
Abstract
Structure and integrity of the mitochondrial network play important roles in many cellular processes. Loss of integrity can lead to the activation of a variety of signalling pathways and affect the cell’s response to infections. The activation of such mitochondria-mediated cellular responses has implications for infection recognition, signal transduction and pathogen control. Although we have a basic understanding of mitochondrial factors such as mitochondrial DNA or RNA that may be involved in processes like pro-inflammatory signalling, the diverse roles of mitochondria in host defence remain unclear. Here we will first summarise the functions of mitochondria in the host cell and provide an overview of the major known mitochondrial stress responses. We will then present recent studies that have contributed to the understanding of the role of mitochondria in infectious diseases and highlight a number of recently investigated models of bacterial and viral infections.
Similar content being viewed by others

Introduction
Even though this is technically still only a hypothesis, it is generally assumed that mitochondria originated as endosymbionts, when an archaeon engulfed a bacterium to be able to develop into a eukaryote [1]. Even if that was not to be the case, mitochondria certainly act as endosymbionts. They form a self-organized, replicating network in the cytosol, they assume responsibility for efficient energy generation and through their metabolism determine a cell’s metabolism, reaction, and behaviour, through their gatekeeping of apoptosis, mitochondria even control death and survival of their host. Given this complex interaction, it may be little surprise that mitochondria can also exert some control over the pro-inflammatory activity of the cell. Probably all cells of the human body can start inflammation, a complex process determining the organism’s response to infection and other types of damage. In addition to ‘cell-autonomous immunity’, the ability of a cell to contain infectious agents, all cells can alert professional immune cells to help combat the invading pathogen. We will here briefly recapitulate the mitochondrial functions relevant to host defence before focussing on their specific inflammatory functions. We believe that these functions in immune defence are one of the features that mitochondria contribute to the defence of a cell, and ultimately the organism, against infection.
Mitochondria—function and organization
The highly dynamic mitochondrial network provides a large intracellular surface, and the mitochondrial proteome encompasses about 1500 proteins in humans (most of them encoded in the nucleus) [2]. Given the massive interface of the mitochondrial network, it may be unsurprising that mitochondria can act as a platform for cellular recognition of intracellular stressors and threats.
Mitochondria are highly structured organelles that have an inner and an outer membrane. The inner membrane is folded and forms the so-called cristae and crista junctions [3]. This structure is important for the export and import of proteins through the inner mitochondrial membrane. Crista junctions, as contact sites of the two mitochondrial membranes, require a highly organised arrangement, the mitochondrial contact site and cristae organising system (MICOS), for their formation and stability (Fig. 1A). MICOS is a protein complex consisting of the Mic60 subcomplex (Mic60 and Mic19) and the Mic10 subcomplex (Mic10, Mic12, Mic26 and Mic27). Within the cristae the main components of the machinery of the mitochondrial oxidative phosphorylation are located and therefore this structure is essential for energy production in cells [4,5,6]. This machinery is built by the electron transport chain, containing 4 different protein complexes and a ATP synthase, also called complex V [7]. The complex I, a NADH:ubiquinone oxidoreductase, reduces quinone by using NADH [8]. The essential NADH for reactions is generated during the chemical reaction cycle called the Krebs cycle [9]. The complex II is a succinate dehydrogenase, and oxidates succinate to fumarate by reducing ubiquinone, also called coenzyme Q, to ubiquinol. [9, 10]. The complex III, a cytochrome bc1 oxidoreductase, uses the ubiquinol produced by complex II to reduce cytochrome c, also called the Q-cycle [11, 12]. The created cytochrome c becomes oxidated by the complex IV, that is a cytochrome c oxidase, thereby the complex IV reduces oxygen to water [13]. The complex I, III and IV in addition serve as proton transporter via the inner membrane of mitochondria, resulting in a proton gradient that is used by the ATP synthase to generate ATP out of ADP and phosphate [14,15,16,17,18] (Fig. 1B).

Mitochondrial organisation and mitochondrial oxidative phosphorylation. A The structure of mitochondria is characteristic, with their two membranes and the folds of their inner membrane, called cristae [3]. Cristae junctions, the contact sites of the two mitochondrial membranes are required for cristae formation. Here, the mitochondrial cristae organising system (MICOS) is required, a protein complex stabilizing the interaction between the membranes. MICOS is containing two large subcomplexes, the Mic60 subcomplex (Mic60 and Mic19) and the Mic10 subcomplex (Mic10, Mic12, Mic26 and Mic27) [4,5,6]. B The mitochondrial oxidative phosphorylation machinery is located within the cristae and is essential for mitochondrial energy production. This machinery contains 4 different protein complexes and an ATP synthase. Complex I reduces quinone using NADH, which origins form the Krebs cycle (metabolic reaction cycle that starts with acetyl-CoA derived from the degradation of glucose in the mitochondrial matrix). Complex II oxidates succinate to fumarate by reducing ubiquinone, also called coenzyme Q (Q), to ubiquinol. Here the succinate is a product coming from the Krebs cycle [9, 10]. Complex III uses the ubiquinol produced by complex II to reduce cytochrome c (CytC) [11, 12]. Complex IV reduces oxygen to water and oxidates cytochrome c [13]. Complex I, III and IV transport protons over the inner mitochondrial membrane and generate a proton gradient [14,15,16,17]. The proton gradient is used by an ATP synthase to generate ATP from ADP and phosphate [18]
The mitochondrial network is a highly dynamic structure within cells and is the product of fission into and fusion of individual mitochondria.
Mitochondrial fission is mainly regulated and initiated by dynamin-related protein 1 (Drp1) [19, 20]. Mitochondrial fission is characterised by the formation of Drp1 spirals around the outer mitochondrial membrane, followed by the processing of GTP, which allows a conformational change of Drp1 that results in the separation of individual mitochondria from the network (Fig. 2A). Mitochondrial fission and fusion are separate cellular processes, each regulated by several proteins. Fission is achieved by post-translational modifications of Drp1, and depending on the phosphorylation site, recruitment of Drp1 to the mitochondrial membrane is initiated or blocked [21,22,23]. Phosphorylation by the extracellular signal-regulated kinases (ERK) of Drp1 for example, results in Drp1 activation and binding to the mitochondria, followed by fission [23], while phosphorylation of Drp1 by protein kinase A (PKA) results in the inhibition of mitochondrial fission [21]. Mitochondrial fusion of the outer membrane is initiated by the interaction of the membrane protein mitofusin 1 or 2 (Mfn1; Mfn2) on two individual juxtaposed mitochondria [24, 25]. The regulation of mitochondrial fusion is not entirely clear, but it has been shown that phosphorylation of Mfn1 by beta-II protein kinase C (βIIPKC) inhibits the process [26]. Once the outer membrane is fused, fusion of the inner membrane takes place, initiated by optic atrophy 1 (OPA1) [6, 27] (Fig. 2B).

