
Abstract
Emerging entities and molecular subgroups in large B-cell lymphomas (LBCLs) were discussed during the 2022 European Association for Haematopathology/Society for Hematopathology workshop in Florence, Italy. This session focused on newly recognized diseases and their diagnostic challenges. High-grade/large B-cell lymphoma with 11q aberration (HG/LBCL-11q) is defined by chromosome 11q-gains and telomeric loss. FISH analysis is recommended for the diagnosis. HG/LBCL-11q can occur in the setting of immunodeficiency, including ataxia-telangiectasia, and predominates in children. The morphological spectrum of these cases is broader than previously thought with often Burkitt-like morphology and coarse apoptotic bodies. It has a Burkitt-like immunophenotype (CD10+, BCL6+, BCL2−) but MYC expression is weak or negative, lacks MYC rearrangement, and is in contrast to Burkitt lymphoma 50% of the cases express LMO2. LBCL with IRF4 rearrangement (LBCL-IRF4) occurs mainly in the pediatric population but also in adults. LBCL-IRF4 has an excellent prognosis, with distinguishing molecular findings. IRF4 rearrangements, although characteristic of this entity, are not specific and can be found in association with other chromosomal translocations in other large B-cell lymphomas. Other molecular subgroups discussed included primary bone diffuse large B-cell lymphoma (PB-DLBCL), which has distinctive clinical presentation and molecular findings, and B-acute lymphoblastic leukemia (B-ALL) with IGH::MYC translocation recently segregated from Burkitt lymphoma with TdT expression. This latter disorder has molecular features of precursor B-cells, often tetrasomy 1q and recurrent NRAS and KRAS mutations. In this report, novel findings, recommendations for diagnosis, open questions, and diagnostic challenges raised by the cases submitted to the workshop will be discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diffuse large B-cell lymphoma (DLBCL) accounts for ~40% of non-Hodgkin lymphomas (NHL), comprising specific subtypes or disease entities [1,2,3]; however, most cases fall into the category of “not otherwise specified” (NOS). DLBCL, NOS represents a heterogeneous group that has been divided based on the gene expression profile (GEP) of B-cells or cell of origin (COO) into two subtypes: germinal center B-cell (GCB), and activated B-cell (ABC) [4]. A small subset of DLBCL is considered “unclassified” and not fitting in any category. Nonetheless, this COO binary classification is insufficient to capture the heterogeneity and the complexity of this disease. The characterization of MYC rearrangement (R) associated with BCL2-R and/or BCL6-R as well as double-hit/dark zone signature has further emphasized the biological diversity of DLBCL [5,6,7]. More recently, the integration of molecular data including structural variants, mutational profile, and somatic copy-number alterations has identified at least 7 DLBCL subgroups with putative clinical relevance, demonstrating the complexity of DLBCL [8,9,10,11]. Among other LBCL with specific sites of involvement and characteristic pathological features are primary mediastinal B-cell lymphoma, and the group of LBCL of the central nervous system, vitreoretinal compartment, and testes, considered immune privileged sites [12, 13]. Others arising in the setting of immune dysfunction or in association with EBV or HHV8 infectious agents have been also identified as specific disease entities [14,15,16].
The presence of specific and recurrent genetic alterations has identified new molecular subgroups of LBCL. Among them, LBCL with IRF4 rearrangement (LBCL-IRF4) [17,18,19], and high-grade/large B-cell lymphoma with 11q aberration (HG/LBCL-11q) with features resembling Burkitt lymphoma (BL) without MYC-R, emerged as new entities in the 2022 International Consensus Classification (ICC) and in the 5th edition of the WHO classification [20,21,22]. However, due to their rarity, some questions regarding their distinct pathological and molecular features or their clinical presentation and outcome need to be addressed. In addition, the issue of what is the best strategy to diagnose these new entities and what technique would be most appropriate in routine diagnosis must be addressed. Other peculiar and rare LBCL such as primary bone diffuse LBCL (PB-DLBCL) or those harboring infrequent and/or complex cytogenetic abnormalities are sometimes difficult to classify accurately.
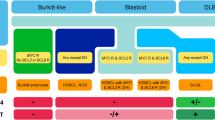
The major theme of the 2022 European association for Haematopathology (EA4HP) and Society for Hematopathology (SH) workshop in Florence, Italy, was “Provisional and emerging disease entities.” A session was dedicated to emerging entities and molecular subgroups in LBCL. Fifty-six cases were submitted, representing the many challenges in the diagnosis of these new recognized lymphomas both in children and adults. The cases were divided into the following thematic groups to illustrate diagnostic dilemmas and/or interesting biological features:
-
1.
High-grade/large B-cell lymphoma with 11q aberration (HG/LBCL-11q)
-
2.
