
Abstract
Primary hyperparathyroidism with parathyroid tumors is a typical manifestation of Multiple Endocrine Neoplasia Type 1 (MEN1) and is historically termed “primary hyperplasia”. Whether these tumors represent a multi-glandular clonal disease or hyperplasia has not been robustly proven so far. Loss of Menin protein expression is associated with inactivation of both alleles and a good surrogate for a MEN1 gene mutation. The cyclin-dependent kinase inhibitor 1B (CDKN1B) gene is mutated in MEN4 and encodes for protein p27 whose expression is poorly studied in the syndromic MEN1 setting.
Here, we analyzed histomorphology and protein expression of Menin and p27 in parathyroid adenomas of 25 patients of two independent, well-characterized MEN1 cohorts. The pattern of loss of heterozygosity (LOH) was assessed by fluorescence in situ hybridization (FISH) in one MEN1-associated parathyroid adenoma. Further, next-generation sequencing (NGS) was performed on eleven nodules of four MEN1 patients.
Morphologically, the majority of MEN1 adenomas consisted of multiple distinct nodules, in which Menin expression was mostly lost and p27 protein expression reduced. FISH analysis revealed that most nodules exhibited MEN1 loss, with or without the loss of centromere 11. NGS demonstrated both subclonal evolution and the existence of clonally unrelated tumors.
Syndromic MEN1 parathyroid adenomas therefore consist of multiple clones with subclones, which supports the current concept of the novel WHO classification of parathyroid tumors (2022). p27 expression was lost in a large fraction of MEN1 parathyroids and must therefore be used with caution in suggesting MEN4.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Multiple Endocrine Neoplasia Type 1 (MEN1) is a rare disorder with autosomal dominant inheritance, characterized by multiple tumors in endocrine organs [1]. Tumors of the parathyroid, the anterior pituitary gland, and the entero-pancreatic system are most frequent in MEN1 [1, 2]. MEN1 is clinically defined as the occurrence of tumors in two of the aforementioned organs or the occurrence of one tumor in combination with a first-degree relative having a known MEN1 disorder.
The first manifestation of the disease is most often primary hyperparathyroidism (PHPT) [3], i.e., hypercalcemia resulting from parathyroid tumors (single- versus multi-glandular disease), both of which are described in this context. As MEN1 patients usually have multi-glandular disease [4], the subtotal or total parathyroidectomy with autotransplantation is associated with the lowest risk of persistence and recurrence [5,6,7]. There appears to be a spectrum of disease aggression in MEN1. Histologically, neither clear-cut morphological nor recurrence-prompting features are reliably reproducible in MEN1 parathyroid tumors [8].
Multiple mutations have been described in the MEN1 gene [9, 10] without a clear genotype–phenotype correlation [11]. MEN type 4 (MEN4), a more recently described MEN syndrome [12, 13], is caused by germline mutations in cyclin-dependent kinase inhibitor 1B (CDKN1B) [14] which encodes protein p27. MEN4 can mimic MEN1 [15], and CDKN1B mutations are implicated in the development of parathyroid adenomas as well [16,17,18].
The MEN1 gene is a tumor suppressor gene, located on the long arm of chromosome 11 (11q13). It encodes the nuclear protein Menin [19], an epigenetic modifier, whose expression in MEN1 parathyroid adenomas is not extensively studied [20, 21]. Further, its role in predisposing to tumor formation is not yet fully understood [22]. The genotype does not always correspond to the same phenotype; the occurrence of tumors, the course of the disease, and the expression of symptoms vary even with the same mutation in the same family [23]. The genotype/phenotype mismatch is still under investigation, but there is some evidence that a variable degree of CpG island methylation of the MEN1 gene, or its gene promoters could have an impact on the phenotype [24]. In duodenal and pancreatic MEN1-associated tumors and microtumors, it has been shown that the wild-type allele is inactivated most frequently by large deletions [25,26,27] and less frequently by point-mutations or promoter hypermethylation.
While MEN1-associated hyperparathyroidism traditionally was described as “primary hyperplasia”, increasing evidence points toward a multi-glandular neoplastic disease rather than a simple hyperplasia of the gland [28]. Some authors found a biological rationale for monoclonality [29], while others postulated independent genetic events within one enlarged parathyroid [30]. Finally, previous evidence by genetic probing suggested a coexistence of different clones in independent evolutionary stages [31].
