withdrawal from the study (Kahl et al. 2019). At doses of 120 μg/kg and above, the ORR was 59.4% (40.6% CR, 18.8% PR), for all doses combined, median OS was 11.6 months, median PFS was 4.8 months and median duration of response 5.5 months. Most patients experienced AEs; grade ≥ 3 fatigue, dyspnoea and abnormalities of haematological or liver function were reported in ≥ 5% of patients (Kahl et al. 2019). In the final analysis of 183 patients, the ORR was 45.6%, with 26.7% achieving CR (Hamadani et al. 2021). Although the maximum tolerated dose was not reached, cumulative toxicity was higher at 200 μg/kg (Hamadani et al. 2021).
Based on these results, a phase II, single-arm trial of loncastuximab tesirine monotherapy (LOTIS-2; NCT03589469), using a starting dose of 150 μg/kg, was initiated in heavily pre-treated patients with relapsed or refractory DLBCL (Table 4) (Caimi et al. 2021). In this trial, the ORR was 48.3%, with equal numbers of patients achieving CR and PR. Median OS and PFS were 9.9 and 4.9 months, respectively (Caimi et al. 2021). In April 2021, the FDA-approved loncastuximab tesirine for the treatment of relapsed or refractory large B-cell lymphomas after 2 or more lines of treatment, including DLBCL (US Food and Drug Administration 2021a).
Ongoing clinical trials include LOTIS-3 (NCT03684694), a two-part dose-escalation and dose-expansion trial of loncastuximab tesirine (in combination with the Bruton’s tyrosine kinase inhibitor ibrutinib, at a dosage of 560 mg/day) in patients with relapsed or refractory DLBCL or mantle cell lymphoma (Table 3). In an interim analysis of the first part of the trial (n = 25), the maximum tolerated dose of loncastuximab tesirine for use in combination with ibrutinib was determined to be 60 μg/kg; at this dosage, seven of the 12 (58.3%) patients who were evaluable for response had CR, and a further two patients had PR, giving an ORR of 75.0% (Depaus et al. 2020). The second part of the LOTIS-3 trial is ongoing.
Loncastuximab tesirine has also undergone preliminary assessment as a treatment for relapsed or refractory B-cell ALL (Table 4) (Jain et al. 2020). A phase I, open-label, single-arm, multicentre study was conducted in 35 patients to evaluate the safety, tolerability and immunogenicity of loncastuximab tesirine 15–150 μg/kg, and to determine its maximum tolerated dose and pharmacokinetic profile (NCT02669264). The maximum tolerated dose was not reached, and anti-drug antibodies were not detected at clinically relevant levels.
Place in therapy
Immunotherapy with monoclonal antibodies (e.g. with the anti-CD20 agent rituximab) has revolutionized treatment in patients with B-cell malignancies. However, treatment options for patients who do not respond to, or have early relapse following, first-line rituximab-based treatment have long been very limited. The recent development of three novel monoclonal antibodies that target the B-cell surface antigen CD19, each of which belongs to a distinct molecular class, will expand the available options for patients with relapsed or refractory disease.
Blinatumomab has been most extensively studied in relapsed or refractory B-cell ALL, and is licensed for this indication in Europe and the US in both adults and children. However, cytokine release syndrome and neurotoxicity, as well as the need for continuous IVI over 4 weeks of each 6-week cycle may limit its clinical usefulness. Less is known about its efficacy and safety in relapsed or refractory NHL. Tafasitamab, on the other hand, has been extensively studied in patients with relapsed or refractory DLBCL, and in the US is approved for use in this indication in combination with lenalidomide in adults who are ineligible for ASCT. Tafasitamab is being studied as part of a first-line treatment regimen for DLBCL, and in the relapsed/refractory setting in other forms of B-cell NHL. Like tafasitamab, loncastuximab tesirine has been studied in heavily pre-treated patients with DLBCL, but there is less experience with its use. Superficially, outcomes appear comparable between the three agents, although long-term follow-up is required to determine the duration of response with some certainty.
Anti-CD19 antibodies have major advantages over CAR T-cell therapy in that they are more readily available and can be used in patients with rapidly evolving disease. Moreover, they are relatively easy to use, as there is now a high level of expertise with monoclonal antibodies in many cancer centres. Monoclonal antibodies are also considerably less expensive and resource-intensive than CAR T-cell therapy, and, additionally, may be safer in older, frailer patients who cannot tolerate high-dose chemotherapy. Anti-CD19 antibodies may also be a useful option in the event of non-response or relapse following CAR T-cell therapy; to date, however, no formal clinical trials have been performed to support their use in this setting.