Large B-cell lymphoma with IRF4 rearrangement (LBCL-IRF4)
-
a.
In pediatric population and young adults (<25 years)
-
b.
In adult patients (>25 years)
-
a.
-
3.
Aggressive B-cell lymphomas with IRF4 and BCL2/MYC/CCND1 rearrangements
-
4.
Other molecular groups in large B-cell lymphomas
High-grade/large B-cell lymphoma with 11q aberrations
HG/LBCL-11q is an aggressive mature B-cell lymphoma with a characteristic chromosome 11q-gain/loss pattern, displaying variable morphological features ranging from a typical BL-like morphology to a more intermediate or blastoid appearance [2, 20, 23]. HG/LBCL-11q cases occurs as a localized nodal (most commonly head and neck; 60–70%) or extranodal disease (gastro-intestinal tract 30–40%). The defining genetic event is a complex aberration involving the long arm of chromosome 11 (11q), showing a gain in 11q23.2-23.3 and a telomeric loss in 11q24.1-qter, in the absence of a MYC-R. Rare cases do not have the gain of 11q23.3 [21, 22, 24]. The 5th WHO lymphoma classification and the 2022 ICC recognize cases with only telomeric loss and/or solely telomeric loss of heterozygosity as bona-fide cases of HG/LBCL-11q [25, 26]. However, further studies are needed to corroborate whether these cases belong to the same disease. The presence of 11q23.3 gains, as the only alteration, is considered non-specific, and therefore not enough for the diagnosis. In contrast to BL, mutations in the ID3-TCF3 pathway, recognized as the biological hallmark of BL, are not detected in HG/LBCL-11q. The mutational landscape of HG/LBCL-11q is closer to that of DLBCL of GCB-type, and GNA13 mutations are observed in about 50% of cases [21, 22]. These new data, together with the absence of the IGH::MYC rearrangement, suggest that HG/LBCL-11q represents a different mature aggressive B-cell lymphoma [20].
In the workshop, 20 cases were submitted with the diagnosis of HG/LBCL-11q (Supplemental Table 1). The diagnosis was confirmed in 18 cases that were considered to represent good examples of HG/LBCL-11q. The clinicopathological features are summarized in Table 1. Male patients predominated (M/F=13/5) with median age at presentation of 22.5 years (range 7–72 years). Thirteen cases were children and young adults (range 7–31 years), whereas 5 patients were >40 years (44–72 years). From the cases submitted, 10 cases showed nodal presentation, whereas 8 cases involved extranodal sites. One patient (LYWS-1073, submitted by Rex Au-Yeng) showed various sites of involvement at different time points. The disease was first diagnosed in a cervical lymph node, and at relapse 2 years later presented in the tonsil. Interestingly, 2 cases occurred in the setting of primary immunodeficiency (LYWS-1125 presented by Olga Balague and LYWS-1442 submitted by Peggy Dartigues). Both cases were known to have ataxia-telangiectasia (A-T) diagnosed at the age of 11 and 13 years, respectively. A-T is the most common DNA repair disorder, caused by the presence of biallelic pathogenic variants in the ATM gene, and characterized by a very high risk of developing hematological malignancies during childhood, especially DLBCL and T-cell acute lymphoblastic leukemia (ALL), but rarely BL [27]. Yet, the association with HG/LBCL-11q has not been reported. This peculiar 11q-gain/loss aberration has been described to be particularly frequent in posttransplant patients, who develop B-cell lymphomas with Burkitt-like morphology [28]. It has also been described in the setting of HIV infection [29,30,31].
The morphological spectrum of the submitted cases varied from blastoid to intermediate to large B-cell lymphomas (Fig. 1). The cases with intermediate morphology displayed cellular pleomorphism, variation in nuclear size, shape, and larger nucleoli ranging from cases very similar to BL to other more akin to DLBCL. Only one case (LYWS-1242 submitted by Snjezana Dotlic) showed large cell morphology. In 10 cases, a starry-sky pattern was present, and in 5 cases, coarse apoptotic debris was observed. In a recent study, coarse apoptotic bodies were reported as characteristic of HG/LBCL-11q, and an important morphological feature to suspect the diagnosis and to prompt the FISH analysis [20, 32]. The cases submitted to the workshop confirmed this contention; however, although a helpful feature to suspect the diagnosis, it was not seen in many cases, and therefore, its absence does not exclude the diagnosis. All cases displayed a GCB phenotype applying the Hans algorithm (CD10+, BCL6+), and a high proliferation index (Ki67>90%). However, 3 cases showed MUM1 and BCL2 expression (LYWS-1129; LYWS-1237; LYWS-1217). A recent study demonstrated that 46% of HG/LBCL-11q expresses LMO2, a germinal center marker, using the monoclonal antibody SP51, as opposed to BL, which is negative [21]. Accordingly, 7/15 (46%) cases investigated were LMO2 positive. The workshop cases confirmed that LMO2 is a useful marker to support HG/LBCL-11q over BL in difficult cases. MYC expression varied from case to case, detected in 20 to 60% of tumor cells but always displaying weak staining. In all cases, MYC-R was excluded by FISH analysis with break-apart probes (BAP). However, BAP cannot identify all MYC-positive cases and 4–20% of cases might remain undetected [33, 34]. The panel tested the cases for IGH::MYC, IGL::MYC, and IGK::MYC translocations. All cases were negative, thus confirming the diagnosis of HG/LBCL-11q. FISH analyses for BCL2-R and BCL6-R with BAP were negative in all cases.

