
Abstract
Castleman disease (CD) is a rare lymphoproliferative disorder of undetermined etiology. Unicentric CD (UCD) and multicentric CD (MCD) are two phenotypes of CD diagnosed by the histopathology of lymph nodes. We attempted to describe a pediatric CD cohort to optimize the management of this disease. We reviewed the medical records of pediatric patients diagnosed with CD between April, 2004, and October, 2022, at the Children’s Hospital of Fudan University. Prognosis information was collected in January, 2023, by telephone inquiry. Twenty-two patients with UCD and 2 patients with MCD were identified, all with hyaline vascular (HV) type. The median ages at diagnosis were 10.75 years (IQR 8, 12.81) for UCD and 14.42 years (IQR 13.42, 15.42) for MCD. The most common lesion location of UCD was the neck (9/22, 40.91%) and abdomen (9/22, 40.91%). Systematic symptoms occurred on 10/22 (45.45%) patients with UCD and 1/2 (50%) patients with MCD, and abnormal laboratory indexes were detected in both. Resection and biopsy were performed on all patients. One out of two patients with MCD also received rituximab for upfront therapy. After a median of 4 years (IQR 1.5, 6) of follow-up time, the overall survival was 100% and the complete remission rate in UCD was 63%. There was no relapse or progression.
Conclusions: Our series demonstrated that HV-UCD was the most common type in children. Resection and biopsy were used for both deterministic diagnoses and treatments. Despite the high possibility to develop systematic inflammation, children with CD showed promising outcomes.
What is Known: • Castleman disease is a rare lymphoproliferative disorder with limited cohort studies, especially in pediatrics. • The ubiquity of delayed confirmations and misdiagnoses points to a lack of knowledge about etiology and characteristics, which is a prerequisite for novel therapeutics. | |
What is New: • We retrospectively reviewed and analyzed the clinical and pathological symptoms, laboratory and imaging features, and treatment outcomes of a Chinese pediatric cohort with Castleman disease. • Our work may improve the recognition and optimize the management of this rare disease in children. |
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Castleman disease (CD) is a lymphoproliferative disease of unidentified etiology, first reported in 1954 and named in 1956 by Castleman et al. [1]. CD has a low incidence of approximately 6500 to 7700 cases per year in the USA [2]; however, definitive reports of childhood morbidity are unavailable. CD can be classified as unicentric CD (UCD) or multicentric CD (MCD) according to its involvement and clinical course [3]. Based on the primary driver of the disease, MCD is further classified as idiopathic MCD (iMCD); human herpes virus 8 (HHV-8)-associated MCD (HHV-8-MCD); and polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes (POEMS) syndrome-associated MCD (POEMS-MCD) [4]. Confirmation of the diagnosis relies on histopathological findings. The hyaline vascular (HV) type is characterized by capsular fibrosis, increased numbers of lymphoid follicles, and regressed germinal centers (GCs) penetrated by pathological hyaline blood vessels [2]. Additionally, hyperplastic mantle zones present a typical “onion skin pattern,” comprising concentric rings of small lymphocytes [2]. The plasma cell (PC) type is characterized by the enrichment of sheetlike PCs in the interfollicular zone, with hyperplastic GCs [3]. Features of both types are present in the mixed type.
The most frequent type of CD in children is HV-UCD, which commonly originates in the neck [5]. The predominant sign is an enlarged, painless lymph node. By contrast, MCD is more aggressive and accompanied by frequent systemic symptoms and laboratory abnormalities. UCD can usually be cured by surgical resection and has a good survival rate, whereas MCD has a worse prognosis because of the risk of relapse or progression. Both misdiagnosis and delayed diagnosis are common in children because of their unclear complaints and inadequate awareness among parents and pediatricians.
We conducted this retrospective study to analyze the clinical and pathological symptoms, laboratory and imaging features, and treatment outcomes of children with CD in an effort to improve the recognition and management of this rare disease.