The availability of several new therapeutic options for relapsed or refractory B-cell malignancies raises the question as to the best sequence in which to use them for maximal clinical benefit. Further controlled studies of sufficiently long duration are needed to determine whether the order in which treatments are used influences OS or other key outcomes.
Conclusions
Blinatumomab, tafasitamab and loncastuximab tesirine are novel monoclonal antibody-based treatments that target CD19, a cell surface protein that is present throughout the B-cell maturation process, and consequently is an important therapeutic target in patients with B-cell malignancies. In particular, the combination of tafasitamab and lenalidomide is associated with synergistic interactions between the two agents and appears to have good activity against B-cell NHLs. This combination has been extensively studied in patients with relapsed or refractory DLBCL, and should be considered, together with loncastuximab tesirine, as a valid alternative to CAR T-cell therapy in patients who are ineligible for ASCT.
Data availability
Not applicable.
Code availability
Not applicable.
Notes
FcγRIIIa is also known as CD16a.
References
Abramson JS, Ghosh N, Smith SM (2020) ADCs, BiTEs, CARs, and small molecules: a new era of targeted therapy in non-Hodgkin lymphoma. Am Soc Clin Oncol Educ Book 40:302–313. https://doi.org/10.1200/edbk_279043
Anderson KC, Bates MP, Slaughenhoupt BL, Pinkus GS, Schlossman SF, Nadler LM (1984) Expression of human B cell-associated antigens on leukemias and lymphomas: a model of human B cell differentiation. Blood 63(6):1424–1433
Awan FT, Lapalombella R, Trotta R, Butchar JP, Yu B, Benson DM Jr et al (2010) CD19 targeting of chronic lymphocytic leukemia with a novel Fc-domain-engineered monoclonal antibody. Blood 115(6):1204–1213. https://doi.org/10.1182/blood-2009-06-229039
Brown PA, Ji L, Xu X, Devidas M, Hogan LE, Borowitz MJ et al (2021) Effect of postreinduction therapy consolidation with blinatumomab vs chemotherapy on disease-free survival in children, adolescents, and young adults with first relapse of B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA 325(9):833–842. https://doi.org/10.1001/jama.2021.0669
Caimi PF, Ai W, Alderuccio JP, Ardeshna KM, Hamadani M, Hess B et al (2021) Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. https://doi.org/10.1016/s1470-2045(21)00139-x
Camicia R, Winkler HC, Hassa PO (2015) Novel drug targets for personalized precision medicine in relapsed/refractory diffuse large B-cell lymphoma: a comprehensive review. Mol Cancer 14:207. https://doi.org/10.1186/s12943-015-0474-2
Cheson BD, Nowakowski G, Salles G (2021) Diffuse large B-cell lymphoma: new targets and novel therapies. Blood Cancer J 11(4):68. https://doi.org/10.1038/s41408-021-00456-w
Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R et al (2002) CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 346(4):235–242. https://doi.org/10.1056/NEJMoa011795
Coyle L, Morley NJ, Rambaldi A, Mason KD, Verhoef G, Furness CL et al (2020) Open-Label, phase 2 study of blinatumomab as second salvage therapy in adults with relapsed/refractory aggressive B-cell non-Hodgkin lymphoma. Leuk Lymphoma 61(9):2103–2112. https://doi.org/10.1080/10428194.2020.1759055
Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J et al (2017) Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood 130(16):1800–1808. https://doi.org/10.1182/blood-2017-03-769620
Depaus J, Ervin-Haynes A, Bryan L, Magagnoli M, Gritti G, Chao G et al. Interim results of a phase 1/2 study of loncastuximab tesirine (Lonca) combined with ibrutinib in advanced diffuse large b-cell lymphoma (DLBCL) or mantle cell lymphoma (MCL) [abstract EP1284].In: 25th Congress of the European Hematology Association; 12 June 2020; Virtual Meeting 2020
Diamantis N, Banerji U (2016) Antibody-drug conjugates–an emerging class of cancer treatment. Br J Cancer 114(4):362–367. https://doi.org/10.1038/bjc.2015.435
Duell J, Lammers PE, Djuretic I, Chunyk AG, Alekar S, Jacobs I et al (2019) Bispecific antibodies in the treatment of hematologic malignancies. Clin Pharmacol Ther 106(4):781–791. https://doi.org/10.1002/cpt.1396
Duell J, Maddocks KJ, Gonzalez-Barca E, Jurczak W, Liberati AM, De Vos S et al (2021) Long-term outcomes from the Phase II L-MIND study of tafasitamab (MOR208) plus lenalidomide in patients with relapsed or refractory diffuse large B-cell lymphoma. Haematologica. https://doi.org/10.3324/haematol.2020.275958
European Medicines Agency. Blincyto (blinatumomab) [summary of product characteristics] 2021. https://www.ema.europa.eu/en/documents/product-information/blincyto-epar-product-information_en.pdf. Accessed 1 May 2021.