The morphological spectrum of high-grade/large B-cell lymphoma with 11q aberration: the morphological spectrum varied from blastoid (A) to cases with intermediate morphology, ranging from cases very similar to BL (B) to other more similar to DLBCL (C); only one case was submitted with typical large cell morphology (D). Starry-sky pattern with conspicuous, coarse apoptotic debris was detected in 5 cases. E FISH analysis showed 11q gain/loss in all cases (F). A–D H&E stain; E Giemsa stain; F FISH analysis with ZytoLight® SPEC 11q gain/loss Triple Color Probe
In 5 cases, the results of next-generation sequencing (NGS) were reported by the submitters. In the remaining cases, for which material was available, the panel performed NGS analysis with a custom panel of 80 genes. The results are reported in Fig. 2 and Supplemental Table 2. The mutational landscape of the workshop cases showed similarities but also differences with previous studies [21, 22]. The previously reported mutations in DDX3X, MYC, KRAS, EZH2, CREBBP, and FOXO1 were confirmed. Typical BL driver mutations (ID3 or TCF3) were not identified; however, there were some differences for genes reported to be more frequently mutated in HG/LBCL-11q. In the workshop cases, mutations in PTEN, TP53, FOXO1, and RHOA were frequently demonstrated. Of note, mutations in ATM, CCND3, and RHOA have not been previously described in HG/LBCL-11q. Of interest, in addition to the two cases with A-T (LYWS-1125 and LYWS-1442), ATM mutations were found in two additional cases (LYWS-1217 and LYWS-1220), with an allele frequency of 49% and 39%, respectively. These mutations may well represent heterozygous germline variants, which have been associated also with increased risk for neoplasia [35, 36]. Thus, these findings further support and expand the association of HG/LBCL-11q with immunodeficiency syndromes. Finally, Fig. 3 shows a comparison of the mutational patterns of BL, HG/LBCL-11q, high-grade B-cell lymphoma (HGBL) with double/triple-hit, and DLBCL. Mutations in GNA13, MYC, and TP53 are shared by BL, HG/LBCL-11q, HGBL, and DLBCL. BTG2 and DYRK1A gene mutations are considered typical for HG/LBCL-11q, engaged in FOXO1 and cyclin regulation, and in the TP53 pathway, respectively.

Mutational profile of HG/LBCL-11q cases submitted to the workshop. The mutational landscape of the workshop cases showed similarities but also difference with previous studies. Mutations in GNA13, DDX3X, MYC, KRAS, EZH2, CREBBP, and FOXO1 were confirmed and new mutations in ATM, CCND3, and RHOA were identified. Typical Burkitt lymphoma driver mutations (ID3 or TCF3 were not detected). The comparison was performed with the studies of Gonzalez-Farre et al. [21] and Wagener et al. [22]

Comparison of the mutational patterns of Burkitt lymphoma (BL), high-grade/large B-cell lymphoma (HG/LBCL)-11q, high-grade B-cell lymphoma (HGBL) wit double/triple hit, and diffuse large B-cell lymphoma (DLBCL). Mutations in GNA13, MYC, and TP53 are shared by BL, HG/LBCL-11q, HGBL, and DLBCL. BTG2 and DYRK1A gene mutations are considered typical for HG/LBCL-11q
Information regarding the outcome was available in 15 cases. Complete remission (CR) was achieved in 11 cases, 8 pediatric, and 3 adult patients; however, treatment was variable (R-CHOP, DA-R-EPOCH, or R-EPOCH). Three cases relapsed, and only one case died of lymphoma after 2 weeks from the diagnosis. Interestingly, all cases in the pediatric group received BL protocols achieving CR, while the three patients that relapsed were adults (LYWS-1224; LYWS-1234; LYWS-1444), received variable treatments, and carried TP53 mutations.TP53 mutations have not been reported in HG/LBCL-11q, so far; nonetheless, it is remarkable that all cases were adults and had a bad prognosis. The question is whether TP53 mutation is a marker of clonal progression or whether cases with TP53 mutations and 11q aberrations in adults are more closely related to DLBCL, NOS. Additional mutations in these cases included HIST1HE1, KMT2D, SETD2, PTEN NSD2, and STAT3, which are more characteristic of DLBCL. However, further studies are warranted to answer this question.