Mitochondrial fission and fusion. A The main factor of mitochondrial fission is dynamin-related protein 1 (Drp1), which forms spirals around the outer mitochondrial membrane [19, 20]. By changing the protein conformation, initiated by its GTPase activity, Drp1 clamps off mitochondria. B Mitochondrial fusion is a two-step process. First, the outer membrane fuses through the interaction of the membrane protein mitofusin 1 or 2 (Mfn1; Mfn2) [24, 25]. Then inner membrane fusion takes place initiated by optic atrophy 1 (OPA1) [6, 27]
Mitochondrial morphology is regulated at a number of levels and has significant effects on cellular functions and signal transduction in a cell. Mitochondrial fragmentation, for example, the result of a relative increase in fission and reduction in fusion, changes the network to more small, individual mitochondria, while dominance of fusion generates large, continuous networks. Mitochondrial fragmentation can lead to loss of mitochondrial network integrity and has been shown to impair Ca2+-induced cell death signalling, and intramitochondrial Ca2+ concentration has been shown to affect autophagy [28,29,30].
The uptake of Ca2+ from the endoplasmic reticulum (ER) is regulated at a contact site between mitochondria and ER [30]. Ca2+ is known to regulate fission and fusion. High cytosolic Ca2+ concentration has been associated with mitochondrial fission triggered by dephosphorylation of Drp1, and elevated intramitochondrial Ca2+ levels appear to be involved in blocking the mitochondrial inner membrane fusion apparatus [31, 32]. Infection processes have frequently been described as stimuli for mitochondrial morphological changes. Infection with Vibrio cholerae, for example, leads to mitochondrial fragmentation triggered by the secreted factor VopE, which targets mitochondria [33]. In Helicobacter pylori infections, vacuolating cytotoxin A (VacA) has been shown to induce mitochondrial fragmentation by activating Drp1 [34]. Here, the mitochondrial fission machinery is used by the toxin expressed by the pathogen to induce fragmentation of the mitochondrial network and subsequent cell death [34]. In addition to destruction of the mitochondrial network, bacterial infections can also lead to mitochondrial elongation, as shown by an infection with Chlamydia trachomatis in which DRP1 levels are downregulated by inducing the expression of a host cell microRNA (miR-30c-5p) [35].
Mitochondrial stress responses
In addition to the essential role of energy generation through oxidative phosphorylation in the mitochondria, some mitochondrial components also have functions in other pathways such as cell death. One example is the role of cytochrome c in mitochondrial apoptosis [36, 37]. During intracellular stress, mitochondrial apoptosis can be activated by the down-regulation of anti-apoptotic members, and the activation of pro-apoptotic members of the Bcl-2 family [38, 39]. The Bcl-2 family plays a crucial role in the regulation of mitochondrial apoptosis. For example, protein synthesis shutdown can cause the loss of MCL-1, a short-lived anti-apoptotic member of the Bcl-2-family, and the loss of MCL-1 enables the activation of mitochondrial apoptosis [39].
The two Bcl-2-family members Bax and Bak are essential for the activation of mitochondrial apoptosis as they function as pore-forming proteins in the outer mitochondrial membrane, permeabilizing the outer mitochondrial membrane (MOMP) [40, 41]. This key event in mitochondrial apoptosis allows the release of cytochrome c into the cytosol, followed by the formation of the apoptosome, a protein complex consisting of APAF-1, cytochrome c and pro-caspase-9 [42, 43]. In the apoptosome, caspase-9 is activated and sets in motion the downstream proteolytic events, especially through caspase-3 [37] (Fig. 3A).

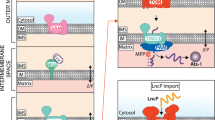
Mitochondrial stress responses. A Mitochondrial apoptosis is linked to intracellular stress. Mitochondrial outer membrane permeabilization (MOMP) is a key event, generating Bax/Bak pores [40, 41]. The permeabilization allows the release of cytochrome c into the cytosol. Cytochrome c together with Apaf-1 and pro-caspase-9 generates the ‘apoptosome’, whose activity activates caspase-9 [42, 43]. Caspase-9 in turn activates the effector caspase-3 [37]. Several reports suggest that mitochondrial DNA (mtDNA) can be released into the cytosol, resulting in activation of the cGAS/STING pathway and cell activation [95,96,97,98,99,100, 105, 109]. B MAVS signalling is an important intracellular pathway, which can originate from the recognition of cytosolic RNA, for example viral RNA. The RNA is recognised by the cytosolic sensors Rig-1 or MDA5 [44,45,46,47,48]. These RNA-receptors bind to the mitochondrial membrane protein MAVS, initiating signalling to activate NF-κB or IRF3 and cell activation (cytokine / interferon production) [44]. C UPRam is activated by protein mistargeting and the accumulation of proteins in the cytosol of the cell. This may be the result of failed import into the mitochondria or retro-translocation, followed by targeting of accumulated proteins for proteasomal degradation [49, 50]. D UPRmt is a reaction to the accumulation of misfolded protein in the mitochondrial matrix. Misfolded proteins are cleaved by the protease CLPP inside mitochondria and transported to the cytosol. Here protein fragments activate the transcription factor ATF5, resulting in transcriptional activation of anti-apoptotic genes (such as Mcl-1 and Bcl-2) and genes related to mitochondrial proteostasis [51,52,53,54,55,56,57]
The mitochondrial apoptotic pathway is an example of mitochondria playing a role in cell signalling in response to cytosolic stress. Another example of mitochondria's ability to recognise and transmit signals is the antiviral response, which relies on the mitochondrial membrane protein MAVS [44]. Viral cytosolic RNA can be recognised by Rig-1 or MDA5, two helicases belonging to the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) [45,46,47,48]. These bind to the ‘adapter’ MAVS, whose aggregation leads to a downstream activation of NF-κB and the production of proinflammatory cytokines, as well as the phosphorylation of IRF3 and interferon production [44] (Fig. 3B).
Mitochondria can also respond to the accumulation of misfolded proteins, either in the cytosol (unfolded protein response, activated by protein mistargeting (UPRam)) or in the mitochondrial matrix (mitochondrial unfolded protein response (UPRmt)) [49, 50]. UPRam is characterised by the fact that accumulated proteins, which have either resulted from failed import into the mitochondria or retro-translocation, are fed to the proteasome for degradation (Fig. 3C). In contrast to UPRam, UPRmt recognises the accumulation of proteins within the mitochondria and activates a stress response. In this process, the misfolded proteins are cleaved by the protease CLPP and exported to the cytosol, where the transcription factor ATF5 gets activated [51,52,53,54], followed by the transcriptional activation of anti-apoptotic genes (such as Mcl-1 and Bcl-2) and genes related to mitochondrial proteostasis [55,56,57] (Fig. 3D).