The aim of this study was to further investigate the molecular background of syndromic MEN1 parathyroid adenomas using fluorescence in situ hybridization, next-generation sequencing with copy number analysis based on the relative number of reads and allelic imbalances, and immunohistochemistry to determine the potential presence of hyperplasia or of multiple independent clones and the amount of Menin loss. As MEN4 may be a clinical differential diagnosis of MEN1, p27 expression was also investigated to assess its value in suggesting a respective germline mutation.
Material and methods
Patient samples and clinical data
This retrospective study includes 25 MEN1 patients from the Medical University of Vienna and Bern University Hospital with surgical removal of parathyroid tumors between 1992 and 2022 (Table 1). MEN1 germline mutations of 23 patients (92.0%) have been confirmed during the diagnostic genetic workup via sequencing in the respective institutions The spectrum of mutations is heterogeneous with only three recurrent mutations in two patients each (mutations summarized in Table S1).
Sex, age at surgery, presence of nephro-/urolithiasis, family history, preoperative osteodensitometry, parathyroid hormone (PTH), calcium, albumin, creatinine, and 25-hydroxyvitamin D levels; the type of surgical exploration, autotransplantation, number of parathyroid glands identified and removed, and synchronous thymectomy were extracted from electronic medical records at both participating institutions. In addition, the follow-up interval, persistence (biochemical hyperparathyroidism within the first 6 months postoperatively), and recurrence (biochemical hyperparathyroidism after 6 months of normal parathyroid function) were documented postoperatively.
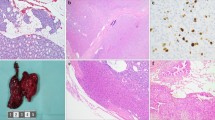
Morphologically distinct intra-glandular nodules and micronodules (< 5 mm, arbitrarily defined in analogy to microtumors of the pancreas [32]) were counted based on one hematoxylin–eosin (H&E) section per patient (compare Fig. 1).

FISH analysis. A MEN1 parathyroid adenoma (H&E) consisting of ten distinct (micro)nodules (N) (extended version in Fig. S1). B FISH analysis of micronodules N3 and N8 reveals a heterogeneity of patterns; while N3 shows mostly MEN1 loss of heterozygosity (LOH), N8 shows centromere 11 loss in most cells. Arrowheads indicate MEN1 LOH. Arrows indicate centromere 11 loss. Red fluorophore indicates MEN1 11q13 locus and green fluorophore indicates CEN11p 11p11.12 locus
The study was conducted in accordance with the Declaration of Helsinki (1964) and ethical approval was obtained from the local ethics committees in Vienna, Austria (reference number “2239/2019”) and Bern, Switzerland (“KEK-BE 105–2015”).
Fluorescence in situ hybridization (FISH)
Sections of two parathyroid adenomas (MEN1 and non-MEN1) were hybridized using a commercial MEN1 FISH probe (Abnova, Taipei, Taiwan; FG0040; MEN1/CEN11p FISH probe, genomic DNA, human origin) according to the manufacturer’s instructions (http://www.abnova.com/support/protocols.asp) and local standard operating procedures (e.g. in [33]). The formalin-fixed, paraffin-embedded (FFPE) FISH “PreTreatment Kit 1” (Abnova) was used for pretreatment. Using 4′,6-diamidino-2-phenylindole (DAPI) as a counterstain, the Texas red fluorophore was used to label the MEN1 11q13 locus (approximately 500 kb), and the green (FITC) fluorophore to label the CEN11p 11p11.12 locus (approximately 630 kb). Tissue regions were photographed with an Olympus XM10 monochrome digital microscope camera using an Olympus BX61VS microscope system (Olympus, Tokyo, Japan). Up to seven regions were examined in each morphologically distinct intra-glandular nodule (“N”) (Fig. 1A, Fig. S1). Per region, all signals per cell were visually counted by one pathologist (KB).
Next-generation sequencing (NGS)
NGS has been performed in parathyroid adenomas of four MEN1 patients. Genomic DNA was extracted from 1-mm tissue punches of two to three nodules per patient (Fig. S2) and sequenced using the “True Sight Oncology (TSO) 500 DNA” panel (Illumina, San Diego, CA) [34] according to manufacturer instructions with adequate coverage depth. The allelic imbalance ratio was calculated for each single nucleotide polymorphism (SNP) from 0 to 1. Tumor cell content per region has been at least 70%. Significant copy number alterations, i.e., large chromosomal aberrations, were identified based on the combination of the number of reads and allelic frequency using in-house standards. Areas of (morphologically) diffuse hyperplasia (with the inclusion of fat cells) were too small to be microdissected and could not be sequenced.