Methods
Patients and materials
This study involved children diagnosed with CD based on the pathological evidence at the Children’s Hospital of Fudan University from April, 2004, to October, 2022. All the diagnoses were reviewed and confirmed by clinical experts, pathologists, and radiologists. The diagnostic delay was defined as the time from the first symptom or identification of a mass to diagnosis. The patients’ demographic information, clinical presentation, pathological findings, and first-line therapy were retrospectively collected from the medical records. Laboratory data and imaging observations prior to surgical resection were chosen for analysis; if several examinations were performed, the results of the first visit were selected.
Follow-up
We were able to access prognostic information for 16 of 22 patients with UCD and 1 of 2 patients with MCD by telephone follow-up in January, 2023. Overall survival (OS) was defined as the time from diagnosis to relapse, death, or the last follow-up.
Statistical analysis
The patients were stratified according to the disease subtype or whether they were asymptomatic. Categorical variables are presented as count and percentage, and continuous variables are presented as median (interquartile range [IQR]) or mean (standard deviation). Wilcoxon’s rank sum test or Student’s t-test was used to calculate the P-value, and P < 0.05 indicated statistical significance. Statistical analyses were performed using Stata/MP version 17.0 (StataCorp, College Station, TX, USA).
Results
We identified 24 patients with UCD (n = 22) and MCD (n = 2) according to biopsy-proven histopathological features. The patients’ characteristics are shown in Tables 1 and 2.
UCD
The sex ratio was 1:1, the median age at diagnosis was 10.75 years (IQR: 8, 12.81), and the median diagnostic delay was 5.25 months (IQR: 1, 15).
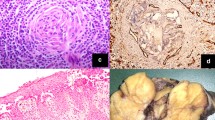
All 22 patients underwent excisional biopsy and the results of the immunohistochemical (IHC) examination of 20 were available. All cases were histologically classified as the HV subtype. Bcl-2 and biomarkers of T lymphocytes, including CD3 and CD5, were detected in interfollicular areas, whereas Bcl-6 and biomarkers of B lymphocytes, including CD20 and CD79a, were detected within follicles. Representative histopathological findings are shown in Fig. 1.

Representative histopathological findings of an involved lymph node in Castleman disease with hyaline vascular type. a Typical manifestations of hyperplastic follicles with regressive germinal centers and concentric proliferating lymphocytes; b a typical lymphatic follicle; c expression patterns of CD5; d expression patterns of CD20
The most common primary lymph nodes were located in the neck (9/22, 41%) and abdomen (9/22, 41%), followed by the mediastinum (2/22, 9%), axilla (1/22, 5%), and groin (1/22, 5%). Twelve (55%) cases were unintentionally palpated and 10 (45%) were incidentally discovered during imaging examinations for various reasons. At diagnosis, systemic symptoms were present in 10/22 (45%) patients, including anemia (10/22, 45%), growth retardation (1/22, 5%), fatigue (1/22, 5%), and anorexia (1/22, 5%). Other complaints were abdominal pain or discomfort (3/22), vomiting (1/22), chest pain (1/22), frequent colds (1/22), and recurrent nosebleeds (1/22).
Among the anemic children, 7/10 (70%) had mild anemia and 3/10 (30%) had moderate anemia, and the median hemoglobin (Hb) level of all patients with UCD was 116 g/L (IQR: 97.55, 132; range: 78–147). A decreased mean corpuscular volume (MCV) was observed in 8/18 (44%) patients overall and 6/10 (60%) patients with anemia. The platelet count was increased in 11/22 (50%) patients, and the median count was 307 × 109/L (IQR: 265.25, 447; range: 178–698). The median albumin level was normal (median: 42.7 g/L [IQR: 34.05, 45.12; range: 27.7–50]), whereas 7/21 (33%) patients had a decreased level. Additionally, 12/21 (57%) patients had an increased globulin level (median: 31.6 g/L [IQR: 25.35, 43.1; range: 20.6–84.9]). C-reactive protein (CRP) was measured in 15 patients and elevated in 5 (33%) (> 8 mg/L). An elevated IgG level was observed in 1/2 (50%) patients. Human immunodeficiency virus (HIV) antibody screening was negative in 19/19 patients. Epstein-Barr virus (EBV) IgG and Cytomegalovirus (CMV) IgG were detected in 6/6 patients and 4/6 patients, respectively, but the relevant lymph node staining was not performed on them. Patient 7 presented with positive anti-streptolysin O (ASO) and rheumatoid factor, and patient 22 had elevated levels of interleukin (IL)-8 and IL-12.
