
Abstract
Podocyte detachment is a major trigger in pathogenesis of focal segmental glomerulosclerosis (FSGS). Detachment via β1 integrin (ITGB1) endocytosis, associated with endothelial cell injury, has been reported in animal models but remains unknown in human kidneys. The objectives of our study were to examine the difference in ITGB1 dynamics between primary FSGS and minimal change nephrotic syndrome (MCNS), among variants of FSGS, as well as between the presence or absence of cellular lesions (CEL-L) in human kidneys, and to elucidate the pathogenesis of FSGS. Thirty-one patients with primary FSGS and 14 with MCNS were recruited. FSGS cases were categorized into two groups: those with CEL-L, defined by segmental endocapillary hypercellularity occluding lumina, and those without CEL-L. The podocyte cytoplasmic ITGB1 levels, ITGB1 expression, and degrees of podocyte detachment and subendothelial widening were compared between FSGS and MCNS, FSGS variants, and FSGS groups with and without CEL-L (CEL-L( +)/CEL-L( −)). ITGB1 distribution in podocyte cytoplasm was significantly greater in CEL-L( +) group than that in MCNS and CEL-L( −) groups. ITGB1 expression was similar in CEL-L( +) and MCNS, but lower in CEL-L( −) compared with others. Podocyte detachment levels were comparable in CEL-L( +) and CEL-L( −) groups, both exhibiting significantly higher detachment than the MCNS group. Subendothelial widening was significantly greater in CEL-L( +) compared with CEL-L( −) and MCNS groups. The findings of this study imply the existence of distinct pathological mechanisms associated with ITGB1 dynamics between CEL-L( +) and CEL-L( −) groups, and suggest a potential role of endothelial cell injury in the pathogenesis of cellular lesions in FSGS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Focal segmental glomerulosclerosis (FSGS) is characterized by a segmental sclerosis in some (focal) glomeruli on light microscopy. Immunofluorescence microscopy typically shows no or low-intensity staining of complement 3 (C3) and/or immunoglobulin M (IgM) in sclerotic areas. FSGS is categorized into primary and secondary forms. Primary FSGS, defined as a podocytopathy, is associated with nephrotic syndrome (NS) and extensive foot process effacement and podocyte detachment on electron microscopy (EM) [1,2,3]. Secondary FSGS, characterized by the absence of NS and segmental foot process effacement on EM, is often due to glomerular hyperfiltration [4, 5].
NS is characterized by massive urinary protein leakage. This leakage results from increased protein permeability due to dysfunction of glomerular filtration barrier. Primary FSGS, a type of NS resistant to steroid therapy, can progress to end-stage renal failure [6]. In contrast, minimal change nephrotic syndrome (MCNS), the most common NS cause in children, responds well to steroids. Light microscopy shows normal glomeruli, and immunofluorescence microscopy reveals no or low-intensity staining for C3 and/or IgM in the mesangial area. While extensive foot process effacement is observed, podocyte detachment is rare [7]. Differences in steroid sensitivity suggest distinct pathogeneses for primary FSGS and MCNS, though the mechanisms are only partially understood. Investigating these mechanisms is crucial, especially for primary FSGS, which is associated with a poor prognosis.
According to the Columbia classification, FSGS is morphologically categorized into five variants: collapsing, tip, cellular, perihilar, and not otherwise specified (NOS) [8]. The prognosis varies by variant, with tip associated with the best and collapsing with the worst outcomes [9,10,11,12]. However, the reasons for these morphological and prognostic differences remain unclear.
Podocyte detachment is a key factor in FSGS pathogenesis. In humans, α3β1 integrin is a crucial adhesion molecule on podocytes [13] and plays an important role in anchoring and signal transduction between podocytes and the extracellular matrix of glomerular basement membrane (GBM) [14]. The glomerular filtration barrier consists of podocytes, GBM, and endothelial cells [15]. Cross-talk between podocytes and endothelial cells is thought to regulate barrier function [16, 17]. Kobayashi et al. reported in a podocyte-specific injury animal model that plasminogen activator inhibitor-1 (PAI-1) from endothelial cells binds urokinase plasminogen activator (uPA) in the blood. This complex then binds to uPA receptor (uPAR) on podocytes, causing β1 integrin (ITGB1) endocytosis and resulting in podocyte detachment [18]. However, this has not been thoroughly validated in human FSGS variants.
Cellular lesions (CEL-L) in FSGS are characterized by segmental endocapillary hypercellularity with mononuclear and/or foam cells occluding the capillary lumina [8]. Thrombotic microangiopathy, characterized by endothelial cell injury, often shows endocapillary hypercellularity with epithelial hyperplasia in the chronic phase, which resembles CEL-L in FSGS. This suggests a potential link between endothelial cells and podocyte injury.
The objectives of this study were to examine the difference in ITGB1 dynamics between primary FSGS and MCNS, among the variants of FSGS, and between the presence or absence of CEL-L in human kidneys and to elucidate the mechanism underlying the pathogenesis of primary FSGS.