Immunohistochemistry (IHC)
Sections (2.5 µm) were deparaffinized in dewax solution (Leica Biosystems, Muttenz, Switzerland) and rehydrated. p27 IHC was performed on a BOND RX (Leica), and Menin IHC on a benchmark automated immunostainer (Ventana, Roche, Arizona, USA). Slides were scanned on a Panoramic 250 Flash scanner (3DHistech, Budapest, Hungary). Staining conditions were as follows: Menin (A300–105A, Bethyl Lab, Fortis Life Sciences, Massachusetts, USA), dilution 1:800, retrieval—citrate buffer, 32 min, 100 °C; antibody incubation—60 min at 37 °C. p27 (SX53G8; #427 M, Cell Marque, Sigma-Aldrich, California, USA), dilution 1:500, retrieval—citrate buffer, 30 min, 100 °C; antibody incubation—15 min at room temperature. Immunohistochemistry was evaluated by two pathologists (KB, AP) on whole slides. For both markers, only nuclear staining was considered positive. According to previous literature [35], p27 is expressed on an average of approximately 60% of neoplastic cells in parathyroid adenomas. Therefore, we defined reduced nuclear expression as expression in less than 60% but more than 20% of neoplastic cells. Menin (p27) expression was considered as lost (absent) only in the presence of an internal positive control. All non-neoplastic cells, i.e., mainly endothelium and stroma (on slide), and a non-MEN1 adenoma were used as positive controls (Fig. 2A). Negative on-slide controls were not available.

Menin and p27 immunohistochemistry. A A non-MEN1 parathyroid adenoma control shows retained and strong nuclear MEN1 expression. Nuclear p27 expression is retained in most cells (> 60%). B An exemplary MEN1 parathyroid adenoma with loss of nuclear menin expression (preserved expression in the endothelium; arrowheads). Focal absence of nuclear p27 expression
Statistics
Statistical analyses were performed using SPSS version 28.0 (SPSS Inc., Chicago, IL, USA). Two-sided Pearson’s χ2 tests were used to calculate contingency tables; p < 0.05 was considered statistically significant. Correlation analyses were conducted using Spearman’s rho coefficients (ρ).
Results
The 25 MEN1 patients included had a mean age at surgery of 40.0 years (SD 9.3) (Table 1). Surgical procedures comprised 14 (56.0%) total, five subtotal (20.0%), and six (24.0%) less than subtotal parathyroidectomies with 13 (52.0%) synchronous thymectomies. The mean follow-up was 88.2 months (SD 76.6).
Syndromic MEN1 adenomas consist of multiple morphologically distinct nodules
While parathyroid hyperplasia shows diffuse, nodular, or mixed growth patterns [36], typical morphological stigmata for MEN1 parathyroid adenomas have not been reported. Reliable differentiation from secondary hyperplasia is difficult, if not impossible, based on histomorphology [37]. Importantly, the majority of MEN1 adenomas (16/25 [64.0%]) in our cohorts revealed at least one distinct large nodule with up to 11 additional smaller (micro)nodules (Table 1). Only four patients (16.0%) did not show any evidence of intra-glandular (micro)nodules.
In total, 17/25 (68.0%) MEN1 adenomas showed at least one fibrous septum, and 19/25 (76.0%) adenomas were at least partially encapsulated (Fig. 1A). The majority were composed mainly of chief cells (18/25 [72.0%]). 5/25 (20.0%) MEN1 adenomas showed at least a focal rim of adjacent non-neoplastic parathyroid tissue. Focal endocrine atypia in terms of larger irregular hyperchromatic nuclei was found in 3/25 (12.0%) MEN1 adenomas. In 15/25 (60.0%) samples focal remaining fat was observed. All 25/25 (100%) MEN1 adenomas showed sparse stromal fat, at least focal cystic configurations, and an absence of calcifications, necrosis, or invasion of adjacent skeletal muscles or vessel invasion.
FISH suggests individual molecular states in syndromic MEN1 adenomas
To further investigate MEN1 loss of heterozygosity (LOH), we compared a MEN1 parathyroid adenoma (“Bern 3”, c.563G > C/p.W188S mutation) with a non-MEN1 parathyroid adenoma using a MEN1 specific and centromere 11 (C11) fluorescence in situ hybridization (FISH) probe.
In the examined non-MEN1 adenoma, three intra-glandular micronodules could be distinguished. The MEN1 adenoma showed ten nodules (Fig. 1A, Fig. S1). Even though there were many non-interpretable signals, we consistently noticed MEN1 loss of heterozygosity (LOH) and/or centromere 11 in the micronodules of the MEN1 adenoma (Fig. 1B). Quantitatively, centromere 11 loss and MEN1 LOH were much more frequent in the MEN1 adenoma compared to the non-MEN1 adenoma.