Mitochondria and infection
Mitochondrial structure and dynamics during infection
Mitochondria are increasingly found to play a role in cell-autonomous immunity. Over time, the interaction between infection dynamics and the integrity of the mitochondrial network has been described for many different infection models. During infections with intracellular or extracellular bacterial pathogens mitochondrial fragmentation has been observed (infections with Shigella, Listeria or Mycobacteria); enhanced fusion has also been observed (Chlamydia) (reviewed in [58, 59]). In the case of viral infections, fragmentation as well as hyper-fusion have been described. In the case of SARS-CoV2- or HIV infection for instance, enhanced fusion has been reported, while for influenza virus or hepatitis B and C viruses mitochondrial fission dominates [59]. In addition, mitochondrial fragmentation has been reported during infection with the parasite Toxoplasma gondii [60].
Furthermore, the differences in the morphology of the mitochondrial network effects the energy generation, but can be in addition a marker for different cell types [59]. It appears that anti-inflammatory macrophages or dendritic cells mainly have elongated mitochondria, while the mitochondrial network of pro-inflammatory macrophages or activated B cells appears to be more on the fragmented side [59]. However, the precise role and effect of the change of the mitochondrial network and the outcome for infection dynamics seem still to be unclear and diverse.
One example is the case of influenza virus infection models: while influenza virus has been more commonly reported to induce mitochondrial fragmentation, a new study has shown that it has, in addition, the ability to induce mitochondrial elongation by blocking the recruitment of the mitochondrial fission protein Drp1 to mitochondria [61]. Interestingly, the pharmacological induction of mitochondrial fragmentation during infection reduced the viral burden, indicating the importance of mitochondrial elongation for viral replication. The importance of this observation in vivo has to be confirmed; however, these results show the complexity in this field and suggest functional relevance [61].
It has been found that the activity of the pore-forming toxin listeriolysin O (LLO) of the intracellular bacterium Listeria monocytogenes [62] leads to fragmentation of the mitochondrial network [63, 64]. L. monocytogenes also caused an increase in Mic10, a MICOS-component [3, 65], in isolated mitochondria. Mic10 deficiency inhibited mitochondrial fragmentation during Listeria infection, while overexpression of Mic10 induced mitochondrial fragmentation independent of infection [65]. It appears that the process of mitochondrial fragmentation supports the infection of epithelial cells with L. monocytogenes in epithelial cells. Mic10-deficient cells showed a decrease in intracellular bacterial numbers; in contrast, infection dynamics were faster upon Mic10 overexpression. Interestingly, ATP levels did not change in Mic10 deficiency, indicating that the reducing effect on bacterial intracellular growth is not related to decreased cellular energy availability. The clinical importance of these results remains unclear, but they identify a new potential mechanism for the control of L. monocytogenes over the host cell.
Mitochondrial metabolism during infection
Cellular energy can be obtained via two main pathways. First, cells can generate energy through glycolysis and lactic acid fermentation, and secondly through mitochondrial respiration. Cells can adjust their utilization of either pathway; most famous is the ‘Warburg effect’ of cancer cells switching to glycolysis [66]. During stimulation of macrophages with for example lipopolysaccharides (LPS), and presumably during infection, macrophages also transform their metabolically activity, and one well-known adjustment is the up- or down-regulation of tricarboxylic acid (TCA) cycle and glycolysis that is associated with the ‘M1’ (pro-inflammatory) vs. ‘M2’(anti-inflammatory) macrophage phenotype [67, 68]. Such changes in mitochondrial and cellular metabolism are likely to impact on many structural and signalling pathways. Some studies have started probing these effects on infections with individual pathogens. A recent study has shown that cells that use mitochondrial respiration for energy production have limited recycling of plasma membrane proteins that may serve as pathogen receptors [69]. Under conditions of high mitochondrial respiration, receptors (the transferrin receptor was measured) accumulated intracellularly, resulting in a reduced presentation of receptors at the surface. As a result, the bacterial invasion may be reduced due to the reduced number of receptors, which translated into a lower intracellular bacterial load during Listeria-infection. Interestingly, deletion of SURF1, an assembly protein of complex IV of the electron transport chain, led to increased intracellular bacterial load. It has previously been shown that deletion of SURF1 in primary fibroblasts is a stimulus for the mitochondrial unfolded protein response (UPRmt), and SURF1−/− mice show increased numbers of mitochondria and UPRmt [70, 71], but also increased maximal mitochondrial respiration [70]. In contrast to these findings, deficiency of SURF1 in epithelial cells results in decreased maximal respiration [69]. These findings establish a link between mitochondrial metabolism and bacterial infection, suggesting that mitochondrial metabolism for energy production has a direct impact on infection success in epithelial cells.
In Mycobacterium tuberculosis infections of mouse bone marrow-derived macrophages (BMDM), a different effect has been observed. A recent study reported that the absence of the Parkinson's disease-associated gene Leucine-Rich Repeat Kinase 2 (LRRK2) leads to an increase in bacterial load [72]. Previously, mutated LRRK2 has been linked to mitochondrial dysfunctions like changes in mitochondrial network morphology and sensitivity to oxidative stress but also to defects in mitophagy [73]. The recent study interestingly shows, that glycolysis in LRRK2-deficient macrophages was reduced, as was mitochondrial respiration [72]. LRRK2 partakes in a variety of cellular processes, but mutations in LRRK2 also cause mitochondrial dysfunction and have been described as a factor in the development of Parkinson's disease [73, 74]. LRRK2-deficient mice infected with M. tuberculosis showed the same bacterial load as controls, while isolated and infected BMDMs under LRRK2-deficient conditions showed increased intracellular replication of bacteria [72]. However, increased inflammation was observed at the site of infection in LRRK2-deficient mice. Furthermore, loss of LRRK2 in macrophages increased the levels of type I interferon (IFN) and interferon-stimulated genes (ISG) in the uninfected state, but the cells were unable to upregulate IFN and ISG expression further after infection, resulting in a less effective IFN response to mycobacterial pathogens and to cytosolic nucleic acids. The most recent study found that cell activation in LRRK2-deficient cells under non-infected conditions was linked to mitochondrial fragmentation leading to the release of mitochondrial DNA into the cytosol and activation of the IFN response via cGAS [72]. The presented findings indicate that the integrity of the mitochondrial network is essential for regulating cell-autonomous immunity and followed inflammation and bacterial control. Therefore, these data represent a link between the LKKR2-mitochondria axis to cellular defence against bacteria.