Materials and methods
Subjects
Renal biopsy specimens were examined at the Department of Pathology, School of Medicine, University of Occupational and Environmental Health (UOEH) between 2006 and 2021. Thirty-one patients with primary FSGS and 14 with MCNS were recruited, consisting of 27 men and 18 women (mean age: 27.9 ± 16.9 years). Twenty-five patients were receiving corticosteroids or immunosuppressants at the time of the study. Diagnoses of FSGS and MCNS were confirmed histopathologically, and FSGS variants were classified according to the Columbia criteria [8]. Primary FSGS was diagnosed in the absence of other glomerular diseases or systemic conditions that could cause secondary segmental glomerular sclerosis. Clinical data were obtained from application forms. Patients with evidence of systemic disease that can cause thrombotic microangiopathy (e.g., thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, malignant hypertension, autoimmune disease, systemic infection), other causes of secondary segmental sclerosis (e.g., reflux nephropathy, pyelonephritis, transplanted kidney, human immunodeficiency virus infection, or congenital disease), or a history of hypertension, obesity, nephrectomy, a solitary kidney, or intravenous drug abuse were excluded. Based on the Columbia classification, representative lesions of each variant often coexist [8]. Coexisting lesions in distinct glomeruli were evaluated based on the Columbia classification, and FSGS cases were categorized into two groups: with and without CEL-L; CEL-L( +); and CEL-L( −) groups. There was a possibility that FSGS cases were hidden among the MCNS cases, so we used 0-h biopsy samples from kidney transplant cases as controls. We examined eight cases without glomerular hypercellularity, interstitial fibrosis, tubular atrophy, or significant arteriosclerosis.
Staining and morphological analysis
Kidney tissues were fixed in 10% neutral-buffered formaldehyde and embedded in paraffin. Sections (3 μm thick) were stained with periodic acid–Schiff, periodic acid–methenamine silver, hematoxylin and eosin, and Masson trichrome. For immunohistochemical staining, Wilms’ tumor 1 (WT1) and double staining for ITGB1 and vimentin were performed. Tissue sections were deparaffinized in xylene for 15 min, rehydrated in ethanol, and washed in distilled water. Endogenous peroxidase was blocked with 3% H2O2. For WT1 staining, after antigen retrieval by autoclaving for 20 min in 10 mM citrate buffer (pH 6.0), sections were incubated with a primary antibody against WT1 (1:300, ab220212; Abcam, Cambridge, UK) for 1 h at room temperature. The sections were visualized using diaminobenzidine (DAB substrate-chromogen system; Dako, Santa Clara, CA, USA). For double staining of ITGB1 and vimentin, antigen retrieval was performed by autoclaving for 20 min in 10 mM citrate buffer (pH 9.0). The sections were incubated with a vimentin antibody (1:20, goat, ab11256; Abcam) for 1 h at room temperature, and an ITGB1 antibody (1:1,000, rabbit, ab52971; Abcam) overnight at 4 °C. Secondary antibodies used were donkey anti-rabbit antibody, Alexa Fluor™ 546 (1:100, A10040; Thermo Fisher Scientific Inc., Waltham, MA, USA) and donkey anti-goat antibody, Alexa Fluor™ 488 (1:100, A11055; Thermo Fisher Scientific Inc.).
The stained tissue sections were digitized using a virtual slide system (VS120; Olympus, Tokyo, Japan) or Nano Zoomer Digital Slide Scanner (Hamamatsu Photonics, Hamamatsu, Japan). To quantify podocyte count, the number of WT1-positive cells per unit area of each glomerulus was measured, and the average was calculated for each case. Double immunofluorescence staining for ITGB1 and vimentin was used to quantify ITGB1 within podocytes, which were identified by vimentin positivity. Up to ten glomeruli were selected per case (detailed numbers are in Supplemental Table 1). The merged area of vimentin and ITGB1 was measured using HALO image analysis software (Indica Labs, Corrales, NM, USA), and the value was divided by the ITGB1-positive area in each glomerulus. The average value per glomerulus was then calculated for each case. Each case was digitized using the same light source intensity and measured after setting a threshold to blacken the background in each staining. Additionally, the number of vimentin-positive cells with merged areas in their cytoplasm was counted in each glomerulus and divided by the total number of WT1-positive cells to determine the proportion of podocytes with cytoplasmic ITGB1 in FSGS cases. The average value per glomerulus was then calculated for each case. The morphology of each glomerulus was also assessed.
Electron microscopic examination
Portions of the tissue were fixed with 2.5% glutaraldehyde for transmission electron microscopy. Ten glomerular tufts were randomly photographed per case. The percentages of podocyte detachment and subendothelial widening relative to the capillary GBM were calculated by summing these lengths across ten images.