In immunohistochemistry, all evaluable (micro)nodules showed complete loss of Menin and reduced p27 protein expression.
Next-generation sequencing (NGS) reveals both clonally distinct tumors and subclonal evolution
NGS confirmed the known MEN1 germline mutations in all investigated (n = 4) patients and nodules (n = 11). No additional driver mutations could be detected in any of the sequenced regions. All the investigated regions showed evidence of clonality and not of hyperplasia, by evidence of LOH of the MEN1 wild-type allele (Fig. S2, Table 2). In two parathyroids, we observed different second hits of the MEN1 region, i.e., copy number–neutral (CNN) LOH and biallelic loss, highly suggestive of the coexistence of clonally independent tumors (Fig. S2A and S2D). We also found evidence of subclonal evolution, i.e., some intra-glandular nodules have acquired additional aberrations (Fig. S2C). One adenoma shows the same aberrations in both investigated nodules (Table 2).
The morphology of subclonal or clonal nodules was not significantly different. Some nodules showed cystic transformation, and other regions were slightly more oncocytic (Fig. S2) without a clear correlation with the genotype.
Tumor mutational burden (TMB) was low in all eleven investigated nodules (each nodule < 4 somatic mutations per megabase), and all were microsatellite stable.
Immunohistochemistry for Menin and p27
In this study, we observed a complete loss of Menin expression in MEN1 parathyroid adenomas (Fig. 2, Fig. S3, Table 3) in all regions evaluable. 9/25 (36.0%) patients showed positive internal controls. Nuclear Menin expression was lost regardless of the type of mutation present. Unfortunately, only 2/25 (8.0%) samples were evaluable in the entire tissue section. 7/25 (28.0%) samples were partially evaluable, mostly in the periphery of the tissue section. As the probes were collected over a time span of 20 years, 16/25 (64.0%) of the samples were not evaluable due to differing preanalytical conditions.
p27 was reduced in 11/25 (44.0%) samples. Six of 25 (24.0%) samples showed a complete absence of expression, while expression was retained in 8/25 (32.0%) of the samples. Among the samples with at least partially evaluable Menin expression, p27 expression was reduced in four (44.4%) and retained in five (55.6%) samples, respectively.
Recurrent PHPT correlates with younger patient age and preoperative PTH levels
Little is known about risk factors for the recurrence of PHPT in MEN1 patients. In all, 5/25 (20.0%) patients had recurrent PHPT, the earliest 8 months after surgery. Neither the number of identified glands (ρ = 0.092; p = 0.675) nor the number of removed glands (ρ = 0.12; p = 0.954) correlated with recurrent PHPT. However, we found a significant correlation between recurrent disease with younger patient age (ρ = − 0.486; p = 0.014) and higher preoperative PTH levels (ρ = 0.571; p = 0.004). The number of intra-glandular (micro)nodules did not show a significant correlation with clinical features or outcomes, respectively.
Discussion
MEN1 parathyroid adenomas consist of multiple morphologically distinct nodules and micronodules. We observed clonal molecular changes in all nodules, consequently we did not detect nodular hyperplastic areas. Therefore, we provide further evidence that parathyroid tumors in MEN1 are multiple monoclonal tumors and not hyperplastic changes, supporting the nomenclature of multi-glandular adenomas as suggested in the novel WHO classification of “Endocrine and Neuroendocrine Tumours” (5th edition, 2022) [38].