Adjunct examinations were performed in 20/22 patients for diagnosis. Ultrasound was most commonly used, followed by computed tomography (CT) (16/20, 80%), magnetic resonance imaging (MRI) (6/20, 30%), and fine-needle aspiration cytology (FNAC) (3/20, 15%). As in adults, lesions imaged with ultrasound always demonstrated homogeneous hypoechoic nodules with rich blood flow signals. Mild to moderate homogeneous enhancement was a typical feature in CT and MRI. The mean CT value before and after enhancement in six patients was 47.6 (9.95) and 132.35 (19.63) HU, respectively. Representative images are displayed in Fig. 2.

Representative imaging pictures of a left axillary mass in unicentric Castleman disease. a Ultrasonography image showed a homogeneous hypoechoic area with a regular shape and sharp border and a thick blood supply vessel passing through the mass (blue arrow); b color Doppler flow imaging detected rich blood flow signals in the mass; c general CT image showed a homogeneous mass with similar density to soft tissue (purple arrow), and the CT value was 55 Hu; d the mass was significantly and homogeneously enhanced after contrast injection, and the CT value was 160 Hu
All patients with UCD underwent surgical resection only. After a median follow-up of 4.08 years (IQR: 1.63, 6.08), all patients had survived and none had developed relapse or progression, 10/16 (63%) were in complete remission, and 6/16 (38%) had stable lymphadenopathy. Patient 10 was prone to colds and slight fevers, and patient 21 complained of fatigue. Patient 8 developed persistent numbness in her right hand after hospital discharge and had an intermittent headache for 5 days, two episodes of vomiting, and one episode of convulsions, which improved after symptomatic treatment.
MCD
Both patients with MCD were male, presented the HV subtype, HIV negative, EBV IgG positive, and CMV IgG positive.
For patient 23, the age at diagnosis was 13 years and the diagnostic delay was 24 months. The primary lesion was a painless mass palpated in the neck. Upon diagnosis, no systemic symptoms were present. Multiple lymphadenopathies were detected in the IB and II regions in the bilateral neck, the V region in the right neck, and the right internal iliac region by positron emission tomography (PET)-CT. The Hb level (159 g/L), MCV (87.3 fL), platelet count (265 × 109/L), CRP level (< 8 mg/L), albumin level (47.1 g/L), and globulin level (27.8 g/L) were normal, while the levels of IgG (15.43 g/L) and inflammatory cytokines including tumor necrosis factor (TNF)-α, interferon (IFN)-α, IFN-γ, and IL-8 were elevated.
For patient 24, the age at diagnosis was 15 years and the diagnostic delay was 5 days. The primary lymph node was located in the axilla and discovered during routine follow-up for connective tissue disorder (CTD). He had an EBV infection and presented EBER-positive. Fever and mild anemia were present at the time of diagnosis. PET-CT revealed involvement of lymph nodes, bones, bone marrow, spleen, and nasopharynx; hepatomegaly was also present. The CRP level was markedly high at 245.87 mg/L. Other laboratory findings were an Hb level of 107 g/L, MCV of 77.9 fL, platelet count of 313 × 109/L, albumin level of 34.21 g/L, globulin level of 32.99 g/L, IL-6 level of 11.3 pg/mL, and IgG level of 18.77 g/L. Serum antinuclear antibody (ANA), ASO, and IL-8 were all increased.