RNA quantitative real-time polymerase chain reaction (qRT-PCR)
Frozen sections of each case were obtained using a cryostat (5 μm thick), mounted onto membrane-coated slides, fixed in acetone for 3 min, washed with RNase-free water 1minuit, stained by 0.05% toluidine blue for 15 s, washed with RNase-free water 2 times for 1minuit, and air-dried for 5 min. Between 9 and 154 glomeruli were isolated via laser capture using an LMD6/7 (Leica Microsystems, Wetzlar, Germany) for each case, regardless of the presence of FSGS lesions. The number of laser-captured glomeruli per case is detailed in Supplemental Table 1. cDNA synthesis was performed using 1 μg of total RNA per case with a High-Capacity cDNA Reverse Transcription Kit (4,368,814; Thermo Fisher Scientific Inc.). The gene expression of ITGB1 (ITGB1; Hs01127536_m1), vascular endothelial growth factor A (VEGFA; Hs00900055_m1), uPAR (PLAUR; Hs00958880_m1), and PAI-1 (serpin family E member 1 [SERPINE1]; Hs00167155_m1) was examined by qRT-PCR using TaqMan® Gene Expression Assays (Thermo Fisher Scientific Inc.). For quantitative analysis, the levels were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Hs02786624_g1) in standard total human kidney RNA (Premium total RNA, 636,529; Clontech, Mountain View, CA, USA) using the ΔΔCt method (Ct indicates the threshold cycle). The results are presented as relative expressions (RQ) in arbitrary units. Unfortunately, mRNA of 0-h biopsy cases could not be extracted due to poor preservation of the frozen samples.
Comparison
The number of WT-1-positive cells, the merged area of ITGB1 and vimentin, mRNA expression levels of ITGB1 and VEGF, as well as uPAR and PAI-1, the degree of podocyte detachment, and subendothelial widening were compared between the 0-h biopsy, MCNS and FSGS groups, among the FSGS variants and between the CEL-L( +) and CEL-L( −) groups. The number of glomeruli analyzed per case is listed in Supplemental Table 1.
Statistical analysis
Continuous data were expressed as median (interquartile range). Group comparisons were conducted using the Wilcoxon rank-sum test. Statistical analyses were performed with JMP version 12 (SAS Institute Inc., Cary, NC, USA), with significance set at p < 0.05. Due to the small sample sizes, no statistical analysis was performed among FSGS variants.
Results
Clinical characteristics of the subjects and Columbia classification
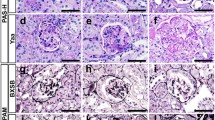
The characteristics of MCNS and FSGS patients are summarized in Table 1. Among the 31 FSGS cases, there was 1 collapsing variant, 8 cellular, 5 tip, 11 perihilar, and 6 NOS variants. CEL-L were present in 4 of 5 tip cases and all 8 cellular cases, leading to their classification into the CEL-L( +) group (12 patients). The remaining 19 cases (1 collapsing, 1 tip, 11 perihilar, and 6 NOS variants) were classified as CEL-L( −). Histological findings for each case are shown in Supplemental Table 2. The median proteinuria was not in the nephrotic range in the MCNS and CEL-L( −) groups. Of the 14 MCNS cases, 10 received immunosuppressive therapy before biopsy, and among the 31 FSGS cases, 15 received similar treatment. As a result, some cases showed improvement in proteinuria by the time of biopsy, leading to a lower median value despite the presence of nephrotic syndrome before treatment. Clinical information for the 0-h biopsy donors was unavailable, as it was not included in the application form.
WT1-positive cells
The number of WT1-positive cells was significantly lower in FSGS compared to MCNS and 0-h biopsy groups (Supplemental Figure S1a). Both CEL-L( +) and CEL-L( −) groups had significantly fewer WT1-positive cells than the MCNS and 0-h biopsy groups (Supplemental Figure S1b). However, no significant difference was observed between the CEL-L( +) and CEL-L( −) groups. Although statistical analysis was not performed due to the small sample size of each variant, the cellular, perihilar, and NOS variants exhibited relatively fewer WT1-positive cells compared to the tip variant (Supplemental Figure S1c).
Merged area of ITGB1 and vimentin
The merged ITGB1 and vimentin area was significantly larger in FSGS compared to MCNS and 0-h biopsy groups (Fig. 1a, b). The CEL-L( +) group had a significantly larger merged area than the CEL-L( −) group, as well as the MCNS and 0-h biopsy groups. In contrast, no significant difference was found between the CEL-L( −), MCNS, and 0-h biopsy groups (Fig. 1c). Among the variants, the merged area appeared larger in the tip and cellular variants compared to others (Supplemental Figure S2). ITGB1 within the cytoplasm of vimentin-positive cells was analyzed in 220 glomeruli (94 and 126 in the CEL-L( +) and CEL-L( −) group). Vimentin-positive cells with cytoplasmic ITGB1 were observed in 37 glomeruli (16.8%). Notably, many of these glomeruli showed minor morphological changes without significant lesions (Fig. 1a and d). The proportion of vimentin-positive cells with cytoplasmic ITGB1 was significantly higher in the CEL-L( +) group compared to the CEL-L( −) group (Fig. 1e). No notable differences among FSGS variants were apparent (Supplemental Figure S3). Data for each glomerulus are shown in Supplemental Table 3.


