
Abstract
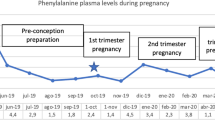
Untreated phenylketonuria (PKU) in pregnancy causes a severe embryopathy called maternal PKU syndrome. Here, we aimed to assess management issues and pregnancy outcomes in the first published series of PKU pregnancies from the developing world. Data were collected retrospectively in a single center from 71 pregnancies and 45 live births of 32 women with PKU, 11 of whom were diagnosed in adulthood after having an affected child. Microcephaly, intellectual disability, and dysmorphic facies were more prevalent in offspring of untreated than treated pregnancies with classical PKU (100% vs. 0%, 91% vs. 0%, and 73% vs. 23% with p < 0.001, p < 0.001, and p = 0.037, respectively). In treated pregnancies, phenylalanine levels were higher during weeks 6–14 than other periods of gestation (4.38 vs. 3.93, 2.00 and 2.28 mg/dl; p < 0.05). Poor compliance correlated with higher phenylalanine levels (ρ = − 0.64, p = 0.019) and fluctuations (ρ = − 0.66, p = 0.014).
Conclusion: More frequent phenylalanine measurements during late first trimester are crucial to improve outcomes in treated pregnancies. In order to prevent untreated pregnancies via detecting undiagnosed adults, countries where significantly many women of childbearing age were not screened as newborns may consider pre-pregnancy PKU screening. Microcephaly in the newborn should prompt screening for PKU in the mother.
What Is Known •Untreated phenylketonuria during pregnancy causes maternal phenylketonuria syndrome in the newborn. •Effective treatment throughout pregnancy can prevent adverse fetal outcomes. | |
What Is New: •Metabolic control is related to frequency of follow-up and worsens during late first trimester. Closer follow-up during this period may improve metabolic control. •In order to prevent untreated pregnancies, pre-pregnancy phenylketonuria screening may be considered if many women of childbearing age were not screened as newborns. |
Similar content being viewed by others


Abbreviations
- HPA:
-
Hyperphenylalaninemia
- ID/DD:
-
Intellectual disability or developmental delay
- IQ:
-
Intelligence quotient
- IQR:
-
Interquartile range
- IUGR:
-
Intrauterine growth restriction
- LMP:
-
Last menstrual period
- NBS:
-
Newborn screening
- PAH:
-
Phenylalanine hydroxylase
- Phe:
-
Phenylalanine
- PKU:
-
Phenylketonuria
References
Ahring K, Belanger-Quintana A, Dokoupil K, Gokmen-Ozel H, Lammardo AM, MacDonald A, Motzfeldt K, Nowacka M, Robert M, van Rijn M (2011) Blood phenylalanine control in phenylketonuria: a survey of 10 European centres. Eur J Clin Nutr 65:275–278
American College of Obstetricians and Gynecologists (2013) ACOG practice bulletin no. 134: fetal growth restriction. Obstet Gynecol 121:1122–1133
Blau N, van Spronsen FJ, Levy HL (2010) Phenylketonuria. Lancet 376:1417–1427
Camp KM, Parisi MA, Acosta PB, Berry GT, Bilder DA, Blau N, Bodamer OA, Brosco JP, Brown CS, Burlina AB, Burton BK, Chang CS, Coates PM, Cunningham AC, Dobrowolski SF, Ferguson JH, Franklin TD, Frazier DM, Grange DK, Greene CL, Groft SC, Harding CO, Howell RR, Huntington KL, Hyatt-Knorr HD, Jevaji IP, Levy HL, Lichter-Konecki U, Lindegren ML, Lloyd-Puryear MA, Matalon K, MacDonald A, McPheeters ML, Mitchell JJ, Mofidi S, Moseley KD, Mueller CM, Mulberg AE, Nerurkar LS, Ogata BN, Pariser AR, Prasad S, Pridjian G, Rasmussen SA, Reddy UM, Rohr FJ, Singh RH, Sirrs SM, Stremer SE, Tagle DA, Thompson SM, Urv TK, Utz JR, van Spronsen F, Vockley J, Waisbren SE, Weglicki LS, White DA, Whitley CB, Wilfond BS, Yannicelli S, Young JM (2014) Phenylketonuria scientific review conference: state of the science and future research needs. Mol Genet Metab 112:87–122
Cragan JD (2016) Surveillence for microcephaly. Centers for Disease Control and Prevention. https://www.cdc.gov/ncbddd/birthdefects/documents/surveillance-microcephaly-webinar-transcript.pdf. Accessed 21 January 2018
Hanley WB (2008) Finding the fertile woman with phenylketonuria. Eur J Obstet Gynecol Reprod Biol 137:131–135
Jacobs IJ, Que J (2013) Genetic and cellular mechanisms of the formation of esophageal atresia and tracheoesophageal fistula. Dis Esophagus 26:356–358
Koch R, Hanley W, Levy H, Matalon K, Matalon R, Rouse B, Trefz F, Guttler F, Azen C, Platt L et al (2003) The maternal phenylketonuria international study: 1984-2002. Pediatrics 112:1523–1529
Lenke RR, Levy HL (1980) Maternal phenylketonuria and hyperphenylalaninemia: an international survey of treated and untreated pregnancies. N Engl J Med 303:1202–1208
Levy HL (2003) Historical background for the maternal PKU syndrome. Pediatrics 112:1516–1518
Levy HL, Waisbren SE, Guttler F, Hanley WB, Matalon R, Rouse B, Trefz FK, de la Cruz F, Azen CG, Koch R (2003) Pregnancy experiences in the woman with mild hyperphenylalaninemia. Pediatrics 112:1548–1552