In two cases (LYWS-1171 submitted by Fang Liu and LYWS-1237 submitted by Julie Bruneau), the panel could not confirm the diagnosis of HG/LBCL-11q. In case LYWS-1171, the FISH analysis showed only gain of 11q23.3 but not loss at 11q24.1 region. As discussed above, the 11q gain is not specific for HG/LBCL-11q because it also occurs in other mature aggressive B-cell lymphomas. In this case, because of lack of material, the panel could not perform additional studies and rendered the final diagnosis of HGBL, NOS. The second case LYWS-1237, with the proposed diagnosis of “BL with unusual BCL2 expression,” showed morphology that was compatible with BL but the strong BCL2 and MUM1 expression, along with a complex karyotype, MYC-R and 11q aberration, were quite atypical for a BL diagnosis. In contrast to HG/LBCL with 11q aberration, this case showed strong, homogeneous MYC expression. The NGS analysis confirmed a mutational profile more compatible with HGBL, NOS. The recommendation of the panel was to classify these cases (MYC-R with 11q aberration) as HGBL, NOS, based on the mutational analysis, although further studies are necessary to answer this question.
Important issues were raised during the discussion of the session, in particular the best technical approach to confirm the diagnosis. The recommendation of the panel was to perform FISH analysis for 11q aberration with commercially available probes in all MYC-R negative cases with an intermediate/blastoid or Burkitt-like morphology. However, in cases where the telomeric loss cannot be demonstrated by FISH, it is necessary to perform additional molecular analysis (Oncoscan/CGH) to confirm or exclude the diagnosis. Another key issue discussed was if FISH analysis for 11q should be performed in all MYC-R BL, particularly in those cases with an atypical phenotype, to identify potential 11q aberrations with MYC-R, as demonstrated in case LYWS-1237. Currently, it is not recommended to do FISH for 11q in typical BL cases with MYC-R; however, this should be encouraged for cases with MYC-R and atypical BL morphology or phenotype.
Large B-cell lymphoma with IRF4 rearrangement
LBCL-IRF4 was introduced, as a provisional entity, in the 2016 revised 4th WHO classification within the group of follicular lymphomas (FLs) because of its frequent follicular growth pattern and excellent prognosis [2, 37], and now is recognized as a definite entity, both in the 2022 International Consensus Classification (ICC), where it remains in the group of FL [25, 38], and in the 5th WHO classification, where it was moved to the group of LBCL [26]. LBCL-IRF4 occurs mainly in children and young adults with distinctive clinical presentation preferentially involving the tonsils and mucosa-associated lymphoid tissue comprising Waldeyer’s ring, head and neck lymph nodes, and less frequently the intestine. It has a slight male predilection and presents as a localized disease (stages I–II) with excellent prognosis, regardless of the large cell cytology and the growth pattern [17, 19, 24]. It has been estimated that in the pediatric population, around 20% of morphologically FL (grade 3) and DLBCL harbor an IRF4 chromosomal translocation but represent only 1–2% of all lymphomas in children and adolescents [24]. Morphologically, LBCL-IRF4 is often follicular and diffuse but it might be purely follicular or diffuse. The tumor cells are medium or large in size and show the morphology of centroblasts or blastoid cells sometimes with a starry-sky pattern and high proliferation rate. The tumor cells express B-cell markers and often germinal center markers including CD10 and BCL6 together with the constitutive expression of MUM1/IRF4. IGH::IRF4 rearrangements are detected in most cases. LBCL-IRF4 lacks BCL2-R and MYC-R; however, many cases carry BCL6-R (35%) [17]. Molecular studies have revealed a GCB-type GEP, despite the constitutive expression of IRF4, and frequent mutations in IRF4 and NF-kB-related genes (CARD11, CD79B, MYD88) [39]. The most frequent chromosomal alteration is loss of 17p13, where TP53 gene is located [39, 40].
It has been shown that biology and pathogenesis in DLBCL is age-dependent and the cutoff of 18 years used in clinical practice seems rather arbitrary and does not reflect DLBCL biology [41, 42]. Molecular features suggest that a better cutoff might be in the mid-thirties [41]. Accordingly, two recent studies have shown that LBCL-IRF4 in young adults (<40 years) share the localization, morphology and genetics of pediatric patients [40, 43]. In the workshop, 16 cases were submitted with the diagnosis of LBCL-IRF4; for practical purposes, an arbitrary cutoff of 25 years was set to separate pediatric and young adults from adult patients to compare the clinico-pathological features of the two groups. All cases were considered to represent good examples of LBCL-IRF4, some with unique features.