Ginaldi L, De Martinis M, Matutes E, Farahat N, Morilla R, Catovsky D (1998) Levels of expression of CD19 and CD20 in chronic B cell leukaemias. J Clin Pathol 51(5):364–369. https://doi.org/10.1136/jcp.51.5.364
Gisselbrecht C, Van Den Neste E (2018) How I manage patients with relapsed/refractory diffuse large B cell lymphoma. Br J Haematol 182(5):633–643. https://doi.org/10.1111/bjh.15412
Goebeler ME, Knop S, Viardot A, Kufer P, Topp MS, Einsele H et al (2016) Bispecific T-cell engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-Hodgkin lymphoma: final results from a phase I study. J Clin Oncol 34(10):1104–1111. https://doi.org/10.1200/jco.2014.59.1586
Gribben JG, Fowler N, Morschhauser F (2015) Mechanisms of action of lenalidomide in B-cell non-Hodgkin lymphoma. J Clin Oncol 33(25):2803–2811. https://doi.org/10.1200/jco.2014.59.5363
Hamadani M, Radford J, Carlo-Stella C, Caimi PF, Reid E, O’Connor OA et al (2021) Final results of a phase 1 study of loncastuximab tesirine in relapsed/refractory B-cell non-Hodgkin lymphoma. Blood 137(19):2634–2645. https://doi.org/10.1182/blood.2020007512
Harris LJ, Patel K, Martin M (2020) Novel therapies for relapsed or refractory diffuse large B-cell lymphoma. Int J Mol Sci. https://doi.org/10.3390/ijms21228553
Hartley JA (2020) Antibody-drug conjugates (ADCs) delivering pyrrolobenzodiazepine (PBD) dimers for cancer therapy. Expert Opin Biol Ther. https://doi.org/10.1080/14712598.2020.1776255
Horton HM, Bernett MJ, Pong E, Peipp M, Karki S, Chu SY et al (2008) Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res 68(19):8049–8057. https://doi.org/10.1158/0008-5472.Can-08-2268
Jain N, Stock W, Zeidan A, Atallah E, McCloskey J, Heffner L et al (2020) Loncastuximab tesirine, an anti-CD19 antibody-drug conjugate, in relapsed/refractory B-cell acute lymphoblastic leukemia. Blood Adv 4(3):449–457. https://doi.org/10.1182/bloodadvances.2019000767
Jamil A, Mukkamalla SKR. Lymphoma. StatPearls Publishing LLC, Treasure Island, FL. 2021. https://www.ncbi.nlm.nih.gov/books/NBK560826/?report=printable. Accessed 1 May 2021.