Merged area of β1 integrin and vimentin. a: β1 integrin (red) and vimentin (green) staining, and morphology of the same glomerulus (PAS staining). Vimentin is positive for podocyte cytoplasm. β1 integrin is seen in the cytoplasm of vimentin-positive cells at FSGS case (arrow), although less observed in MCNS case. Left-upper: MCNS case. Left-lower and right-upper: Glomerulus of FSGS case exhibiting minor change morphology. Right-lower: Glomerulus of FSGS case exhibiting cellular lesion. b Merged area per β1 integrin area comparing with FSGS, MCNS, and 0-h biopsy groups. c: Merged area comparing with CEL-L( +), CEL-L( −), MCNS, and 0-h biopsy groups. d: Number of glomeruli with merged area in vimentin-positive cells. Many of the glomeruli containing vimentin/ITBG1 double-positive cells exhibited minor change morphology. e: Number of glomeruli with merged area in vimentin-positive cells comparing with CEL-L( +) and CEL-L( −). Sample size: FSGS (n = 31), CEL-L( +) (n = 12), CEL-L( −) (n = 19), MCNS (n = 14), 0-h biopsy (n = 8). Abbreviations: MCNS, minimal change nephrotic syndrome. FSGS, focal segmental glomerulosclerosis. CEL-L( +), FSGS with cellular lesions. CEL-L( −), FSGS without cellular lesions. MGA, minor glomerular abnormality. COL, collapsing lesion. CEL, cellular lesion. TIP, tip lesion. PH, perihilar lesion. NOS, NOS lesion. Ad, tuft adhesion. MES, Mild mesangial hypercellularity. “Mild mesangial hypercellularity” refers to an increase in mesangial cells in less than 25% of the glomerular area. * p < 0.05, ** p < 0.01
RT-PCR
FSGS exhibited significantly lower ITGB1 expression than MCNS (Fig. 2a). Although ITGB1 expression was lower in CEL-L( −) group than in CEL-L( +) and MCNS groups, it was similar between CEL-L( +) and MCNS groups (Fig. 2b). There were no differences among the FSGS variants (Supplemental Figure S4). VEGF expression was significantly lower in the CEL-L( −) group than in the MCNS group. However, there was no significant difference between the CEL-L( +) and CEL-L( −) groups. RT-PCR analysis of uPAR and PAI-1 did not reveal significant differences between the CEL-L( +) and CEL-L( −) groups (Fig. 2c–e). mRNA from 0-h biopsy cases could not be extracted due to poor sample preservation.

mRNA expression level of β1 integrin, VEGF, UPAR, and PAI-1. a: β1 integrin mRNA expression level comparing with FSGS and MCNS. b: β1 integrin mRNA expression level comparing with CEL-L( +), CEL-L( −), and MCNS. c: VEGF mRNA expression level comparing with CEL-L( +), CEL-L( −), and MCNS. d: uPAR mRNA expression level comparing with CEL-L( +), CEL-L( −), and MCNS. e: PAI-1 mRNA expression level comparing with CEL-L( +), CEL-L( −), and MCNS. Sample size: FSGS (n = 31), CEL-L( +) (n = 12), CEL-L( −) (n = 19), MCNS (n = 14). Abbreviations: MCNS, minimal change nephrotic syndrome. FSGS, focal segmental glomerulosclerosis. CEL-L( +), FSGS with cellular lesions. CEL-L( −), FSGS without cellular lesions. * p < 0.05, ** p < 0.01
Electron microscopic examination of podocyte detachment and subendothelial widening
Electron microscopy images of podocyte detachment are shown in Fig. 3a. FSGS exhibited significantly more extensive podocyte detachment compared to the MCNS and 0-h biopsy groups (Fig. 3b). The extent of podocyte detachment was similar between the CEL-L( +) and CEL-L( −) groups, with both showing significantly greater detachment compared to the MCNS and 0-h biopsy groups (Fig. 3c). Among FSGS variants, the perihilar variant appeared to have relatively more podocyte detachment than the tip variant (Supplemental Figure S5).