LBCL-IRF4 in patients < 25 years
The cases submitted illustrated the clinical, morphological, and genetic spectrum described in this disease (Table 2 and Supplemental Table 3) [17,18,19, 24, 44]. There were 5 males and 2 females with a median age of 8 years (range 6–25 years). Four cases presented in the tonsil, and one each in a cervical lymph node, intestine, and spleen. The spleen presentation (Case LYWS-1163, submitted by E. Shuyu) is exceptional and occurred in an 18-year-old male, who presented with a single 10-cm mass (Fig. 4A–D). Importantly all cases were in stage I or II and all the patients were in CR, four after systemic chemotherapy. One case without systemic chemotherapy (Case LYWS-1112, submitted by Dehua Wang) was a 7-year-old boy with a 2.2-cm mass in terminal ileum that was surgically resected due to intussusception (Fig. 4E–L). Morphologically, the lymphoma was follicular with typical phenotype and genotype. This case together with case LYWS-1395 and other cases reported in the literature that achieved CR without systemic treatment raise the question of whether chemotherapy can be reduced or a watch-and-wait strategy can be followed for those cases with follicular growth pattern and localized disease after excision [18, 19, 45]. Accordingly, in the pediatric cohort published by the NHL-Berlin-Frankfurt-Münster group, because of the excellent prognosis of these patients, it was recommended therapy de-escalation in future clinical trials [24]. Morphologically, 3 cases were follicular and diffuse, 2 cases diffuse, and 2 cases follicular. In 6 cases, the immunophenotype revealed co-expression of CD10, BCL6, and MUM1. The abnormal co-expression of these three markers should prompt to investigate the diagnosis of LBCL-IRF4 [40]. Only one case was CD10 negative (LYWS-1049). As previously reported [17], 3 cases (3/7, 43%) showed weak CD5 expression (LYWS-1279, LYWS-1112, LYWS-1395), and 6 cases (6/7, 86%) expressed BCL2 without t(14;18) translocation. FISH analyses demonstrated in 6/7 cases an IRF4 break indicating an IRF4 translocation. In one case, presented by Rachel Mariani (case LYWS-1279), with typical localization, morphology, and immunophenotype (Fig. 5A–I), FISH BAP analysis was negative for IRF4 break. NGS demonstrated an IGH::IRF4 juxtaposition supporting the presence of a cryptic IRF4 translocation. The failure to demonstrate an IRF4 break with available FISH probes has been estimated to occur in approximately 10% of LBCL-IRF4 [18, 19, 37, 43, 45]. In cryptic cases, the presence of IGH/IGK/IGL rearrangement helps to support the diagnosis. Furthermore, case LYWS-1279 had, in addition, an IRF4 mutation. The presence of one or multiple mutations affecting the IRF4 gene with an aberrant somatic hypermutation pattern is a hallmark of the IRF4 translocation [37, 39, 40]. Therefore, the presence of IRF4 mutations in the highly conserved N-terminal DNA-binding domain in exon 2, in the correct context, might be used as a surrogate marker for the presence of IRF4 rearrangement [39, 40]. Mutational analysis performed by the panel demonstrated the presence of multiple IRF4 mutations in 4 of 5 cases analyzed (80%) (Supplemental Table 4). The only case without IRF4 mutations was submitted by Eric Hsi (LYWS-1202). This case is unique in that the IRF4 rearrangement was with an unknown partner on chromosome 21, demonstrated by RNA sequencing. In all cases reported until now, the IRF4 translocation partner is either IGH or less frequently IGL or IGK [17, 39]. The significance of a non-IG partner is not known. Surprisingly, three cases (3/5, 60%) showed pathogenic TP53 mutations (LYWS-1279, LYWS-1163, and LYWS-1202); nevertheless, the presence of TP53 mutation did not seem to convey a bad prognosis, since all three cases were in CR 18, 27, and 38 months after diagnosis. In one of the first studies of LBCL-IRF4, TP53 mutations were identified in 3 of 6 cases with 17p loss [46]. A subsequent study in pediatric population demonstrated 17p/TP53 deletions in 25% of the cases; however, no TP53 mutations were identified [39]. Similar findings were reported in an adult cohort [40]. The incidence of TP53 mutations in this disease is unknown and its prognostic significance remains to be established. FISH analyses for BCL2, BCL6, and MYC were performed by the panel in all cases without evidence of translocations. The four cases investigated with GEP revealed a GCB-type signature.