Both patients with MCD underwent excisional biopsy and IHC examinations. Patient 23 was a consultation patient without access to subsequent treatment. Patient 24 received rituximab therapy once a week for 1 month postoperatively and then switched to thalidomide and dexamethasone. Some inflammatory indexes were abnormal during follow-up, including IL-6 (8.8 pg/mL), CRP (53.52 mg/L), and erythrocyte sedimentation rate (27 mm/h); however, no symptoms developed.
Discussion
Our patients with UCD had a balanced sex ratio, consistent with previous reports [5, 6]. Although both patients with MCD were male, more research is needed to determine the sex distribution of MCD. CD can occur at any age, but the median age of patients with UCD is usually younger than that of patients with MCD [7, 8]. This is also supported by our results.
CD was originally described as HV and PC types with an intermediate mixed type. However, experts have proposed that the features present in the different subtypes occur along a spectrum of pathologies rather than fitting into three easily definable clusters [3]. UCD commonly presents as the HV subtype, whereas MCD commonly presents as the PC and mixed subtypes [3]. Nevertheless, both of our patients with MCD exhibited typical characteristics of the HV subtype, and up to 67% of patients with iMCD in a two-site pediatric cohort presented the HV subtype [5]. IHC has high sensitivity and is valuable for the differential diagnosis and histological profiling of lymphoproliferative diseases. Expressions of CD138 and VS38C can facilitate verifying the polyclonal nature of CD [9], discriminating it from neoplastic diseases. Especially, the proliferative plasmacytoid cells in HHV-8-MCD express monotypic but not monoclonal IgM-λ, due to up-regulation of recombination activation protein-mediated V(D)J recombination and Igλ expression in HHV-8-infected Igκ-naïve B lymphocytes [2]. Although the categorization can be helpful for the histopathological diagnosis of CD, it has an ambiguous clinical impact and cannot be utilized alone for clinical management.
UCD is generally considered to present a homogenous phenotype as asymptomatic isolated lymph nodes or symptoms associated with a mass effect. In our cohort, up to 45% of patients with UCD presented with systemic symptoms, similar to another pediatric cohort [5]. However, the proportion was significantly higher than that in adults at 18% [7]. One of our patients with UCD exhibited growth retardation, and a previous report also described a patient with UCD whose only symptom was short stature [10]. Growth inhibition may result from chronic inflammation mediated by cytokines, which is also attributed to delayed diagnosis. This might occasionally be the primary reason for hospitalization and could be a crucial presentation of CD in children.
Among our 22 patients with UCD, 9 had detectable symptoms including pallor, abdominal pain or discomfort, chest pain, fatigue, anorexia, and growth retardation, whereas the other 13 were asymptomatic. As shown in Table 3, the primary lymph nodes were located internally in all symptomatic patients but in only 2/13 (15%) of asymptomatic patients. Overall, peripherally located masses were easily detected, resulting in earlier access to medical care, shorter diagnostic delays, and less likely to be symptomatic. Conversely, our data suggested median diagnostic delays of 6 months (IQR: 1, 18) and 4.5 months (IQR: 0.85, 24) for the asymptomatic and symptomatic patients, respectively, but the difference was not statistically significant (z = 0.00, P = 1.00). The mean maximum dimension of single lesions in the two groups was 3.98 (0.96) and 5.23 (1.13) cm, respectively, and was larger in the symptomatic group (t = − 2.82, P = 0.01).
MCD can be subdivided into HHV-8-MCD, iMCD, and POEMS-MCD. HHV-8-MCD is typically accompanied by immune impairment, most commonly HIV infection [4]. Both of our patients with MCD were HIV negative, but their HHV-8 infection status was unavailable. By comparison, adults with MCD have an 86.2% chance of being HHV-8 positive [7], whereas the proportion in children with MCD is always 0% [5, 6, 9]. This may be attributed to the varying prevalence of HIV and HHV-8 in different populations.