Jurczak W, Bryk AH, Mensah P, Gałązka K, Trofimiuk-Müldner M, Wyrobek Ł et al (2016) Single-agent MOR208 salvage and maintenance therapy in a patient with refractory/relapsing diffuse large B-cell lymphoma: a case report. J Med Case Rep 10(1):123. https://doi.org/10.1186/s13256-016-0875-x
Jurczak W, Zinzani PL, Gaidano G, Goy A, Provencio M, Nagy Z et al (2018) Phase IIa study of the CD19 antibody MOR208 in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma. Ann Oncol 29(5):1266–1272. https://doi.org/10.1093/annonc/mdy056
Jurczak W, Zinzani PL, Hess G, Gaidano G, Provencio M, Nagy Z et al (2019) A phase IIa, open-label, multicenter study of single-agent tafasitamab (MOR208), an Fc-optimized anti-CD19 antibody, in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma: long-term follow-up, final analysis. Blood 134(Suppl 1):4078. https://doi.org/10.1182/blood-2019-124297
Kahl BS, Hamadani M, Radford J, Carlo-Stella C, Caimi P, Reid E et al (2019) A phase I study of ADCT-402 (loncastuximab tesirine), a novel pyrrolobenzodiazepine-based antibody-drug conjugate, in relapsed/refractory B-cell non-Hodgkin lymphoma. Clin Cancer Res 25(23):6986–6994. https://doi.org/10.1158/1078-0432.Ccr-19-0711
Kantarjian H, Stein A, Gökbuget N, Fielding AK, Schuh AC, Ribera JM et al (2017) Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med 376(9):836–847. https://doi.org/10.1056/NEJMoa1609783
Katz BZ, Herishanu Y (2014) Therapeutic targeting of CD19 in hematological malignancies: past, present, future and beyond. Leuk Lymphoma 55(5):999–1006. https://doi.org/10.3109/10428194.2013.828354
Kellner C, Zhukovsky EA, Pötzke A, Brüggemann M, Schrauder A, Schrappe M et al (2013) The Fc-engineered CD19 antibody MOR208 (XmAb5574) induces natural killer cell-mediated lysis of acute lymphoblastic leukemia cells from pediatric and adult patients. Leukemia 27(7):1595–1598. https://doi.org/10.1038/leu.2012.373
Kellner C, Otte A, Cappuzzello E, Klausz K, Peipp M (2017) Modulating cytotoxic effector functions by Fc engineering to improve cancer therapy. Transfus Med Hemother 44(5):327–336. https://doi.org/10.1159/000479980
Locatelli F, Zugmaier G, Rizzari C, Morris JD, Gruhn B, Klingebiel T et al (2021) Effect of blinatumomab vs chemotherapy on event-free survival among children with high-risk first-relapse B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA 325(9):843–854. https://doi.org/10.1001/jama.2021.0987
Narkhede M, Cheson BD (2020) Tafasitamab. Monoclonal antibody targeting CD19, treatment of B-cell malignancies. Drugs Fut 45(9):641. https://doi.org/10.1358/dof.2020.45.9.3176880
Nowakowski GS, Rodgers TD, Marino D, Frezzato M, Barbui AM, Castellino C et al (2020) RE-MIND study: a propensity score-based 1:1 matched comparison of tafasitamab + lenalidomide (L-MIND) versus lenalidomide monotherapy (real-world data) in transplant-ineligible patients with relapsed/refractory (R/R) diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 38(15_suppl):8020. https://doi.org/10.1200/JCO.2020.38.15_suppl.8020
Olejniczak SH, Stewart CC, Donohue K, Czuczman MS (2006) A quantitative exploration of surface antigen expression in common B-cell malignancies using flow cytometry. Immunol Invest 35(1):93–114. https://doi.org/10.1080/08820130500496878
Patriarca A, Gaidano G (2021) Investigational drugs for the treatment of diffuse large B-cell lymphoma. Expert Opin Investig Drugs 30(1):25–38. https://doi.org/10.1080/13543784.2021.1855140
Poe JC, Minard-Colin V, Kountikov EI, Haas KM, Tedder TF (2012) A c-Myc and surface CD19 signaling amplification loop promotes B cell lymphoma development and progression in mice. J Immunol 189(5):2318–2325. https://doi.org/10.4049/jimmunol.1201000
Rafiq S, Cheney C, Mo X, Jarjoura D, Muthusamy N, Byrd JC (2012) XmAb-5574 antibody demonstrates superior antibody-dependent cellular cytotoxicity as compared with CD52- and CD20-targeted antibodies in adult acute lymphoblastic leukemia cells. Leukemia 26(7):1720–1722. https://doi.org/10.1038/leu.2012.40
Roßkopf S, Eichholz KM, Winterberg D, Diemer KJ, Lutz S, Münnich IA et al (2020) Enhancing CDC and ADCC of CD19 antibodies by combining Fc protein-engineering with Fc glyco-engineering. Antibodies (basel) 9(4):63. https://doi.org/10.3390/antib9040063
Salles G, Duell J, González Barca E, Tournilhac O, Jurczak W, Liberati AM et al (2020) Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol 21(7):978–988. https://doi.org/10.1016/s1470-2045(20)30225-4
Sapkota A, Shaikh H. Non-Hodgkin lymphoma. StatPearls Publishing LLC, Treasure Island, FL. 2020. https://www.ncbi.nlm.nih.gov/books/NBK559328/?report=printable. Accessed 1 May 2021.