Histologic and immunophenotypic features of large B-cell lymphoma with IRF4-rearrangement presenting in extranodal sites. A–D Case LYWS-1163 courtesy of E. Shuyu. A Spleen section showing a well-circumscribed nodular infiltration. B The tumor is composed of large centroblastic lymphoid cells with open chromatin, several large nuclei, and abundant cytoplasm. C The tumor cells are CD10 and D IRF4/MUM1 strongly positive. E–L Case LYWS-1112 courtesy of D. Wang. E Terminal ileum with a 2.2-cm large polypoid mass. F The intestinal crossed section shows a white soft mass infiltrating the mucosa, submucosa, and the muscularis propria. G H&E section reveals a follicular lymphoid infiltrate with large, back-to-back follicles. The follicles are composed of medium to large-sized centroblasts. H The tumor cells are positive for CD79a, I CD10, J BCL6, and K IRF4/MUM1. L Interphase FISH analysis using break apart probes for IRF4. Most cells have 3 fusion signals (yellow) and 2 red signals with loss of the green signals indicating an IRF4 translocation

Histologic and immunophenotypic features of large B-cell lymphoma with IRF4-rearrangement presenting in the tonsil. Case LYWS-1279 courtesy of R. Mariani. A Panoramic view of a tonsil showing residual normal tonsil areas and lymphoma infiltration with follicular and diffuse pattern. B Higher magnification shows a lymphoid infiltrate composed of large-sized centroblasts. C IRF4/MUM1 stain shows in the normal residual lymphoid tissue few positive plasma cells. The left side shows a follicular growth pattern whereas the left side reveals a diffuse growth pattern. D The tumor cells show an aberrant CD5 expression. Note the strong CD5 expression of the reactive T cells. E The tumor cells are positive for BCL6, and F BCL2. G The MIB1 stain shows the normal polarization of residual germinal centers, whereas the tumor shows a proliferation of approximately 80% in both the follicular and in the diffuse areas. H The CD10 stain is strong and homogeneous positive in the residual germinal center whereas the stain is weak in the follicular areas and partially lost in the diffuse areas. I IGV screenshots of chimeric pairs in chromosomes 6 and 14 supporting the presence of IGH::IRF4 juxtaposition
Case LYWS-1276 submitted by Francisco Llamas-Gutierrez was an interesting case of a 14-year-old boy who presented with a right tonsillar tumor. Although the morphology and phenotype (CD10+, BCL6+ MUM1+) were suggestive of the diagnosis of LBCL-IRF4, no IRF4 break was identified by FISH, and RNAseq analysis also failed to prove the presence of an IRF4 rearrangement. Instead, a BCL6 translocation was demonstrated. Mutational analysis revealed typical mutations of DLBCL without IRF4 mutations, and GEP showed an ABC-type signature. The panel agreed with the diagnosis of DLBCL, NOS, ABC-type with BCL6 translocation, mimicking a LBCL-IRF4. This case highlights the importance of FISH and molecular analysis to render the correct diagnosis.
LBCL-IRF4 in patients >25 years
Nine cases were submitted with this diagnosis in adult patients (Table 2 and Supplemental Table 4). There were 7 males and 2 females with a median age of 69 years (range 37–87 years). In two cases, probably because of the age of the patients, a composite lymphoma was found; one case with lymphoplasmacytic lymphoma (LPL) (LYWS-1076), and one case with a marginal zone lymphoma (LYWS-1320). In contrast to the pediatric cases, adult cases were more often nodal (5 cases) with only two tonsillar presentation, one case with forearm soft tissue involvement, and one oral lesion in an HIV+ patient. Interestingly, a recent study suggests that involvement of skin/soft tissue is relatively frequent in elderly patients, a presentation previously unrecognized [43]. Although cases in adults presented more often with high clinical stages (stages III/IV) (4/9; 44%), all patients achieved CR with R-CHOP (rituximab-cyclophosphamide, hydroxydaunorubicin, oncovine, and prednisone). Morphologically, 3 cases showed FL3B-like morphology, and 6 cases were diffuse. The immunophenotype was similar to the pediatric population (CD10+, BCL6+, BCL2+); however, CD5 expression was not demonstrated in adult patients and in two cases CD10 was negative. Case LYWS-1076 was presented by Dominik Nann and corresponded to a 74-year-old patient in clinical stage III with FL3B-like morphology, and bone marrow (BM) infiltration by a clonally unrelated LPL. This case, in addition to an IRF4 translocation, also carried a BCL6 translocation. BCL6 translocations were reported in 35% (8/23) of the cases in the original description of the disease, six of them in adult patients [17]. However, out of all of the cases submitted to the workshop, both children and adults, only this case carried a BCL6 translocation (1/14; 7%). In general, in adult patients with morphology of FL3B, as demonstrated also by case LYWS-1370, submitted by Bettina Bisig (Fig. 6A–F), it is recommended to stain for MUM1/IRF4, and if positive, to perform FISH analysis for IRF4 [25, 47]. These cases, in contrast to FL3B cases, have an excellent prognosis as demonstrated by the cases submitted to the workshop with complete remission up to 10 years. The two cases that presented in the tonsil corresponded to a 57-year-old man (LYWS-1142, submitted by Austin Gray) and a 79-year-old woman (LYWS-1238, submitted by Konnie Hebeda), with clinical stage I or stage II, respectively. These two cases were clinically, morphologically, and genetically indistinguishable from the cases in the pediatric population confirming the existence of LBCL-IRF4 in elderly patients indistinguishable from the pediatric counterpart [40, 43, 47]. It may also indicate that cases arising in tonsils, Waldeyer’s ring or bowel may represent a distinct clinicopathologic entity regardless of the age of the patient at disease presentation.