To the best of our knowledge, multiple individual clones in MEN1 parathyroid adenomas have not been robustly proven to date. Using restriction fragment length polymorphism of the x chromosome–linked PGK gene and differential methylation of cytosine residues in 14 non-MEN1 patients, nodular parathyroid hyperplasia was suggested to be monoclonal and diffuse parathyroid hyperplasia to be polyclonal [29]. Genetic analysis of 14 MEN1 parathyroid adenomas revealed evidence of monoclonality as evidenced by allelic loss [31]. It has been hypothesized that inactivation of the MEN1 gene leads to tumor initiation and progression. FISH is a reliable method to detect LOH [39]. We show evidence of clonality in all examined (micro)nodules using FISH in one patient. The nodules showed MEN1 LOH, and in all those nodules, we could prove MEN1 inactivation by loss of immunohistochemical Menin expression with retained immunoreactivity in internal positive controls. Our observations are comparable to those in tumors of the endocrine pancreas where LOH was shown for tumors but not for islet hyperplasia in MEN1 [32]. NGS confirmed all known germline MEN1 mutations, no additional oncogenic driver mutations were detected in > 500 investigated genes. We found evidence for both subclonal evolution as well as clearly clonally unrelated nodules in the same parathyroid indicated by different patterns of MEN1 LOH and chromosomal imbalances. Morphologically, we could not distinguish these molecular patterns. The chromosomal imbalances observed in clones and subclones have been previously described in sporadic parathyroid adenomas, i.e., copy number neutral LOH of MEN1 [10]; 1q and 15q loss [40]; partial loss of chromosome 9, 3q, 16q, and 17p gains [41]; as well as abnormal ploidy [42]. One of our patients demonstrated a reproducible 19q loss, while only 19q gains have been described in (“large”) parathyroid tumors [41]. Interestingly, there was little concordance regarding the observed aberrations (only 16q loss observed in two patients), underlining not only an intra-tumoral but also inter-patient genetic heterogeneity, indicating different clonal evolutions in individual patients.
mRNA analyses have demonstrated that Menin expression is downregulated in MEN1 parathyroid tumors [43]. This is consistent with immunohistochemistry, where Menin expression was lost in the nucleus of tumor cells. The reported sensitivity and specificity of Menin immunohistochemistry for MEN 1 are 86% and 87%, respectively [20]. Its interpretation is difficult because MEN1 does not necessarily correlate with a complete loss of protein expression [37, 44]. Here, Menin immunohistochemistry was only evaluable in the entire tissue of two and at the well-fixed periphery of seven samples. A recent study [21] suggests that Menin immunohistochemistry is a useful screening tool for MEN1 especially when genetic testing is inconclusive or inaccessible. The authors describe nuclear Menin loss in parathyroid tumors in 16/16 (100.0%) MEN1 patients of their single-center cohort, with MEN4 not taken into account. Our findings are in line with this, as we could confirm Menin loss in all, at least partially evaluable, samples. However, while MEN1 stainings work reproducibly with recently standardized pre-analytics, in this archival series over many years, reliable results could be detected only in 36.0% of cases.
The absence of p27 protein expression has been described in primary hyperparathyroidism and MEN1 mutant parathyroids [45, 46]. Here, immunohistochemistry suggested that the p27 protein is mostly downregulated in MEN1 with a concomitant absence of expression in MEN1 patients. In general, parathyroid adenomas show reduced nuclear p27 positivity compared to hyperplasia or normal parathyroid glands [35]. As p27 can be (partially) lost in sporadic parathyroid adenomas [35, 37, 45], interpretation is difficult and does not prove a syndromic background. MEN4 is clinically a differential diagnosis of MEN1, our results show that loss of p27 expression cannot be used as potential evidence for this disease as p27 is frequently negative in MEN1 parathyroids as well.
Finally, the clinical management of MEN1 parathyroid adenomas is an ongoing debate [7]. In this study, we observed few significant clinicopathologic correlations. Histologically, a rim of adjacent atrophic parathyroid tissue is described to occur in the majority of (non-syndromic) parathyroid adenomas [47,48,49]. Here, only a fifth of our MEN1 samples showed this histomorphological feature.
Our study has several limitations. First, although we study a large cohort of MEN1 parathyroid adenomas from two academic centers, our study relies mainly on archived FFPE material. Although comparatively large, the number of patients included is still small for statistical analysis. Second, the Menin antibody used was only satisfactory on parts of the sections of in total of seven (28.0%) patients. Differences in fixation and processing procedures may explain the heterogeneity of protein expression, as our experience is much better in samples processed in recent years. Moreover, reproducible cutoffs for p27 immunohistochemistry in MEN1 have not been defined so far with conflicting observations. Third, some micronodules were lost in deeper sections, making direct comparison of staining difficult. Fourth, the FISH procedure led to relatively weak signals (frequently not interpretable) and could only be performed on a single MEN1 adenoma. Therefore, we interpret the results of our FISH analysis with caution. Fifth, CDKN1B germline mutation analysis to rule out MEN4 has not been performed, but respective mutations were excluded in the somatic NGS analysis. Copy-number calls derived from relative read numbers are semi-quantitative and insensitive for short genetic alterations.