Infections with Zika virus, a virus of the Flaviviridae family that can infect cells in the cerebral cortex but also astrocytes and replicates in the endoplasmic reticulum [75], can cause an imbalance of reactive oxygen species and defects in mitochondrial respiration and ATP synthesis [76]. Recent analyses of mitochondrial ATP synthesis by oxidative phosphorylation during Zika virus infections of astrocytes showed no difference in basal respiration at the early stages of infection, but at later time points basal respiration decreased, possibly indicating mitochondrial damage. It has already been shown that infections with hepatitis C virus (HCV), which belongs to the same virus family, can lead to Ca2+ release from the endoplasmic reticulum, which can represent a signal for mitochondria to increase ROS production [77]. These early studies suggest that mitochondrial metabolism will affect infections with different pathogens in a multitude of ways, and probably in different ways depending on the type of the infected host cell.
Apart from such direct effects on intracellular replication, changes to the mitochondrial metabolism will also affect inflammation in a non-cell-autonomous manner. In the report quoted above, ZIKA virus infection led to reactive astrogliosis, a situation in which astrocytes remodel their gene expression and morphology in response to stressors [76, 78]. Reactive astrocytes are an important component of the innate immune response of the central nervous system and can respond to a variety of different stressors such as various cytokines, damage-associated molecular patterns (DAMP) and pathogen-associated molecular patterns (PAMP) [79].
It is at least conceivable that the previously described increase in proinflammatory cytokines in infected astrocytes [80] is a consequence of mitochondrial dysfunction and activation of the immune response leading to differentiation into reactive astrocytes. Because mitochondrial metabolism can determine the secretion of inflammatory products, the response to infection is thus likely to affect inflammation and immune response beyond the infected cell.
Mitochondria and cell death pathways during infection
The function of mitochondria in apoptosis is very well established. Mitochondria host in their outer membrane the Bcl-2-protein family, whose function is to organize the release of the intermembrane space proteins cytochrome c and Smac, which in the cytosol activate caspases and initiate apoptosis. Much is known about this regulation, and the reader is referred to specialist articles on this aspect [39, 81]. In a summary, the interplay of the many members of Bcl-2-family proteins controls the activation of the mitochondrial effectors Bax and Bak, whose activity causes the release of the intermembrane space proteins, cytochrome c and Smac.
Beta-barrel proteins in the regulation of mitochondrial apoptosis events
Although the Bcl-2-family can regulate membrane permeabilization in the absence of all other proteins, as shown in synthetic membranes, in intact cells they are controlled by the mitochondrial porin VDAC2 [82]. This is of particular interest in infections with Gram-negative bacteria. These bacteria and mitochondria very likely have a common evolutionary origin, and they certainly share the features of two membranes and β-barrel-proteins that regulate flux over the outer membrane, known as porins. At least one porin, PorB from Neisseria gonorrhoeae can have pro-apoptotic activity [83]. It is a relatively new and largely unexplained finding that small vesicles appear to be able to traffic through mammalian cells and for instance fuse with intracellular organelles [84,85,86]. Outer membrane vesicles (OMVs) are constantly generated by gram-negative bacteria, and OMVs appear to be able to enter mammalian cells and deliver cargo to various subcellular locations [85, 86]. In macrophage-pathogen interaction models, OMVs from N. gonorrhoeae, uropathogenic E. coli (UPEC) and Pseudomonas aeruginosa have been shown to be able to induce mitochondrial apoptosis and NLRP3 inflammasome activation [87, 88].
A sensing, or sentinel function of mitochondria
The pro-apoptotic effects of infection have been illustrated in many situations, and at least most cases involve mitochondrial apoptosis. This may illustrate that mitochondria have a sentinel function: the recognition of pathogens can lead to the activation of the mitochondrial apoptosis pathway. How these signals are conveyed to mitochondria is unclear in most cases – as indeed is the signalling upstream of mitochondria in many cases. Candidates are pattern recognition receptors, which in experimental situations can induce apoptosis [89,90,91]: sensing of microbes by these receptors can signal to activate the sentinels and activate sub-lethal apoptosis signals at mitochondria. The importance of this defence mechanism is suggested by the multiple identifications of apoptosis-inhibitors in viruses [92]. An intriguing example is found in the infection with the obligate intracellular Chlamydiae. Bacteria of the Chlamydiaceae, most notably Chlamydia trachomatis, infect human cells. Chlamydia has a very strong anti-apoptotic activity [93]. A related bacterium, Parachlamydia acanthamoebae, a symbiont of free-living amoebae, cannot grow in human cells. Instead, P. acanthamoeba induces apoptosis when infecting the epithelial cells site [94]. However, under conditions where mitochondrial apoptosis is experimentally blocked, P. acanthamoeba can replicate intracellularly in human epithelial cells, suggesting that mitochondrial apoptosis not only prevents infection but also plays a role in intracellular inhibition of bacterial replication.
More recently, a pro-inflammatory function of mitochondrial apoptosis has been suggested. This is a novel concept: traditionally, apoptosis has been seen as non-stimulatory cell death, and in most situations, it will be just that. However, when mitochondrial apoptosis is induced and the cytosolic effectors of apoptosis, the caspase proteases, are inhibited, the cell triggered to undergo apoptosis secretes pro-inflammatory mediators [95,96,97]. This is curious: there are no cells known not to express caspases, so it may well be asked when such a potential pro-inflammatory function of the apoptosis pathway may be called into action. We hypothesize that the function of this system—i.e., pro-inflammatory signalling of mitochondria—in fact lies in a process we will refer to as sub-lethal signalling in the apoptosis system.
Mitochondria, sub-lethal apoptosis, and inflammation
For a long time, the field had assumed that there was a comprehensive step of mitochondrial outer membrane permeabilization (MOMP), which defined the point of no return in apoptotic cell death. Retrospectively, earlier work can be identified showing that this is not the case; the cell-biologically clearest study was a publication in 2015, which identified ‘minority MOMP’, a process where only parts of the mitochondrial network became permeabilized and the signalling remained sub-lethal [98]. We have found that sub-lethal signals are regularly generated during the infection of human cells with any of the tested bacterial, viral or protozoan pathogens and have reported that these mitochondrial signals contribute to chemokine/cytokine-secretion [99]. The precise nature of the pro-inflammatory signal is unclear: candidates are mtDNA or RNA, the intermembrane space protein Smac (which is known to activate alternative NF-κB) and perhaps even caspases themselves. One candidate signalling pathway involves signalling through STING: the induction of sub-lethal apoptosis signals led to cytokine-secretion that depended on STING [99]. STING may be activated through mtDNA-recognition by cGAS [95, 99]. mtDNA can also trigger the activation of the NLRP3 inflammasome [100] although this pathway is more likely to be active in myeloid cells.
Consequences of sub-lethal signals for control of microbes and inflamamtion
The consequences of mitochondrial apoptosis apparatus-dependent signals in cell activation and cytokine secretion, for infection dynamics and pathogen clearance need to be investigated in the future. In two examples of bacterial infections, the lack of a functional mitochondrial apoptosis pathway led to defects in intracellular bacterial control, as shown by enhanced intracellular growth of C. trachomatis and Salmonella Typhimurium [99].