Histologic and immunophenotypic features of large B-cell lymphoma with IRF4-rearrangement in an adult patient. Case LYWS-1370 courtesy of B. Bisig. A Panoramic view of a lymph node with preserved capsule and effacement of the architecture by the presence of multiple, large confluent nodules. No diffuse areas or necrosis are observed. B Higher magnification demonstrates that the nodules are composed of large cells with fine chromatin, multiple nucleolei, abundant cytoplasm typical of centroblasts. Some apoptotic figures and some mitosis are observed. C The tumor cells were CD10 positive as well as D BCL6 and E IRF4/MUM1. F Interphase FISH analysis using break apart probes for IRF4 revealed 1 fusion signal (yellow arrow), one red signal (red arrow), and one green signal (green arrow) indicative of an IRF4 rearrangement. G Diagram of the relative positions of IRF4 driver mutations. The approximate location of somatic mutations identified is indicated. The analysis was performed by next generation sequencing. IRF4 mutations are mainly in the DNA binding domain (DBD). Domains of the protein are represented according to the Uniprot database (www.uniprot.org)
Of the 16 cases submitted with the diagnosis of LBCL-IRF4, the panel performed mutational analysis (Ion GeneStudio S5, ThermoFisher Scientific) and GEP (HTG EdgeSeq System, HTG Molecular Diagnostics) in those cases where material was available for molecular studies. Multiple IRF4 mutations in exon 2 were demonstrated in eight cases (8/10; 80%) (Fig. 6G, Supplemental Table 6), whereas GEP demonstrated a GCB-type signature in these cases (8/8; 100%).
Aggressive B-cell lymphomas with IRF4 and BCL2/MYC/CCND1 rearrangements
Seven cases with IRF4-R associated with other genetic alterations, namely BCL2-R, MYC-R, and CCND1-R, were submitted to the workshop (Table 2 and Supplemental Table 5). There were 5 males and 2 females with a median age of 75 years (range 14–92). The only pediatric patient was a 14-year-old girl with a plasmablastic lymphoma (PBL) with IGK::IRF4 and IGH::MYC rearrangements. The other 6 cases were elderly patients. Case LYWS-1024 was presented by Holly Berg and corresponded to a 92-year-old woman who presented with a scalp mass in stage IIIA. The case was CD10+, BCL6+, MUM1+, BCL2+, and CD5 weak positive. FISH analyses demonstrated IRF4 and BCL2 rearrangements. Mutational analysis revealed gene mutations characteristic of DLBCL including KMT2D, CREBBP, BCL2, CCND3, NOTCH2, and DNMT3A. No IRF4 mutations were identified. A similar case was submitted by James Cook (case LYWS-1086). Recent studies have suggested that large B-cell lymphomas with IRF4 and BCL2 rearrangements in elderly patients show mutational profiles closer to DLBCL [40, 43]. Therefore, cases with both IRF4 and BCL2 rearrangements should be diagnosed as DLBCL, NOS. Case LYWS-1408 submitted by Maria Rodriguez-Pinilla is a unique case of 75-year-old man, who presented in stage I disease with tonsillar involvement and is in complete remission after 2 years only with rituximab. The immunophenotype was CD10+, BCL6+, MUM1+, BCL2+, cyclin D1+, and SOX11 negative. This case harbored an IRF4-R together with a CCND1-R. The panel performed mutational analysis and identified three CD70 mutations, and SOCS1 and TMSB4X mutations. Clinically, the case behaved indolently, unlike mantle cell lymphoma. The panel rendered the diagnosis of DLBCL, NOS with IRF4 and CCND1 rearrangements. There were two cases of FL (Cases LYWS-1099 and LYWS-1133), one grade 3A with BCL2 and IRF4 rearrangements, and one transformed FL to high-grade lymphoma that in the transformation acquired MYC and IRF4 rearrangements. The meaning of IRF4 rearrangements in other aggressive lymphomas is not well understood and warrants further studies. Importantly, these cases should not be diagnosed as LBCL-IRF4.