In summary, we provide molecular evidence that enlarged MEN1 parathyroids consist of multiple subclones as well as clonally unrelated tumors in an autonomous evolutionary trajectory and that the term “primary hyperplasia” is therefore biologically incorrect. Complete nuclear Menin loss is frequent in MEN1 parathyroids, which may be of help in separation from secondary hyperparathyroidism. Ultimately, as p27 protein expression was at least partially absent in the majority of our MEN1 samples, the use of p27 immunohistochemistry cannot reliably suggest MEN4 in patients.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR et al (2012) Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 97(9):2990–3011
Marx SJ (2018) Recent topics around multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 103(4):1296–1301
Herath M, Parameswaran V, Thompson M, Williams M, Burgess J (2019) Paediatric and young adult manifestations and outcomes of multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 91(5):633–638
Schreinemakers JMJ, Pieterman CRC, Scholten A, Vriens MR, Valk GD, Borel Rinkes IHM (2011) The optimal surgical treatment for primary hyperparathyroidism in MEN1 patients: a systematic review. World J Surg 35(9):1993–2005
Pieterman CRC, van Hulsteijn LT, den Heijer M, van der Luijt RB, Bonenkamp JJ, Hermus ARMM et al (2012) Primary hyperparathyroidism in MEN1 patients: a cohort study with longterm follow-up on preferred surgical procedure and the relation with genotype. Ann Surg 255(6):1171
Wilhelm SM, Wang TS, Ruan DT, Lee JA, Asa SL, Duh QY et al (2016) The American Association of Endocrine Surgeons guidelines for definitive management of primary hyperparathyroidism. JAMA Surg 151(10):959–968
Nastos C, Papaconstantinou D, Kofopoulos-Lymperis E, Peppa M, Pikoulis A, Lykoudis P et al (2021) Optimal extent of initial parathyroid resection in patients with multiple endocrine neoplasia syndrome type 1: a meta-analysis. Surgery 169(2):302–310
Erickson LA, Mete O (2018) Immunohistochemistry in diagnostic parathyroid pathology. Endocr Pathol 29(2):113–129
Lemos MC, Thakker RV (2008) Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 29(1):22–32
Cromer MK, Starker LF, Choi M, Udelsman R, Nelson-Williams C, Lifton RP et al (2012) Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. J Clin Endocrinol Metab 97(9):E1774-1781
Sharretts JM, Simonds WF (2010) Clinical and molecular genetics of parathyroid neoplasms. Best Pract Res Clin Endocrinol Metab 24(3):491–502
Alrezk R, Hannah-Shmouni F, Stratakis CA (2017) MEN4 and CDKN1B mutations: the latest of the MEN syndromes. Endocr Relat Cancer 24(10):T195-208
Singeisen H, Renzulli MM, Pavlicek V, Probst P, Hauswirth F, Muller MK, Adamczyk M, Weber A, Kaderli RM, Renzulli P (2023) Multiple endocrine neoplasia type 4: a new member of the MEN family. Endocr Connect Jan 24;12(2):e220411. https://doi.org/10.1530/EC-22-0411
Schernthaner-Reiter MH, Trivellin G, Stratakis CA (2016) MEN1, MEN4, and Carney Complex: Pathology and Molecular Genetics. Neuroendocrinology 103(1):18–31
Seabrook A, Wijewardene A, De Sousa S, Wong T, Sheriff N, Gill AJ et al (2022) MEN4, the MEN1 mimicker: a case series of three phenotypically heterogenous patients with unique CDKN1B mutations. J Clin Endocrinol Metab 107(8):2339–2349
Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Höfler H et al (2006) Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci 103(42):15558–15563
Agarwal SK, Mateo CM, Marx SJ (2009) Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab 94(5):1826–1834
Costa-Guda J, Arnold A (2014) Genetic and epigenetic changes in sporadic endocrine tumors: parathyroid tumors. Mol Cell Endocrinol 386(1):46–54
Thakker RV (2010) Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab 24(3):355–370
Grolmusz VK, Borka K, Kövesdi A, Németh K, Balogh K, Dékány C et al (2017) MEN1 mutations and potentially MEN1-targeting miRNAs are responsible for menin deficiency in sporadic and MEN1 syndrome-associated primary hyperparathyroidism. Virchows Arch 471(3):401–411