The diagnosis of MCD in patient 23 required differentiation from IgG4-related disease (IgG4RD) because IHC showed a high number of IgG4-positive cells (> 100/hpf) and elevation of serum IgG4. In addition, the lymphadenopathy and increased levels of serum inflammatory biomarkers observed in patient 23 were shared by iMCD and IgG4RD [11]. It is essential to distinguish between these conditions because of their different clinical courses and therapeutic strategies. The serum IgG4/IgG ratio is a more reliable discriminator than the absolute IgG4-positive cell count, which may be distinctive for IgG4RD when > 40% [11]. By contrast, a persistent rise in IgA, IgM, and CRP is an exclusive criterion for IgG4RD [11]. In our patient, the IgG4/IgG ratio was < 40% and the histopathologic findings were characteristic for the HV type; thus, the diagnosis of MCD was confirmed.
At diagnosis, patient 24 presented overlapping manifestations between MCD and CTD, including fever, anemia, and positive autoantibody (ANA 1:320 +). Autoimmune appearances can occur at any period in the course of MCD. In previous studies, 28% of patients with systemic lupus erythematosus [12] and almost all patients with rheumatoid arthritis [13] showed histopathological features comparable with those of MCD. Additionally, 30% of patients with iMCD showed autoantibody positivity or autoimmune hemolytic anemia [14]. These findings demonstrate that autoimmune abnormalities have a potential relationship with MCD, and clinicians should be aware of the possibility of the coexistence of autoimmune diseases and CD to improve the accuracy of diagnosis.
The underlying pathogenesis of CD has not been fully elucidated. Some data have demonstrated that UCD may be a neoplastic cloning process involving stromal cells, predominately follicular dendritic cells [2]. PDGFRB mutations located in CD45− stromal cells were found in 17% of patients with UCD [15], and the allele frequency increased with the proliferation of stromal cells [16]. Uncontrolled HHV-8 infection and inflammatory storms involving cytokines such as IL-6 are crucial drivers of HHV-8-MCD and iMCD, respectively [2]. However, transcriptome sequencing showed that IL-6 expression was not significantly higher in iMCD-involved lymph nodes than in normal lymph nodes [17]. Notably, other cytokines including TNF-α, IFN-α, IFN-γ, and IL-8 were elevated in our patients, and their role in the disease course needs further verification. Among them, IFN-I has been proven to be involved in the pathogenesis of iMCD by increasing JAK/STAT-dependent activation of mTOR signaling [18]. Mutations in genes associated with autoinflammatory diseases may also be involved in the pathogenesis of iMCD [6]. The next step should be deeper investigation into the mechanism level.
There is no consensus regarding whether FNAC can serve as a definitive diagnostic modality. In the present study, 3/22 patients with UCD and 1/2 patients with MCD underwent FNAC, but none were conclusively diagnosed by FNAC alone. Distinguishing morphological features of CD include lymphocyte-dominant cell smears, an admixture of GC cells, PCs, and hyalinized capillary fragments with adhered lymphocytes [19]. Deeper insight into the subtle cytomorphological features of CD is essential to discriminate it from its mimics.
Regardless of the pathology, the gold standard treatment for UCD is surgical resection, which has a complete remission rate of 90%. For unresectable disease, chemotherapy, radiotherapy, or embolization should be considered [3]. For patients with HHV-8-MCD, a rituximab-based therapy regimen results in a 5-year OS rate of 90% [3]; however, there is no standard treatment regimen for iMCD. IL-6 inhibitors are the only first-line therapy for iMCD approved by the FDA [20], but up to two-thirds of patients have no response [21]. High IgG and fibrinogen levels imply a high response potential, whereas the opposite is true for high CRP and Hb levels [22]. A 17% reduction in CXCL13 by 8 days after siltuximab therapy is also a predictive indicator [23]. Therefore, serum index quantification can help guide and modify the treatment strategies. Likewise, rituximab was ineffective for patient 24 after just two doses, and a combination of thalidomide and dexamethasone kept him in stable condition. Recent clinical trials involving the thalidomide-cyclophosphamide-prednisone (TCP) regimen and the bortezomib-cyclophosphamide-dexamethasone (BCD) regimen have shown promising outcomes [24, 25]. The mTOR inhibitor sirolimus [26] and the JAK inhibitor ruxolitinib [27] have also benefited some patients. Such clinical trials in children are urgently needed to determine the efficacy and safety of these drugs. A clinical trial of sirolimus involving adults and children is ongoing (NCT03933904).