Tilly H, Gomes da Silva M, Vitolo U, Jack A, Meignan M, Lopez-Guillermo A et al (2015) Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 26(Suppl 5):v116–v125. https://doi.org/10.1093/annonc/mdv304
Topp MS, Gökbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S et al (2014) Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol 32(36):4134–4140. https://doi.org/10.1200/jco.2014.56.3247
Topp MS, Gökbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC et al (2015) Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol 16(1):57–66. https://doi.org/10.1016/s1470-2045(14)71170-2
US Food and Drug Administration. Zynlonta™(loncastuximab tesirine-lpyl) for injection, for intravenous use [prescribing information]. 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761196s000lbl.pdf. Accessed 1 June 2021.
US Food and Drug Administration. Blincyto® (blinatumomab) for injection, for intravenous use [prescribing information]. 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125557s008lbl.pdf. Accessed 1 May 2021.
van der Horst HJ, Nijhof IS, Mutis T, Chamuleau MED (2020) Fc-engineered antibodies with enhanced Fc-effector function for the treatment of B-cell malignancies. Cancers (basel). https://doi.org/10.3390/cancers12103041
Viardot A, Goebeler ME, Hess G, Neumann S, Pfreundschuh M, Adrian N et al (2016) Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 127(11):1410–1416. https://doi.org/10.1182/blood-2015-06-651380
Viardot A, Hess G, Bargou RC, Morley NJ, Gritti G, Goebeler ME et al (2020) Durability of complete response after blinatumomab therapy for relapsed/refractory diffuse large B-cell lymphoma. Leuk Lymphoma 61(11):2767–2770. https://doi.org/10.1080/10428194.2020.1783442
Woyach JA, Awan F, Flinn IW, Berdeja JG, Wiley E, Mansoor S et al (2014) A phase 1 trial of the Fc-engineered CD19 antibody XmAb5574 (MOR00208) demonstrates safety and preliminary efficacy in relapsed CLL. Blood 124(24):3553–3560. https://doi.org/10.1182/blood-2014-08-593269
Zalevsky J, Leung IW, Karki S, Chu SY, Zhukovsky EA, Desjarlais JR et al (2009) The impact of Fc engineering on an anti-CD19 antibody: increased Fcgamma receptor affinity enhances B-cell clearing in nonhuman primates. Blood 113(16):3735–3743. https://doi.org/10.1182/blood-2008-10-182048
Zammarchi F, Corbett S, Adams L, Tyrer PC, Kiakos K, Janghra N et al (2018) ADCT-402, a PBD dimer-containing antibody drug conjugate targeting CD19-expressing malignancies. Blood 131(10):1094–1105. https://doi.org/10.1182/blood-2017-10-813493
Zinzani PL, Barbui AM, Castellino C, Meli E, Fowler N, Salles G et al (2021) RE-MIND: comparing tafasitamab + lenalidomide (L-MIND) with a real world lenalidomide monotherapy cohort in relapsed or refractory diffuse large B-cell lymphoma. Clin Cancer Res. https://doi.org/10.1158/1078-0432.CCR-21-1471
Acknowledgements
We would like to thank Lorenza Lanini and Richard Crampton, who wrote the outline and first draft, respectively, on behalf of Springer Healthcare Communications. This medical writing assistance was funded by Incyte.
Funding
Development of this review was funded by Incyte.
Author information
Authors and Affiliations
Contributions
Both authors contributed to the conceptualization and drafting of this manuscript, commented on previous versions of the manuscript and read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Pier Luigi Zinzani has participated in the speaker bureau for Verastem, Celltrion, Gilead, Janssen-Cilag, BMS, Servier, MSD, TG Therapeutics, Takeda, Roche, Eusapharma, Kyowa Kirin, Novartis, Incyte, Beigene, has participated in the advisory boards for Verastem, Celltrion, Gilead, Janssen-Cilag, BMS, Servier, MSD, TG Therapeutics, Takeda, Roche, Eusapharma, Kyowa Kirin, Novartis, Incyte, Beigene, Sandoz, ADC Therapeutics and served as a consultant to Verastem, MSD, Eusapharma and Novartis. Giorgio Minotti declares no conflict of interest.
Ethical approval
Not applicable.
Informed consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zinzani, P.L., Minotti, G. Anti-CD19 monoclonal antibodies for the treatment of relapsed or refractory B-cell malignancies: a narrative review with focus on diffuse large B-cell lymphoma. J Cancer Res Clin Oncol 148, 177–190 (2022). https://doi.org/10.1007/s00432-021-03833-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-021-03833-x