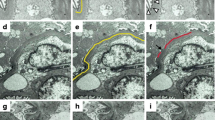
Degree of podocyte detachment by electron microscopic examination. The percentages of the length of podocyte detachment relative to the length of the capillary GBM were calculated. a: Electron microscopic image of MCNS, CEL-L( +), and CEL-L( −). Podocyte detachment (yellow circle) is prominent in CEL-L( +) and CEL-L( −) compared with MCNS. Degree of podocyte detachment is similar between CEL-L( +) and CEL-L( −). Swelling of endothelial cells, reduction of its fenestration, and subendothelial widening are associated with podocyte detachment. b: Ratio of podocyte detachment comparing with FSGS, MCNS and 0-h biopsy groups. c: Ratio of podocyte detachment comparing with CEL-L( +), CEL-L( −), MCNS, and 0-h biopsy groups. Sample size: FSGS (n = 31), CEL-L( +) (n = 12), CEL-L( −) (n = 19), MCNS (n = 14), 0-h biopsy (n = 8). Abbreviations: MCNS, minimal change nephrotic syndrome. FSGS, focal segmental glomerulosclerosis. CEL-L( +), FSGS with cellular lesions. CEL-L( −), FSGS without cellular lesions. * p < 0.05, ** p < 0.01
Electron microscopy images of subendothelial widening are shown in Fig. 4a. FSGS exhibited significantly greater subendothelial widening compared to the MCNS and 0-h biopsy groups (Fig. 4b). The CEL-L( +) group showed significantly more subendothelial widening than the CEL-L( −) group, with both groups showing significantly more widening than the MCNS and 0-h biopsy groups (Fig. 4c). Among FSGS variants, the cellular variant appeared to have greater subendothelial widening than the perihilar variant (Supplemental Figure S6).

Degree of subendothelial widening by electron microscopic examination. The percentages of the length of subendothelial widening relative to the length of the capillary GBM were calculated. a: Electron microscopic image of MCNS, CEL-L( +) and CEL-L( −). Subendothelial widening (arrowhead) is prominent in CEL-L( +), comparing with CEL-L( −) or MCNS. Along with the subendothelial widening, swelling of endothelial cells, reduction of its fenestration, and new basement membrane formation are observed. Additionally, foot process effacement is noted in areas corresponding to the subendothelial widening. b: Ratio of subendothelial widening comparing with FSGS, MCNS, and 0-h biopsy groups. (c): Ratio of subendothelial widening comparing with CEL-L( +), CEL-L( −), MCNS, and 0-h biopsy groups. Sample size: FSGS (n = 31), CEL-L( +) (n = 12), CEL-L( −) (n = 19), MCNS (n = 14), 0-h biopsy (n = 8). Abbreviations: MCNS, minimal change nephrotic syndrome. FSGS, focal segmental glomerulosclerosis. CEL-L( +), FSGS with cellular lesions. CEL-L( −), FSGS without cellular lesions. * p < 0.05, ** p < 0.01
Discussion
In this study, ITGB1 distribution in podocytes was higher in FSGS than in MCNS, particularly in the CEL-L( +) group compared to MCNS and CEL-L( −) groups. Notably, ITGB1 expression was similar between CEL-L( +) and MCNS groups but significantly lower in the CEL-L( −) group than in both MCNS and CEL-L( +) groups.
ITGB1 plays a crucial role in podocyte adhesion to the GBM [14]. Podocyte detachment in primary FSGS may be linked to reduced ITGB1 expression [19]. Conversely, ITGB1 upregulation has been linked to podocyte detachment in early-stage diabetic nephropathy [20]. Previous studies on ITGB1 and podocyte detachment have shown conflicting results, with some indicating increased and others decreased ITGB1 levels. Moreover, no studies have explored differences among FSGS variants. This study is the first to show that ITGB1 expression is not reduced in FSGS with CEL-L.
Podocyte detachment may result from ITGB1 endocytosis through the PAI-1/uPA complex binding to podocyte uPAR [18]. In this study, the degree of podocyte detachment was similar in the CEL-L( +) and CEL-L( −) groups. However, ITGB1 distribution in podocyte cytoplasm was significantly greater in CEL-L( +) group than in CEL-L( −) group. These findings suggest different mechanisms for podocyte detachment between FSGS with and without CEL-L. Higher degree of ITGB1 in podocytes in CEL-L( +) group may indicate that podocyte detachment may occur through endocytosis of ITGB1 in FSGS with CEL-L. In contrast, lower ITGB1 expression and lower degree of ITGB1 within podocytes in CEL-L( −) suggest that downregulation of ITGB1 may be the primary mechanism of podocyte detachment in FSGS without CEL-L. Additionally, many glomeruli with ITGB1 in the cytoplasm of podocytes displayed minor morphological changes without FSGS lesions. This interesting finding suggests that ITGB1 endocytosis into podocyte cytoplasm may indicate an early change linked to lesion formation.
Kobayashi et al. suggested the involvement of endothelial cell injury in the ITGB1 endocytosis using a podocyte-specific injury mouse model [18]. Our study found no significant differences in mRNA levels of uPAR, VEGF, and PAI-1 between FSGS and MCNS or between FSGS with and without CEL-L. However, morphological evaluation using electron microscopy revealed greater subendothelial widening in the CEL-L( +) group compared to both the CEL-L( −) and MCNS groups. Ultrastructural alterations representing endothelial cell injury include subendothelial widening [21], endothelial swelling [17], decrease of endothelial cell number, or reduction in its fenestration [22]. Subendothelial widening is the most common glomerular lesion associated with endothelial damage [23]. Previous study reported a significant increase in subendothelial widening in the FSGS group, particularly in collapsing, cellular, and NOS variants [24]. However, no study has focused on the presence or absence of CEL-L. Our findings suggest a potential involvement of endothelial cell injury in the development of CEL-L in FSGS.