The importance of the mitochondrial apoptotic apparatus in the activation of pro-inflammatory cellular responses was recently also found in macrophage infection with severe fever with thrombocytopenia virus (SFTSV), a tick-borne virus (RNA virus) [101,102,103]. Here, activation of Bax/Bak was shown to be essential for the release of mtDNA into the cytosol during infection, followed by the formation of the NLRP3 inflammasome and activation of IL-1β secretion [104]. Comparative transcriptomic and proteomic analysis revealed activation of inflammatory pathways and mitochondrial damage. Caspase-1-dependent secretion of IL-1β was reported, based on activation of the MyD88/NF-κB axis during SFTSV infection. The authors hypothesised that recognition of mtDNA leads to the expression of pro-inflammatory cytokines. A recent study also indicated that the release of mitochondrial DNA and its detection by the cGAS/STING pathway plays a role in SARS-CoV-2 detection in IFN-signalling [105]. It appeared that treatment of infected mice with a STING-inhibitor can reduce inflammatory response and results in increased survival of the infected mice. Interestingly, the expression of Bak in SFTSV-infected patients corresponded with the viral load in the patients' serum [104], and infection experiments with mice suggested that inhibition of caspase-1 activity can lead to a better outcome for the infected individuals. The results suggest that Bax/Bak pore formation during activation of mitochondrial apoptosis is important for inflammasome-activation, which leads to a pro-inflammatory response. Inhibition of this process increased the survival rate of infected mice, suggesting that down-regulation of mitochondrial apoptosis may be one way of future treatments of infectious diseases.
Besides the previously described intracellular DNA recognition machinery of cGAS/STING, the cytosolic RNA recognition machinery of helicases, signalling through MAVS contributes to cellular immunity [106, 107], and the reciprocal effect—i.e. inflammatory signals acting on mitochondrial physiology—has been described in this axis. In epithelial cell infections with measles virus (MeV), a negative-sense single-stranded RNA (-ssRNA) virus, recent findings showed that mitochondrial biogenesis is downregulated and mitochondrial elongation can be observed [108]. In addition, the release of mtDNA into the cytosol has been detected, and activation of the cGAS/STING axis led to an antiviral response. MAVS-deficient cells as well as cGAS-deficient cells show a delayed IFN-β response upon infection. Interestingly, in infections with vesicular stomatitis virus (VSV, an RNA virus), reduced IFN-β mRNA expression was not observed in the absence of cGAS, but MAVS was essential for the viral response. Consistent with this, depletion of mtDNA had no effect on IFN-β mRNA expression of cells infected with VSV. In vivo studies found that the presence of MAVS and cGAS contributed to the survival of mice after MeV infection. Increased viral load was observed in cGAS deficiency, suggesting that viral RNA and mtDNA recognition supports the antiviral response and overall outcome of infection dynamics. The effect of mtDNA recognition and the impact on IFN-β response does not only apply to RNA viruses. The expression of IFN-β mRNA was also reduced in infections with the poxvirus Modified Vaccinia Virus Ankara (MVA), a DNA virus, when mitochondrial apoptosis was disabled. The effect of mtDNA in the activation of cells and immune responses during viral infections has already been reported by the demonstration that during infections with MVA, the induced IL-6 secretion was reduced under mtDNA-depleted conditions [99].
These results highlight the complexity of the pathways that can be activated, and how the interplay between them can lead to inflammatory responses. Reprogramming these signalling pathways to control infection may be a future goal.
Perspectives
The interaction between pathogens and the mitochondrial network appears to be multifaceted, and its outcome in the dynamics of the infection is often difficult to predict. What is clear is that the integrity or disruption of mitochondria plays an important role in immunity and the cell-autonomous responses to pathogens. Morphological changes in the mitochondrial network in a cell can be associated with alterations in energy metabolism or cell activation, as illustrated by the appearance of elongated mitochondria in anti-inflammatory macrophages, and fragmented mitochondria in pro-inflammatory macrophages. The roles of mitochondria in mammalian cells are manifold, and the question of how the mitochondrial network orchestrates the activation of different cell-autonomous signalling pathways is of great interest to understand their role in immunity against infectious diseases.
How mitochondrial morphology relates to mitochondrial function, in what way it drives cellular immunity, or how mitochondrial protein synthesis and stress responses influence immunity to pathogens, are all open questions. Recent studies have shown that mitochondrial respiration or glycolysis affects host–pathogen interactions. We need to understand what impact this has on the infected cell, on the organism and to what extent these results are pathogen-specific. Studies comparing the role of mitochondria in different cell types during infection are important to understand the whole picture and regulating/manipulating mitochondria-dependent regulation of immunity could lead to new treatments of infectious disease.