Maillot F, Cook P, Lilburn M, Lee PJ (2007) A practical approach to maternal phenylketonuria management. J Inherit Metab Dis 30:198–201
Matalon KM, Acosta PB, Azen C (2003) Role of nutrition in pregnancy with phenylketonuria and birth defects. Pediatrics 112:1534–1536
Matthews A, Dowswell T, Haas DM, Doyle M, O’Mathuna DP (2010) Interventions for nausea and vomiting in early pregnancy. Cochrane Database Syst Rev:CD007575
Ozalp I, Coskun T, Tokatli A, Kalkanoglu HS, Dursun A, Tokol S, Koksal G, Ozguc M, Kose R (2001) Newborn PKU screening in Turkey: at present and organization for future. Turk J Pediatr 43:97–101
Rouse B, Azen C, Koch R, Matalon R, Hanley W, de la Cruz F, Trefz F, Friedman E, Shifrin H (1997) Maternal Phenylketonuria Collaborative Study (MPKUCS) offspring: facial anomalies, malformations, and early neurological sequelae. Am J Med Genet 69:89–95
Teissier R, Nowak E, Assoun M, Mention K, Cano A, Fouilhoux A, Feillet F, Ogier H, Oger E, de Parscau L (2012) Maternal phenylketonuria: low phenylalaninemia might increase the risk of intra uterine growth retardation. J Inherit Metab Dis 35:993–999
van Wegberg AMJ, MacDonald A, Ahring K, Belanger-Quintana A, Blau N, Bosch AM, Burlina A, Campistol J, Feillet F, Gizewska M et al (2017) The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis 12:162–217
Vockley J, Andersson HC, Antshel KM, Braverman NE, Burton BK, Frazier DM, Mitchell J, Smith WE, Thompson BH, Berry SA (2014) Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med 16:188–200
Wiedemann A, Leheup B, Battaglia-Hsu SF, Jonveaux P, Jeannesson E, Feillet F (2013) Undiagnosed phenylketonuria in parents of phenylketonuric patients, is it worthwhile to be checked? Mol Genet Metab 110:S62–S65
Yano S, Moseley K, Bottiglieri T, Arning E, Azen C (2014) Maternal Phenylketonuria International Collaborative Study revisited: evaluation of maternal nutritional risk factors besides phenylalanine for fetal congenital heart defects. J Inherit Metab Dis 37:39–42
Yildiz Y, Dursun A, Tokatli A, Coskun T, Sivri S (2017) Partial hydatidiform mole in a phenylketonuria patient treated with sapropterin dihydrochloride. Gynecol Endocrinol 33:19–20
Acknowledgments
The authors would like to acknowledge İmran Özalp for establishing the first metabolic center and newborn screening in Turkey; metabolic physicians Turgay Coşkun, Ayşegül Tokatlı, Ali Dursun, Halil İbrahim Aydın, Mustafa Kılıç, Özlem Ünal, Burcu Öztürk Hişmi, Berrak Bilginer Gürbüz, and Emine Pektaş; metabolic dietitians Gülden Köksal, Hülya Gökmen-Özel, Sabriye Saruhan, and others for the diagnosis and management of the patients in this paper; Talat Demirsöz, Gökçen Düzgün Konuşkan, Yamaç Karaboncuk, Zeynep Tüzün, Berge Velibaşoğlu, and Tuğba Kaya for performing neuropsychometric testing; Esin Göksun and Rıza Köksal Özgül for the genetic analyses; Özgür Özyüncü for providing obstetric care; our laboratory staff; and our patients and their families. This work was prepared as a residency thesis in pediatrics.
Author information
Authors and Affiliations
Contributions
The study was conceptualized by HSS. Both authors (YY and HSS) contributed to study design and interpretation of data. YY was involved in acquisition and analysis data and writing the draft of the manuscript, which was critically edited by HSS. Both authors approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by Peter de Winter
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Online Resource 1
Detailed Characteristics of Children Born to Mothers with Phenylalanine Hydroxylase Deficiency. (PDF 527 kb)
Online Resource 2
Medians and interquartile ranges of blood Phe levels and weekly frequencies of Phe measurements in classical PKU pregnancies treated with Phe-restricted diet. (PDF 181 kb)
Online Resource 3
Blood Phe Measurements in Classical PKU Pregnancies Treated with Phe-Restricted Diet. Weeks 14 and 28 mark the start of second and third trimesters, respectively. Week 6 divides the first trimester into two parts. “0” marks the last menstrual period. The accepted upper limit of blood Phe level during pregnancy is 6 mg/dl. Blood Phe levels are higher and fluctuate more in especially the second part of the first trimester, after which Phe levels drop and remain stable for the most part. Phe: phenylalanine, PKU: phenylketonuria. (PDF 106 kb)
Rights and permissions
About this article

Cite this article
Yıldız, Y., Sivri, H.S. Maternal phenylketonuria in Turkey: outcomes of 71 pregnancies and issues in management. Eur J Pediatr 178, 1005–1011 (2019). https://doi.org/10.1007/s00431-019-03387-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-019-03387-8