The prognosis of CD is more favorable in children than adults [5], and as expected, the progression-free survival rate in our study was 100%. In a CD cohort with the largest sample size to date, the 5-year OS rate was 95% and 74% for patients with UCD and MCD, respectively [8]. The PC type, hepatomegaly and/or splenomegaly, Hb of ≤ 80 g/L, and albumin of ≤ 30 g/L were recently confirmed as independent prognostic factors for OS, especially in patients with iMCD [8].
This study had some limitations. Because of its single-center, retrospective nature, bias in sample selection and data collection was inevitable. The panel of IHC was incomplete. Given the lack of samples, the calculation of mean values in the patients with MCD, and the comparison between two phenotypes were meaningless. However, our work effectively summarized the characteristics of children with CD to a certain degree, aiming to provide a reference for optimal management.
Conclusion
We retrospectively analyzed a pediatric cohort with CD, in which HV-UCD was the most prevalent type. The children’s histopathological manifestations were relatively homogeneous. Diagnostic delay remains an unresolved issue. Surgical excision is the first-line treatment for UCD and has been proven quite beneficial. A standard therapeutic regimen for iMCD is needed, especially for patients who do not respond to IL-6 inhibitors. Although there was a high proportion of systemic symptoms among children with UCD, OS was still considerably high.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- ANA:
-
Antinuclear antibody
- ASO:
-
Anti-streptolysin O
- CD:
-
Castleman disease
- CMV:
-
Cytomegalovirus
- CRP:
-
C-reactive protein
- CTD:
-
Connective tissue disorder
- EBV:
-
Epstein-Barr virus
- FNAC:
-
Fine-needle aspiration cytology
- GC:
-
Germinal center
- Hb:
-
Hemoglobin
- HHV-8:
-
Human herpes virus 8
- HIV:
-
Human immunodeficiency virus
- HV:
-
Hyaline vascular
- IFN:
-
Interferon
- IgG4RD:
-
IgG4-related disease
- IHC:
-
Immunohistochemical
- IL:
-
Interleukin
- iMCD:
-
Idiopathic multicentric Castleman disease
- IQR:
-
Interquartile range
- MCD:
-
Multicentric Castleman disease
- MCV:
-
Mean corpuscular volume
- OS:
-
Overall survival
- PC:
-
Plasma cell
- POEMS:
-
Polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes
- TNF:
-
Tumor necrosis factor
- UCD:
-
Unicentric Castleman disease
References
Castleman B, Iverson L, Menendez VP (1956) Localized mediastinal lymphn-ode hyperplasia resembling thymoma. Cancer 9(4):822–830. https://acsjournals.onlinelibrary.wiley.com/doi/10.1002/1097-0142(195607/08)9:4%3C822::AID-CNCR2820090430%3E3.0.CO;2-4
Carbone A, Borok M, Damania B, Gloghini A, Polizzotto MN, Jayanthan RK, Fajgenbaum DC, Bower M (2021) Castleman disease. Nat Rev Dis Primers 7(1):84. https://doi.org/10.1038/s41572-021-00317-7
Dispenzieri A, Fajgenbaum DC (2020) Overview of Castleman disease. Blood 135(16):1353–1364. https://doi.org/10.1182/blood.2019000931
Fajgenbaum DC, Uldrick TS, Bagg A et al (2017) International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 129(12):1646–1657. https://doi.org/10.1182/blood-2016-10-746933
Sopfe J, Endres A, Campbell K, Hayes K, Trout AT, Liang X, Lorsbach R, O’Brien MM, Cost CR (2019) Castleman disease in pediatrics: insights on presentation, treatment, and outcomes from a two-site retrospective cohort study. Pediatr Blood Cancer 66(5):e27613. https://doi.org/10.1002/pbc.27613