References
Roger AJ, Muñoz-Gómez SA, Kamikawa R (2017) The origin and diversification of mitochondria. Curr Biol 27:R1177–R1192. https://doi.org/10.1016/j.cub.2017.09.015
Song J, Herrmann JM, Becker T (2021) Quality control of the mitochondrial proteome. Nat Rev Mol Cell Biol 22:54–70. https://doi.org/10.1038/s41580-020-00300-2
Rampelt H, Zerbes RM, van der Laan M, Pfanner N (2017) Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim Biophys Acta Mol Cell Res 1864:737–746. https://doi.org/10.1016/j.bbamcr.2016.05.020
Davies KM, Strauss M, Daum B et al (2011) Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci USA 108:14121–14126. https://doi.org/10.1073/pnas.1103621108
Mühleip AW, Joos F, Wigge C et al (2016) Helical arrays of U-shaped ATP synthase dimers form tubular cristae in ciliate mitochondria. Proc Natl Acad Sci USA 113:8442–8447. https://doi.org/10.1073/pnas.1525430113
Cogliati S, Frezza C, Soriano ME et al (2013) Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155:160–171. https://doi.org/10.1016/j.cell.2013.08.032
Guo R, Gu J, Zong S et al (2018) Structure and mechanism of mitochondrial electron transport chain. Biomed J 41:9–20. https://doi.org/10.1016/j.bj.2017.12.001
Leif H, Sled VD, Ohnishi T et al (1995) Isolation and characterization of the proton-translocating NADH:ubiquinone oxidoreductase from Escherichia coli. Eur J Biochem 230:538–548. https://doi.org/10.1111/j.1432-1033.1995.tb20594.x
Martínez-Reyes I, Chandel NS (2020) Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun 11:1–11. https://doi.org/10.1038/s41467-019-13668-3
Hägerhäll C (1997) Succinate: quinone oxidoreductases. Variations on a conserved theme. Biochim Biophys Acta Bioenerg 1320:107–141. https://doi.org/10.1016/S0005-2728(97)00019-4
Xia D, Esser L, Tang W-K et al (2013) Structural analysis of cytochrome bc1 complexes: implications to the mechanism of function. Biochim Biophys Acta 1827:1278–1294. https://doi.org/10.1016/j.bbabio.2012.11.008
Alcázar-Fabra M, Navas P, Brea-Calvo G (2016) Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim Biophys Acta Bioenerg 1857:1073–1078. https://doi.org/10.1016/j.bbabio.2016.03.010
Konstantinov AA (2012) Cytochrome c oxidase: intermediates of the catalytic cycle and their energy-coupled interconversion. FEBS Lett 586:630–639. https://doi.org/10.1016/j.febslet.2011.08.037
Kampjut D, Sazanov LA (2020) The coupling mechanism of mammalian respiratory complex. Science. https://doi.org/10.1126/SCIENCE.ABC4209
Mitchell P (1975) Protonmotive redox mechanism of the cytochrome b-c1 complex in the respiratory chain: protonmotive ubiquinone cycle. FEBS Lett 56(1):1–6. https://doi.org/10.1016/0014-5793(75)80098-6
Jones AJY, Blaza JN, Varghese F, Hirst J (2017) Respiratory complex i in Bos taurus and Paracoccus denitrificans pumps four protons across the membrane for every NADH oxidized. J Biol Chem 292:4987–4995. https://doi.org/10.1074/jbc.M116.771899
Vilhjálmsdóttir J, Albertsson I, Blomberg MRA et al (2020) Proton transfer in uncoupled variants of cytochrome c oxidase. FEBS Lett 594:813–822. https://doi.org/10.1002/1873-3468.13679
Klusch N, Murphy BJ, Mills DJ et al (2017) Structural basis of proton translocation and force generation in mitochondrial ATP synthase. Elife 6:1–16. https://doi.org/10.7554/eLife.33274
Smirnova E, Griparic L, Shurland DL, Van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–2256. https://doi.org/10.1091/mbc.12.8.2245
Otera H, Mihara K (2011) Discovery of the membrane receptor for mitochondrial fission GTPase Drp1. Small GTPases 2:167–172. https://doi.org/10.4161/sgtp.2.3.16486
Chang CR, Blackstone C (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282:21583–21587. https://doi.org/10.1074/jbc.C700083200
Cribbs JT, Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8:939–944. https://doi.org/10.1038/sj.embor.7401062
Kashatus JA, Nascimento A, Myers LJ et al (2015) Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell 57:537–551. https://doi.org/10.1016/j.molcel.2015.01.002
Chen H, Detmer SA, Ewald AJ et al (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200. https://doi.org/10.1083/jcb.200211046
Pagliuso A, Cossart P, Stavru F (2018) The ever-growing complexity of the mitochondrial fission machinery. Cell Mol Life Sci 75:355–374. https://doi.org/10.1007/s00018-017-2603-0
Ferreira JCB, Campos JC, Qvit N et al (2019) A selective inhibitor of mitofusin 1-βIIPKC association improves heart failure outcome in rats. Nat Commun 10:329. https://doi.org/10.1038/s41467-018-08276-6
Cipolat S, De Brito OM, Dal Zilio B, Scorrano L (2004) OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA 101:15927–15932. https://doi.org/10.1073/pnas.0407043101
Szabadkai G, Simoni AM, Chami M et al (2004) Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell 16:59–68. https://doi.org/10.1016/j.molcel.2004.09.026
Decuypere JP, Bultynck G, Parys JB (2011) A dual role for Ca2+ in autophagy regulation. Cell Calcium 50:242–250. https://doi.org/10.1016/j.ceca.2011.04.001
Rizzuto R, De Stefani D, Raffaello A, Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13:566–578. https://doi.org/10.1038/nrm3412
Cereghetti GM, Stangherlin A, Martins De Brito O et al (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 105:15803–15808. https://doi.org/10.1073/pnas.0808249105
Ishihara N, Maeda M, Ban T, Mihara K (2017) Cell-free mitochondrial fusion assay detected by specific protease reaction revealed Ca2+ as regulator of mitofusin-dependent mitochondrial fusion. J Biochem 162:287–294. https://doi.org/10.1093/jb/mvx029
Suzuki M, Danilchanka O, Mekalanos JJ (2014) Vibrio cholerae T3SS effector VopE modulates mitochondrial dynamics and innate immune signaling by targeting Miro GTPases. Cell Host Microbe 16:581–591. https://doi.org/10.1016/j.chom.2014.09.015
Jain P, Luo ZQ, Blanke SR (2011) Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc Natl Acad Sci USA 108:16032–16037. https://doi.org/10.1073/pnas.1105175108
Chowdhury SR, Reimer A, Sharan M et al (2017) Chlamydia preserves the mitochondrial network necessary for replication via microRNA-dependent inhibition of fission. J Cell Biol 216:1071–1089. https://doi.org/10.1083/jcb.201608063
Zhou M, Li Y, Hu Q et al (2015) Atomic structure of the apoptosome: mechanism of cytochrome c- and dATP-mediated activation of Apaf-1. Genes Dev 29:2349–2361. https://doi.org/10.1101/gad.272278.115
Li P, Nijhawan D, Budihardjo I et al (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91:479–489. https://doi.org/10.1016/S0092-8674(00)80434-1
Norbury CJ, Zhivotovsky B (2004) DNA damage-induced apoptosis. Oncogene 23:2797–2808. https://doi.org/10.1038/sj.onc.1207532