Podocytes and endothelial cells are believed to cross-talk each other, but whether podocyte or endothelial cell injury occurs first in primary FSGS remains unclear. Recent studies suggest that circulating permeability factors may cause podocyte injury. Wei et al. found that soluble uPAR is elevated in FSGS patients’ serum and on podocyte surfaces, where it activates β3 integrin (ITGB3) and disrupts the actin cytoskeleton, leading to foot process effacement [25, 26]. This mechanism may lead to podocyte detachment and PAI-1 induction in endothelial cells. Additionally, suppression of ITGB1 activates ITGB3 via Rac1 and ERK [27]. Taken together with the findings of Kobayashi et al., it is hypothesized that primary podocyte injury induced by circulating permeability factors (e.g., soluble uPAR) leads to foot process effacement through ITGB3 activation and subsequent PAI-1 release from endothelial cells. This release induces ITGB1 endocytosis, causing podocyte detachment and activating ITGB3, which perpetuates a vicious cycle worsening disease progression. While our study does not pinpoint the primary cause or detailed mechanisms of podocyte injury, it suggests that ITGB1 endocytosis and endothelial cell damage play a key role in FSGS, particularly in CEL-L( +).
This study included eight cellular variant cases and four tip variant cases in the CEL-L( +) group. Given that the tip variant generally has a better outcome compared to the cellular variant [11], there may be distinct pathophysiological mechanisms between the two. However, the relationship between FSGS variants and kidney outcomes is controversial. Several reports have suggested that prognosis is influenced by responsiveness to immunosuppressive therapy irrespective of the variant. [28,29,30]. Kawaguchi et al. reported that there were no significant difference in prognosis among variants in primary FSGS patients using Japanese nationwide registry of kidney biopsies [31]. Additionally, Tsuchimoto et al. reported that the administration of steroids and/or immunosuppressive therapy led to good prognosis in cellular variants and suggested that differences in treatment approaches, particularly in the case of the cellular variant, can impact outcomes [9]. The pathophysiological differences among FSGS variants remain unclear. Considering the results of this study, comparison of tip variants with and without CEL-L may be warranted; however, the small sample size precluded such an analysis. Further investigations are required.
This study has several limitations. First, it was a retrospective single-center study involving only Japanese patients. Second, the small number of cases for each primary FSGS variant, particularly the collapsing variant, may limit the validity of comparisons. Consequently, we classified FSGS cases into two groups—those with and without CEL-L—and compared ITGB1 dynamics, expression, and endothelial cell injury between these groups. Third, vimentin can be expressed in parietal cells within glomeruli that are either aged or have podocyte injury. Initially, we used synaptopodin for specific podocyte staining, but antigen retrieval with autoclaving and protease treatment needed for ITGB1 double staining damaged the specimens and made evaluation impossible. Therefore, we used vimentin, which can be retrieved using the same method as ITGB1. To avoid parietal cells, we excluded those lining the Bowman’s capsule basement membrane. Although FSGS lesions may include parietal cells undergoing epithelial-to-mesenchymal transition (EMT), many glomeruli with vimentin/ITGB1 double-positive cells showed minor morphological changes, suggesting these cells are primarily podocytes. Fourth, RT-PCR data from entire glomeruli included mRNA from podocytes, mesangial cells, and endothelial cells, which may not precisely reflect podocyte-specific expression. However, no reports indicate increased ITGB1 expression in mesangial or endothelial cells in FSGS. Though three cases showed mild mesangial cell proliferation, the proportions were minimal (case 5: 1/31 glomeruli, case 9: 2/38 glomeruli, case 24: 1/25 glomeruli). Thus, the observed differences likely reflect podocyte expression.
In conclusion, the results of the present study indicated the possibility of distinct pathological mechanisms involving the dynamics of ITGB1 between FSGS with and without CEL-L and the potential involvement of endothelial cell injury in the development of CEL-L.
Data Availability
All data generated or analyzed during this study are included in this article and its online supplementary material files. Further inquiries can be directed to the corresponding author.