Borocco C, Ballot-Schmit C, Ackermann O et al (2020) The French paediatric cohort of Castleman disease: a retrospective report of 23 patients. Orphanet J Rare Dis 15(1):95. https://doi.org/10.1186/s13023-020-1345-5
Oksenhendler E, Boutboul D, Fajgenbaum D et al (2018) The full spectrum of Castleman disease: 273 patients studied over 20 years. Br J Haematol 180(2):206–216. https://doi.org/10.1111/bjh.15019
Liu W, Cai Q, Yu T et al (2022) Clinical characteristics and outcomes of Castleman disease: a multicenter Consortium study of 428 patients with 15-year follow-up. Am J Cancer Res 12(9):4227–4240
Hu C, Zou Y, Pan J, Yang J, Yang T, Tan T, Li J (2020) Analysis of clinical characteristics, pathological changes and changes of interleukin-6 (IL-6) and C-reactive protein (CRP) in children with Castleman’s disease. Med Sci Monit 26:e924783. https://doi.org/10.12659/MSM.924783
Sonoda K, Kaneko U, Hiura M, Kinoshita Y, Umezu H, Ito S, Saitoh A, Imai C (2022) Short stature as an initial presenting presentation of unicentric castleman disease in a child: a case report with long-term follow-up and a literature review. Mod Rheumatol Case Rep 7(1):261–266. https://doi.org/10.1093/mrcr/rxac034
Cheng CC, Chen YC, Hsu YH, Su KY (2022) Idiopathic multicentric castleman disease with strikingly elevated IgG4 concentration in the serum and abundant IgG4-positive cells in the tissue: a case report. Diagnostics (Basel) 12(9):2261. https://doi.org/10.3390/diagnostics12092261
Kojima M, Nakamura S, Morishita Y et al (2000) Reactive follicular hyperplasia in the lymph node lesions from systemic lupus erythematosus patients: a clinicopathological and immunohistological study of 21 cases. Pathol Int 50(4):304–312. https://doi.org/10.1046/j.1440-1827.2000.01052.x
Kojima M, Motoori T, Nakamura S (2006) Benign, atypical and malignant lymphoproliferative disorders in rheumatoid arthritis patients. Biomed Pharmacother 60(10):663–672. https://doi.org/10.1016/j.biopha.2006.09.004
Liu AY, Nabel CS, Finkelman BS et al (2016) Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol 3(4):e163–175. https://doi.org/10.1016/S2352-3026(16)00006-5
Li Z, Lan X, Li C et al (2019) Recurrent PDGFRB mutations in unicentric Castleman disease. Leukemia 33(4):1035–1038. https://doi.org/10.1038/s41375-018-0323-6
Singh KI, Gollapudi S, Kumar J et al (2022) Case report: Castleman disease with an associated stromal spindle cell proliferation, PDGFRB mutation and p53 expression: clonal origins of a rare disease. Front Oncol 12:857606. https://doi.org/10.3389/fonc.2022.857606
Horna P, King RL, Jevremovic D, Fajgenbaum DC, Dispenzieri A (2022) The lymph node transcriptome of unicentric and idiopathic multicentric Castleman disease. Haematologica 108(1):207–218. https://doi.org/10.3324/haematol.2021.280370
Pai RAL, Japp AS, Gonzalez M et al (2020) Type I IFN response associated with mTOR activation in the TAFRO subtype of idiopathic multicentric Castleman disease. JCI Insight 5(9):e135031. https://doi.org/10.1172/jci.insight.135031
Singh N, Chowdhury N, Pal S, Goyal JP, Bhakhri BK, Rao S (2021) Hyaline vascular type of Castleman disease: diagnostic pitfalls on cytology and its clinical relevance. Cureus 13(8):e17174. https://doi.org/10.7759/cureus.17174