Chipuk JE, Moldoveanu T, Llambi F et al (2010) The BCL-2 family reunion. Mol Cell 37:299–310
Dewson G, Kluck RM (2009) Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci 122:2801–2808. https://doi.org/10.1242/jcs.038166
Kalkavan H, Green DR (2018) MOMP, cell suicide as a BCL-2 family business. Cell Death Differ 25:46–55. https://doi.org/10.1038/cdd.2017.179
Bao Q, Shi Y (2007) Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ 14:56–65. https://doi.org/10.1038/sj.cdd.4402028
Cain K, Bratton SB, Cohen GM (2002) The Apaf-1 apoptosome: a large caspase-activating complex. Biochimie. https://doi.org/10.1016/S0300-9084(02)01376-7
Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 122:669–682. https://doi.org/10.1016/j.cell.2005.08.012
Hägele H, Allam R, Pawar RD, Anders HJ (2009) Double-stranded RNA activates type i interferon secretion in glomerular endothelial cells via retinoic acid-inducible gene (RIG)-1. Nephrol Dial Transplant 24:3312–3318. https://doi.org/10.1093/ndt/gfp339
Pichlmair A, Schulz O, Tan Choon P et al (2006) RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 314:997–1001. https://doi.org/10.1126/science.1132998
Kato H, Takeuchi O, Sato S et al (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. https://doi.org/10.1038/nature04734
Berke IC, Modis Y (2012) MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J 31:1714–1726. https://doi.org/10.1038/emboj.2012.19
Melber A, Haynes CM (2018) UPR mt regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res 28:281–295. https://doi.org/10.1038/cr.2018.16
Callegari S, Dennerlein S (2018) Sensing the stress: a role for the UPRmt and UPRam in the quality control of mitochondria. Front Cell Dev Biol 6:1–10. https://doi.org/10.3389/fcell.2018.00031
Deepa SS, Bhaskaran S, Ranjit R et al (2016) Down-regulation of the mitochondrial matrix peptidase ClpP in muscle cells causes mitochondrial dysfunction and decreases cell proliferation. Free Radic Biol Med 91:281–292. https://doi.org/10.1016/j.freeradbiomed.2015.12.021
Torres-Odio S, Lei Y, Gispert S et al (2021) Loss of mitochondrial protease CLPP activates type I IFN responses through the mitochondrial DNA–cGAS–STING signaling axis. J Immunol 206:1890–1900. https://doi.org/10.4049/jimmunol.2001016
Haynes CM, Petrova K, Benedetti C, et al (2007) ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell 13:467–480. https://doi.org/10.1016/j.devcel.2007.07.016
Fiorese CJ, Schulz AM, Lin Y-F et al (2016) The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol 26:2037–2043. https://doi.org/10.1016/j.cub.2016.06.002
Sears TK, Angelastro JM (2017) The transcription factor ATF5: role in cellular differentiation, stress responses, and cancer. Oncotarget 8:84595–84609. https://doi.org/10.18632/oncotarget.21102
Sheng Z, Li L, Zhu LJ et al (2010) A genome-wide RNA interference screen reveals an essential CREB3L2-ATF5-MCL1 survival pathway in malignant glioma with therapeutic implications. Nat Med 16:671–677. https://doi.org/10.1038/nm.2158
Dluzen D, Li G, Tacelosky D et al (2011) BCL-2 is a downstream target of ATF5 that mediates the prosurvival function of ATF5 in a cell type-dependent manner. J Biol Chem 286:7705–7713. https://doi.org/10.1074/jbc.M110.207639
Spier A, Stavru F, Cossart P (2020) Interaction between intracellular bacterial pathogens and host cell mitochondria. Bact Intracellularity 2:3–13. https://doi.org/10.1128/9781683670261.ch1
Cervantes-Silva MP, Cox SL, Curtis AM (2021) Alterations in mitochondrial morphology as a key driver of immunity and host defence. EMBO Rep 22:1–18. https://doi.org/10.15252/embr.202153086
Syn G, Anderson D, Blackwell JM, Jamieson SE (2017) Toxoplasma gondii infection is associated with mitochondrial dysfunction in-vitro. Front Cell Infect Microbiol 7:1–17. https://doi.org/10.3389/fcimb.2017.00512
Pila-Castellanos I, Molino D, McKellar J et al (2021) Mitochondrial morphodynamics alteration induced by influenza virus infection as a new antiviral strategy. PLoS Pathog 17:1–20. https://doi.org/10.1371/JOURNAL.PPAT.1009340
WHO (1988) Foodborne listeriosis. World Health Organ 66:421–428
Stavru F, Bouillaud F, Sartori A et al (2011) Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc Natl Acad Sci USA 108:3612–3617. https://doi.org/10.1073/pnas.1100126108
Stavru F, Palmer AE, Wang C et al (2013) Atypical mitochondrial fission upon bacterial infection. Proc Natl Acad Sci 110:1600316008. https://doi.org/10.1073/pnas.1315784110
Carvalho F, Spier A, Chaze T et al (2020) Listeria monocytogenes exploits mitochondrial contact site and cristae organizing system complex subunit mic10 to promote mitochondrial fragmentation and cellular infection. MBio. https://doi.org/10.1128/mBio.03171-19
Liberti MV, Locasale JW (2016) The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41:211–218. https://doi.org/10.1016/j.tibs.2015.12.001
Ryan DG, O’Neill LAJ (2020) Krebs cycle reborn in macrophage immunometabolism. Annu Rev Immunol 38:289–313. https://doi.org/10.1146/annurev-immunol-081619-104850
Diskin C, Pålsson-McDermott EM (2018) Metabolic modulation in macrophage effector function. Front Immunol 9:1–17. https://doi.org/10.3389/fimmu.2018.00270
Spier A, Connor MG, Steiner T et al (2021) Mitochondrial respiration restricts Listeria monocytogenes infection by slowing down host cell receptor recycling. Cell Rep. https://doi.org/10.1016/j.celrep.2021.109989
Pharaoh G, Pulliam D, Hill S et al (2016) Ablation of the mitochondrial complex IV assembly protein Surf1 leads to increased expression of the UPRMT and increased resistance to oxidative stress in primary cultures of fibroblasts. Redox Biol 8:430–438. https://doi.org/10.1016/j.redox.2016.05.001
Pulliam DA, Deepa SS, Liu Y et al (2014) Complex IV-deficient Surf1(-/-) mice initiate mitochondrial stress responses. Biochem J 462:359–371. https://doi.org/10.1042/BJ20140291
Weindel CG, Bell SL, Vail KJ et al (2020) LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. Elife. https://doi.org/10.7554/eLife.51071
Singh A, Zhi L, Zhang H (2019) LRRK2 and mitochondria: recent advances and current views. Brain Res 1702:96–104. https://doi.org/10.1016/j.brainres.2018.06.010
Kim CY, Alcalay RN (2017) Genetic forms of Parkinson’s disease. Semin Neurol 37(2):135–146
Pierson TC, Diamond MS (2018) The emergence of Zika virus and its new clinical syndromes. Nature 560:573–581. https://doi.org/10.1038/s41586-018-0446-y
Ledur PF, Karmirian K, da Pedrosa CSG et al (2020) Zika virus infection leads to mitochondrial failure, oxidative stress and DNA damage in human iPSC-derived astrocytes. Sci Rep 10:1–14. https://doi.org/10.1038/s41598-020-57914-x