Verschuur AVD, Kok ASM, Morsink FHM, de Leng WWJ, van den Broek MFM, Koudijs MJ et al (2023) Diagnostic utility of menin immunohistochemistry in patients with multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol 47(7):785
Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR et al (1997) Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 276(5311):404–407
Concolino P, Rossodivita A, Carrozza C, Raffaelli M, Lombardi CP, Rigante D et al (2008) A novel MEN1 frameshift germline mutation in two Italian monozygotic twins. Clin Chem Lab Med 46(6):824–826
De Paoli-Iseppi R, Prentice L, Marthick JR, Thomson R, Holloway AF, Dickinson JL et al (2018) Multiple endocrine neoplasia type 1: clinical correlates of MEN1 gene methylation. Pathology (Phila) 50(6):622–628
Cebrián A, Ruiz-Llorente S, Cascón A, Pollán M, Díez JJ, Picó A et al (2003) Mutational and gross deletion study of the MEN1 gene and correlation with clinical features in Spanish patients. J Med Genet 40(5):e72–e72
Giacché M, Panarotto A, Mori L, Daffini L, Tacchetti MC, Pirola I et al (2012) A novel menin gene deletional mutation in a little series of Italian patients affected by apparently sporadic multiple endocrine neoplasia type 1 syndrome. J Endocrinol Invest 35(2):124–128
Raef H, Zou M, Baitei EY, Al-Rijjal RA, Kaya N, Al-Hamed M et al (2011) A novel deletion of the MEN1 gene in a large family of multiple endocrine neoplasia type 1 (MEN1) with aggressive phenotype. Clin Endocrinol (Oxf) 75(6):791–800
Doherty GM, Lairmore TC, DeBenedetti MK (2004) Multiple endocrine neoplasia type 1 parathyroid adenoma development over time. World J Surg 28(11):1139–1142
Tominaga Y, Kohara S, Namii Y, Nagasaka T, Haba T, Uchida K et al (1996) Clonal analysis of nodular parathyroid hyperplasia in renal hyperparathyroidism. World J Surg 20(7):744–752
Dwight T, Nelson AE, Theodosopoulos G, Richardson AL, Learoyd DL, Philips J et al (2002) Independent genetic events associated with the development of multiple parathyroid tumors in patients with primary hyperparathyroidism. Am J Pathol 161(4):1299–1306
Friedman E, Sakaguchi K, Bale AE, Falchetti A, Streeten E, Zimering MB et al (1989) Clonality of Parathyroid tumors in familial multiple endocrine neoplasia type 1. N Engl J Med 321(4):213–218
Perren A, Anlauf M, Henopp T, Rudolph T, Schmitt A, Raffel A et al (2007) Multiple endocrine neoplasia type 1 (MEN1): loss of one MEN1 allele in tumors and monohormonal endocrine cell clusters but not in islet hyperplasia of the pancreas. J Clin Endocrinol Metab 92(3):1118–1128
Wartenberg M, Centeno I, Haemmig S, Vassella E, Zlobec I, Galván JA et al (2016) PTEN alterations of the stromal cells characterise an aggressive subpopulation of pancreatic cancer with enhanced metastatic potential. Eur J Cancer 1(65):80–90
Conroy JM, Pabla S, Glenn ST, Seager RJ, Roey EV, Gao S et al (2021) A scalable high-throughput targeted next-generation sequencing assay for comprehensive genomic profiling of solid tumors. PLoS ONE 16(12):e0260089
Erickson LA, Jin L, Wollan P, Thompson GB, van Heerden JA, Lloyd RV (1999) Parathyroid hyperplasia, adenomas, and carcinomas: Differential Expression of p27: Kip1: protein. Am J Surg Pathol 23(3):288
Duan K, Hernandez KG, Mete O (2015) Clinicopathological correlates of hyperparathyroidism. J Clin Pathol 68(10):771–787
Turchini J, Gill AJ (2020) Hereditary parathyroid disease: sometimes pathologists do not know what they are missing. Endocr Pathol 31(3):218–230
Erickson LA, Mete O, Juhlin CC, Perren A, Gill AJ (2022) Overview of the 2022 WHO classification of parathyroid tumors. Endocr Pathol 33(1):64–89
Scheie D, Andresen PA, Cvancarova M, Bø AS, Helseth E, Skullerud K et al (2006) Fluorescence in situ hybridization (FISH) on Touch preparations: a reliable method for detecting loss of heterozygosity at 1p and 19q in oligodendroglial tumors. Am J Surg Pathol 30(7):828
Palanisamy N, Imanishi Y, Rao PH, Tahara H, Chaganti RSK, Arnold A (1998) Novel chromosomal abnormalities identified by comparative genomic hybridization in parathyroid adenomas. J Clin Endocrinol Metab 83(5):1766–1770
Sulaiman L, Nilsson IL, Juhlin CC, Haglund F, Höög A, Larsson C et al (2012) Genetic characterization of large parathyroid adenomas. Endocr Relat Cancer 19(3):389–407