Deisseroth A, Ko CW, Nie L et al (2015) FDA approval: siltuximab for the treatment of patients with multicentric Castleman disease. Clin Cancer Res 21(5):950–954. https://doi.org/10.1158/1078-0432.CCR-14-1678
van Rhee F, Wong RS, Munshi N et al (2014) Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 15(9):966–974. https://doi.org/10.1016/S1470-2045(14)70319-5
Morra DE, Pierson SK, Shilling D, Nemat S, Appiani C, Guilfoyle M, Tendler C, van Rhee F, Fajgenbaum DC (2019) Predictors of response to anti-IL6 monoclonal antibody therapy (siltuximab) in idiopathic multicentric Castleman disease: secondary analyses of phase II clinical trial data. Br J Haematol 184(2):232–241. https://doi.org/10.1111/bjh.15588
Pierson SK, Katz L, Williams R et al (2022) CXCL13 is a predictive biomarker in idiopathic multicentric Castleman disease. Nat Commun 13(1):7236. https://doi.org/10.1038/s41467-022-34873-7
Zhang L, Zhao AL, Duan MH et al (2019) Phase 2 study using oral thalidomide-cyclophosphamide-prednisone for idiopathic multicentric Castleman disease. Blood 133(16):1720–1728. https://doi.org/10.1182/blood-2018-11-884577
Zhang L, Zhang MY, Cao XX, Zhou DB, Fajgenbaum DC, Dong YJ, Li J (2022) A prospective, multicenter study of bortezomib, cyclophosphamide, and dexamethasone in relapsed/refractory iMCD. Leuk Lymphoma 63(3):618–626. https://doi.org/10.1080/10428194.2021.1999437
Fajgenbaum DC, Langan RA, Japp AS et al (2019) Identifying and targeting pathogenic PI3K/AKT/mTOR signaling in IL-6-blockade-refractory idiopathic multicentric Castleman disease. J Clin Invest 129(10):4451–4463. https://doi.org/10.1172/JCI126091
Lust H, Gong S, Remiker A, Rossoff J (2021) Idiopathic multicentric Castleman disease with TAFRO clinical subtype responsive to IL-6/JAK inhibition: a pediatric case series. Pediat Blood Cancer 68(10):e29261. https://doi.org/10.1002/pbc.29261
Author information
Authors and Affiliations
Contributions
Xiaowen Zhai conceptualized and designed the study. Shiwen Hu, Zifeng Li, Hongsheng Wang, and Xiaowen Zhai conducted data collection, analyses, and interpretation. Shiwen Hu drafted the initial manuscript. Lian Chen, Yangyang Ma, Xiaohua Zhu, Jun Li, Rui Dong, Wei Yao, Chenbin Dong, Huifeng Zhang, Kai Li, and Kuiran Dong provided advice on the study design and contributed to the data acquisition. All authors critically reviewed and revised the manuscript. All authors approved the final manuscript and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Ethics approval
This retrospective study was conducted on already available data and was approved by the Medical Ethics Committee of the Children’s Hospital of Fudan University institutional review board and conducted under the Declaration of Helsinki principles (approval reference: No (2023) 47; February 28, 2023).
Consent to participate
Informed consent was obtained from the legal guardians of all participants included in the study.
Competing interests
The authors declare no competing interests.
Additional information
Communicated by Peter de Winter
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, S., Li, Z., Wang, H. et al. Clinical features and treatment outcomes of Castleman disease in children: a retrospective cohort in China. Eur J Pediatr 182, 5519–5530 (2023). https://doi.org/10.1007/s00431-023-05235-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-023-05235-2