Medvedev R, Ploen D, Hildt E (2016) HCV and oxidative stress: implications for HCV life cycle and HCV-associated pathogenesis. Oxid Med Cell Longev. https://doi.org/10.1155/2016/9012580
Moulson AJ, Squair JW, Franklin RJM et al (2021) Diversity of reactive astrogliosis in CNS pathology: heterogeneity or plasticity? Front Cell Neurosci 15:1–32. https://doi.org/10.3389/fncel.2021.703810
Sofroniew MV (2020) Astrocyte reactivity: subtypes, states, and functions in CNS innate immunity. Trends Immunol 41:758–770. https://doi.org/10.1016/j.it.2020.07.004
Stefanik M, Formanova P, Bily T et al (2018) Characterisation of Zika virus infection in primary human astrocytes. BMC Neurosci 19:1–8. https://doi.org/10.1186/s12868-018-0407-2
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59
Yuan Z, Dewson G, Czabotar PE, Birkinshaw RW (2021) VDAC2 and the BCL-2 family of proteins. Biochem Soc Trans 49:2787–2795. https://doi.org/10.1042/BST20210753
Deo P, Chow SH, Hay ID et al (2018) Outer membrane vesicles from Neisseria gonorrhoeae target PorB to mitochondria and induce apoptosis. PLoS Pathog 14:1–30. https://doi.org/10.1371/journal.ppat.1006945
Sen WT, Coppens I, Saorin A et al (2020) Endolysosomal targeting of mitochondria is integral to BAX-mediated mitochondrial permeabilization during apoptosis signaling. Dev Cell 53:627-645.e7. https://doi.org/10.1016/j.devcel.2020.05.014
Bielaszewska M, Rüter C, Bauwens A et al (2017) Host cell interactions of outer membrane vesicle-associated virulence factors of enterohemorrhagic Escherichia coli O157: intracellular delivery, trafficking and mechanisms of cell injury. PLoS Pathog 13(2):e1006159
Bielaszewska M, Rüter C, Kunsmann L et al (2013) Enterohemorrhagic Escherichia coli hemolysin employs outer membrane vesicles to target mitochondria and cause endothelial and epithelial apoptosis. PLoS Pathog 9:1–30. https://doi.org/10.1371/journal.ppat.1003797
Deo P, Chow SH, Han M et al (2020) Mitochondrial dysfunction caused by outer membrane vesicles from Gram-negative bacteria activates intrinsic apoptosis and inflammation. Nat Microbiol 5:1418–1427
Dhital S, Deo P, Stuart I, Naderer T (2021) Bacterial outer membrane vesicles and host cell death signaling. Trends Microbiol 29:1106–1116. https://doi.org/10.1016/j.tim.2021.04.003
Ruckdeschel K, Pfaffinger G, Haase R et al (2004) Signaling of apoptosis through TLRs critically involves Toll/IL-1 receptor domain-containing adapter inducing IFN-β, but not MyD88, in bacteria-infected murine macrophages. J Immunol 173:3320–3328. https://doi.org/10.4049/jimmunol.173.5.3320
Besch R, Poeck H, Hohenauer T et al (2009) Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest 119:2399–2411. https://doi.org/10.1172/JCI37155
Gulen MF, Koch U, Haag SM et al (2017) Signalling strength determines proapoptotic functions of STING. Nat Commun. https://doi.org/10.1038/s41467-017-00573-w
Galluzzi L, Brenner C, Morselli E et al (2008) Viral control of mitochondrial apoptosis. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1000018
Fan T, Lu H, Hu H et al (1998) Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med 187:487–496. https://doi.org/10.1084/jem.187.4.487
Brokatzky D, Kretz O, Häcker G (2020) Apoptosis functions in defence against infection of mammalian cells with environmental chlamydiae. Infect Immun 88:1–14. https://doi.org/10.1128/iai.00851-19
Riley JS, Quarato G, Cloix C et al (2018) Mitochondrial inner membrane permeabilisation enables mt DNA release during apoptosis. EMBO J. https://doi.org/10.15252/embj.201899238
Riley JS, Tait SW (2019) Mitochondria and pathogen immunity: from killer to firestarter. EMBO J 38:2–4. https://doi.org/10.15252/embj.2019102325
Giampazolias E, Zunino B, Dhayade S et al (2017) Mitochondrial permeabilization engages NF-κB-dependent anti-tumour activity under caspase deficiency. Nat Cell Biol 19:1116–1129. https://doi.org/10.1038/ncb3596
Ichim G, Lopez J, Ahmed SU et al (2015) Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell 57:860–872. https://doi.org/10.1016/j.molcel.2015.01.018
Brokatzky D, Dörflinger B, Haimovici A et al (2019) A non-death function of the mitochondrial apoptosis apparatus in immunity. EMBO J 38:e2018100907. https://doi.org/10.15252/embj.2018100907
Youle RJ (2019) Mitochondria—Striking a balance between host and endosymbiont. Science. https://doi.org/10.1126/science.aaw9855
Wang LF, Crameri G (2014) Emerging zoonotic viral diseases. OIE Rev Sci Tech 33:569–581. https://doi.org/10.20506/rst.33.2.2311
Yu X-J, Liang M-F, Zhang S-Y et al (2011) Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med 364:1523–1532. https://doi.org/10.1056/NEJMoa1010095
Casel MA, Park SJ, Choi YK (2021) Severe fever with thrombocytopenia syndrome virus: emerging novel phlebovirus and their control strategy. Exp Mol Med 53:713–722. https://doi.org/10.1038/s12276-021-00610-1
Li S, Li H, Zhang YL et al (2020) SFTSV infection induces BAK/BAX-dependent mitochondrial DNA release to trigger NLRP3 inflammasome activation. Cell Rep 30:4370-4385.e7. https://doi.org/10.1016/j.celrep.2020.02.105
Di DJ, Gulen MF, Saidoune F et al (2022) The cGAS–STING pathway drives type I IFN immunopathology in COVID-19. Nature 603:145–151. https://doi.org/10.1038/s41586-022-04421-w
Chow KT, Gale M, Loo YM (2018) RIG-I and other RNA sensors in antiviral immunity. Annu Rev Immunol 36:667–694. https://doi.org/10.1146/annurev-immunol-042617-053309
Li T, Chen ZJ (2018) The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215:1287–1299. https://doi.org/10.1084/jem.20180139
Sato H, Hoshi M, Ikeda F et al (2021) Downregulation of mitochondrial biogenesis by virus infection triggers antiviral responses by cyclic GMP-AMP synthase. PLOS Pathog 17:e1009841. https://doi.org/10.1371/journal.ppat.1009841
McArthur K, Whitehead LW, Heddleston JM et al (2018) BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. https://doi.org/10.1126/science.aao6047
Acknowledgements
D.B. is supported by the Deutsche Forschungsgemeinschaft (DFG) Walter Benjamin Programme (BR 6637/1-1). The work of G.H. on mitochondria is supported by the DFG (HA2128/29-1).
Funding
Deutsche Forschungsgemeinschaft, BR 6637/1-1, Dominik Brokatzky, HA2128/29-1, Georg Häcker.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflicts of interest.
Additional information
Edited by: Volkhard A.J. Kempf.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Brokatzky, D., Häcker, G. Mitochondria: intracellular sentinels of infections. Med Microbiol Immunol 211, 161–172 (2022). https://doi.org/10.1007/s00430-022-00742-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-022-00742-9