References
Fogo AB, Lusco MA, Najafian B, Alpers CE (2015) AJKD Atlas of renal pathology: focal segmental glomerulosclerosis. Am J Kidney Dis 66:e1–e2. https://doi.org/10.1053/j.ajkd.2015.04.007
(2012) Chapter 6: idiopathic focal segmental glomerulosclerosis in adults. Kidney Int Suppl 2:181–185. https://doi.org/10.1038/kisup.2012.19
Grishman E, Churg J (1975) Focal glomerular sclerosis in nephrotic patients: an electron microscopic study of glomerular podocytes. Kidney Int 7:111–122. https://doi.org/10.1038/ki.1975.16
Sethi S, Glassock RJ, Fervenza FC (2015) Focal segmental glomerulosclerosis: towards a better understanding for the practicing nephrologist. Nephrol Dial Transplant 30:375–384. https://doi.org/10.1093/ndt/gfu035
De Vriese AS, Sethi S, Nath KA, Glassock RJ, Fervenza FC (2018) Differentiating primary, genetic, and secondary FSGS in adults: a clinicopathologic approach. J Am Soc Nephrol JASN 29:759–774. https://doi.org/10.1681/ASN.2017090958
Nishi S, Ubara Y, Utsunomiya Y, Okada K, Obata Y, Kai H, Kiyomoto H, Goto S, Konta T, Sasatomi Y, Sato Y, Nishino T, Tsuruya K, Furuichi K, Hoshino J, Watanabe Y, Kimura K, Matsuo S (2016) Evidence-based clinical practice guidelines for nephrotic syndrome 2014. Clin Exp Nephrol 20:342–370. https://doi.org/10.1007/s10157-015-1216-x
(2012) Chapter 5: minimal-change disease in adults. Kidney Int Suppl 2:177–180. https://doi.org/10.1038/kisup.2012.18
D’Agati VD, Fogo AB, Bruijn JA, Jennette JC (2004) Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis 43:368–382. https://doi.org/10.1053/j.ajkd.2003.10.024
Tsuchimoto A, Matsukuma Y, Ueki K, Tanaka S, Masutani K, Nakagawa K, Mitsuiki K, Uesugi N, Katafuchi R, Tsuruya K, Nakano T, Kitazono T (2020) Utility of Columbia classification in focal segmental glomerulosclerosis: renal prognosis and treatment response among the pathological variants. Nephrol Dial Transplant 35:1219–1227. https://doi.org/10.1093/ndt/gfy374
D’Agati VD, Alster JM, Jennette JC, Thomas DB, Pullman J, Savino DA, Cohen AH, Gipson DS, Gassman JJ, Radeva MK, Moxey-Mims MM, Friedman AL, Kaskel FJ, Trachtman H, Alpers CE, Fogo AB, Greene TH, Nast CC (2013) Association of histologic variants in FSGS clinical trial with presenting features and outcomes. Clin J Am Soc Nephrol 8:399–406. https://doi.org/10.2215/CJN.06100612
Stokes MB, Valeri AM, Markowitz GS, D’Agati VD (2006) Cellular focal segmental glomerulosclerosis: clinical and pathologic features. Kidney Int 70:1783–1792. https://doi.org/10.1038/sj.ki.5001903
Kwon YE, Han SH, Kie JH, An SY, Kim YL, Park KS, Nam KH, Leem AY, Oh HJ, Park JT, Chang TI, Kang EW, Kang S-W, Choi KH, Lim BJ, Jeong HJ, Yoo T-H (2014) Clinical features and outcomes of focal segmental glomerulosclerosis pathologic variants in Korean adult patients. BMC Nephrol 15:52. https://doi.org/10.1186/1471-2369-15-52
Korhonen M, Ylänne J, Laitinen L, Virtanen I (1990) Distribution of beta 1 and beta 3 integrins in human fetal and adult kidney. Lab Investig J Tech Methods Pathol 62:616–625
Sachs N, Sonnenberg A (2013) Cell-matrix adhesion of podocytes in physiology and disease. Nat Rev Nephrol 9:200–210. https://doi.org/10.1038/nrneph.2012.291
D’Agati VD, Kaskel FJ, Falk RJ (2011) Focal segmental glomerulosclerosis. N Engl J Med 365:2398–2411. https://doi.org/10.1056/NEJMra1106556
Daehn I, Casalena G, Zhang T, Shi S, Fenninger F, Barasch N, Yu L, D’Agati V, Schlondorff D, Kriz W, Haraldsson B, Bottinger EP (2014) Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest 124:1608–1621. https://doi.org/10.1172/JCI71195
Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber H-P, Ferrara N, Barisoni L, Alpers CE, Quaggin SE (2008) VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358:1129–1136. https://doi.org/10.1056/NEJMoa0707330
Kobayashi N, Ueno T, Ohashi K, Yamashita H, Takahashi Y, Sakamoto K, Manabe S, Hara S, Takashima Y, Dan T, Pastan I, Miyata T, Kurihara H, Matsusaka T, Reiser J, Nagata M (2015) Podocyte injury-driven intracapillary plasminogen activator inhibitor type 1 accelerates podocyte loss via uPAR-mediated β 1 -integrin endocytosis. Am J Physiol-Ren Physiol 308:F614–F626. https://doi.org/10.1152/ajprenal.00616.2014