Chryssochoos JT, Weber CJ, Cohen C, Moore J, DeRose PB, Hagler M et al (1995) DNA index and ploidy distinguish normal human parathyroids from parathyroid adenomas and primary hyperplastic parathyroids. Surgery 118(6):1041–1050
Bhuiyan MMR, Sato M, Murao K, Imachi H, Namihira H, Takahara J (2000) Expression of menin in parathyroid tumors. J Clin Endocrinol Metab 85(7):2615–2619
Juhlin CC, Erickson LA (2021) Genomics and epigenomics in parathyroid neoplasia: from bench to surgical pathology practice. Endocr Pathol 32(1):17–34
Borsari S, Pardi E, Pellegata NS, Lee M, Saponaro F, Torregrossa L et al (2017) Loss of p27 expression is associated with MEN1 gene mutations in sporadic parathyroid adenomas. Endocrine 55(2):386–397
Sengul Aycicek G, Aydogan BI, Sahin M, Cansız Ersoz C, Sak SD, Baskal N (2018) Clinical impact of p27Kip1 and CaSR expression on primary hyperparathyroidism. Endocr Pathol 29(3):250–258
van der Walt J (2012) Pathology of the parathyroid glands. Diagn Histopathol 18(6):221–233
Baloch ZW, LiVolsi VA (2013) Pathology of the parathyroid glands in hyperparathyroidism. Semin Diagn Pathol 30(3):165–177
Akerström G, Malmaeus J, Bergström R (1984) Surgical anatomy of human parathyroid glands. Surgery 95(1):14–21
Acknowledgements
The authors acknowledge the Translational Research Unit at the Institute of Tissue Medicine and Pathology, University of Bern, Switzerland, for their excellent technical support.
Funding
Open access funding provided by University of Bern. The study was supported by the “Fondation Rolf Gaillard pour la recherche en endocrinologie, diabétologie et métabolisme”.
Author information
Authors and Affiliations
Contributions
KB, CN, AP, and RK designed the study, provided, analyzed and interpreted the data, and wrote the manuscript. AD and MN established and performed fluorescence in situ hybridization. TG performed and analyzed next-generation sequencing. PR, CS, BN, PM, RT, and NK provided and reviewed data. All authors revised the manuscript and approved its final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Figure S1
(Micro-)Nodules in a MEN1 parathyroid adenoma. A: Ten morphologically identifiable nodules (N1) and micronodules (N2 to N10); overview. B: (Micro-)Nodules in high resolution. (PDF 4686 KB)
Supplementary Figure S2
Next-generation sequencing (NGS) of eleven nodules (“N”) in four MEN1 patients. A (patient “Vienna 16”, Table S1): N3 demonstrates biallelic MEN1 loss and is probably a different tumor, while N1 and N2 show conventional MEN1 LOH. N1 and N2 have different gains supporting subclonal evolution. B (patient “Vienna 9”): N4 and N5 demonstrate the same gains and losses, and represent most probably the same tumor. C (patient “Bern 1”): All three nodules share a CNN LOH of chromosome 11, as well as 16q and 19q losses. N6 and N8 have identical additional aberrations, suggesting N7 to be the primary clone with subclonal evolution in N6 and N8. Interestingly, N7 and N8 have cystic components. D (“Bern 4”): N9, N10 and N11 show different types of MEN1 inactivation supporting the concept of different tumors. In addition, N9 has further losses, N10 and N11 do not have.
Bars (y-axis): number of reads; CNN: copy number neutral; LOH: Loss of heterozygosity; scatter plot: allelic frequency/single nucleotide polymorphisms (SNPs); x-axis: chromosomes (on the left q-arm, on the right p-arm). (PDF 7888 KB)
Supplementary Figure S3
Menin loss in a MEN1 parathyroid adenoma. A: MEN1 parathyroid adenoma (H&E), on the right Menin immunohistochemistry (overview). B: Loss of Menin expression in a larger clone, adjacent non-neoplastic parathyroid tissue (arrow) and intermingled non-neoplastic cells (arrowhead) with retained Menin expression. Inset: 40x magnification.(PDF 287 KB)
Supplementary Table S1
(DOCX 16.1 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bräutigam, K., Nesti, C., Riss, P. et al. Syndromic MEN1 parathyroid adenomas consist of both subclonal nodules and clonally independent tumors. Virchows Arch 484, 789–798 (2024). https://doi.org/10.1007/s00428-023-03730-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-023-03730-3