Chen C-A, Hwang J-C, Guh J-Y, Chang J-M, Lai Y-H, Chen H-C (2006) Reduced podocyte expression of α3β1 integrins and podocyte depletion in patients with primary focal segmental glomerulosclerosis and chronic PAN-treated rats. J Lab Clin Med 147:74–82. https://doi.org/10.1016/j.lab.2005.08.011
Sawada K, Toyoda M, Kaneyama N, Shiraiwa S, Moriya H, Miyatake H, Tanaka E, Yamamoto N, Miyauchi M, Kimura M, Wada T, Fukagawa M (2016) Upregulation of α 3 β 1-Integrin in Podocytes in Early-Stage Diabetic Nephropathy. J Diabetes Res 2016:1–7. https://doi.org/10.1155/2016/9265074
Miki Y, Shimizu H, Danbara A, Fujita Y, Ito Y, Matsuo S (2002) A case of mesangial proliferative glomerulonephritis with endothelial damage. Nihon Jinzo Gakkai Shi 44:547–551
Toyoda M, Najafian B, Kim Y, Caramori ML, Mauer M (2007) Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes 56:2155–2160. https://doi.org/10.2337/db07-0019
Leontsini M (2003) Mesangiolysis. HIPPOKRATIA 7:147–151
Taneda S, Honda K, Ohno M, Uchida K, Nitta K, Oda H (2015) Podocyte and endothelial injury in focal segmental glomerulosclerosis: an ultrastructural analysis. Virchows Arch 467:449–458. https://doi.org/10.1007/s00428-015-1821-9
Wei C, Möller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, Xie L, Henger A, Schmid H, Rastaldi MP, Cowan P, Kretzler M, Parrilla R, Bendayan M, Gupta V, Nikolic B, Kalluri R, Carmeliet P, Mundel P, Reiser J (2008) Modification of kidney barrier function by the urokinase receptor. Nat Med 14:55–63. https://doi.org/10.1038/nm1696
Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, Maiguel D, Karumanchi SA, Yap H-K, Saleem M, Zhang Q, Nikolic B, Chaudhuri A, Daftarian P, Salido E, Torres A, Salifu M, Sarwal MM, Schaefer F, Morath C, Schwenger V, Zeier M, Gupta V, Roth D, Rastaldi MP, Burke G, Ruiz P, Reiser J (2011) Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med 17:952–960. https://doi.org/10.1038/nm.2411
Hayashida T, Jones JCR, Lee CK, Schnaper HW (2010) Loss of β1-integrin enhances TGF-β1-induced collagen expression in epithelial cells via increased αvβ3-Integrin and Rac1 activity *. J Biol Chem 285:30741–30751. https://doi.org/10.1074/jbc.M110.105700
Chun MJ, Korbet SM, Schwartz MM, Lewis EJ (2004) Focal segmental glomerulosclerosis in nephrotic adults: presentation, prognosis, and response to therapy of the histologic variants. J Am Soc Nephrol 15:2169–2177. https://doi.org/10.1097/01.ASN.0000135051.62500.97
Troost JP, Trachtman H, Nachman PH, Kretzler M, Spino C, Komers R, Tuller S, Perumal K, Massengill SF, Kamil ES, Oh G, Selewski DT, Gipson P, Gipson DS (2018) An outcomes-based definition of proteinuria remission in focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 13:414–421. https://doi.org/10.2215/CJN.04780517
Korbet SM (2012) Treatment of Primary FSGS in Adults. J Am Soc Nephrol 23:1769–1776. https://doi.org/10.1681/ASN.2012040389
Kawaguchi T, Imasawa T, Kadomura M, Kitamura H, Maruyama S, Ozeki T, Katafuchi R, Oka K, Isaka Y, Yokoyama H, Sugiyama H, Sato H (2022) Focal segmental glomerulosclerosis histologic variants and renal outcomes based on nephrotic syndrome, immunosuppression and proteinuria remission. Nephrol Dial Transplant 37:1679–1690. https://doi.org/10.1093/ndt/gfab267
Acknowledgements
I dedicate this paper to my late mentor, Dr. Hisano, who unfortunately passed away midway, with gratitude for his guidance and support.
Author information
Authors and Affiliations
Contributions
E.K., S.H., and T.N. designed the study; E.K., S.K., K.M., N.U., T.M., Y.H., and S.S. acquired and analyzed data; E.K. and T.N. interpreted the data; E.K., R.K., and T.N. drafted the article; E.K., S.K., K.M., N.U., T.M., Y.H., S.S., R.K., and T.N. approved the final version.
Corresponding author
Ethics declarations
Ethics approval
This study was approved by the Human Ethics Review Committee of UOEH (IRB number: UOEHCR23-035).
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Katafuchi, E., Hisano, S., Kurata, S. et al. Aberrant localization of β1 integrin in podocyte cytoplasm of primary FSGS with cellular lesion. Virchows Arch (2024). https://doi.org/10.1007/s00428-024-03919-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00428-024-03919-0