Other molecular groups in large B-cell lymphomas
The 13 cases included in this group were very heterogeneous and represented single examples of different molecular subgroups (Supplemental Table 7). Three cases were thoroughly discussed during the workshop representing novel concepts in lymphoma biology and classification. The case presented by Gabriel Caponetti (LYWS-1026) corresponded to a 69-year-old male who presented with 2-week history of fatigue and shortness of breath. Peripheral blood analysis demonstrated leukocytosis (57.3/L), thrombocytopenia (68,000/L) without anemia, and 28% blasts with Burkitt-like morphology (Fig. 7A–O). The patient had lymphadenopathy and splenomegaly. The BM aspirate revealed 81% blasts. Although the progenitor markers CD34 and TdT were absent, the neoplasm lacked CD20 and surface immunoglobulin (sIg) findings that can be seen with immaturity. The tumor cells expressed CD19, PAX5, CD10, MUM1, and MYC, and showed a proliferation rate of 100%. The karyotype revealed an abnormal male karyotype: 47,XY,+i(1)(q10), t(8;14)(q24.2;q32)[8]/46,XY[12]. FISH analysis confirmed the MYC rearrangement. Mutational analysis demonstrated a NRAS mutation and two TP53 mutations. Cases similar to this have been reported in the literature as BL with supernumerary isochromosome 1q resulting in tetrasomy 1q [48,49,50]. However, recent studies have demonstrated that these cases are better classified as “B-acute lymphoblastic leukemia (B-ALL) with MYC rearrangement.” [25, 51, 52] B-ALL with MYC rearrangement is recognized in the 2022 ICC as a specific entity [25] and is included in the 5th WHO classification within the group of “B-ALL with other rearrangements.” [26] The disease affects mostly male patients (range 3–75 years), and the tumor cells have an immature phenotype, often with expression of TdT and CD10 without CD20, BCL6 and sIg; however lack CD34. The lack of TdT expression, as in this case, does not preclude this diagnosis. The IG::MYC translocation derives from an aberrant (out-of-frame) VDJ recombination in an immature B-cell, and not in a germinal center B-cell like BL [53]. Molecularly, these cases are also distinct from BL and show frequent gains in chromosome 1q21.1-q44 and mutations in NRAS and KRAS [51, 54]. The prognosis is poor, as demonstrated in this case; the patient died 5 months after the initial diagnosis. The panel agreed with the final diagnosis of B-ALL with MYC rearrangement.

Histologic, immunophenotype, and cytogenetic features of a B-cell acute lymphoblastic leukemia with IGH::MYC according to the 2022 ICC. Case LYWS-1026 courtesy of G. Caponetti. A CT scan demonstrates supradiaphragmatic lymphadenopathy (red arrow). B CT scan reveals a soft tissue lesion expanding the posterior aspect of the left iliacus muscle (red arrow) C PET scan shows intense FDG uptake by the soft tissue mass (red arrow). D The bone marrow aspirate shows 81% blasts with vacuolated cytoplasm typical of Burkitt lymphoma. E The bone marrow biopsy is hypercellular for age and shows a diffuse infiltrate with relatively large blastoid cells. F The tumor cells are positive for CD19. G CD10, H TdT, and I CD34 are negative. J CD10 is strongly positive in the tumor cells. K BCL2 is negative, whereas L MYC is strongly positive. M The MIB1 stain shows a 100% proliferation. N The karyotype analysis reveals an abnormal male karyotype with a supernumerary isochromosome for the entire long arm of chromosome 1, which results in tetrasomy 1q (black arrow head). Additionally, a balanced translocation between chromosome 8 and 14 is observed with breakpoints at bands 8q24.2 and 14q32 resulting in a IGH::MYC translocation. O Interphase FISH analysis using break apart probes for MYC, BCL2, and BCL6 reveals in MYC 1 fusion signal (yellow arrow), one red signal (red arrow), and one green signal (green arrow) indicative of MYC rearrangement. In contrast, the analyses of BCL2 and BCL6 demonstrate two normal fusion signals (yellow arrows)
Another interesting case (LYWS-1231) presented by Yen-Chun Liu corresponded to a 24-year-old female who presented with left knee pain for 3 years (Fig. 8). Imaging studies demonstrated in the left femur a lobulated mass extended from the mid-diaphysis into the distal epiphysis including both the medial and lateral femoral condyles (Fig. 8A–B). No additional lesions were demonstrated. The bone biopsy was diagnostic of a DLBCL with CD20 and BCL6 positivity but negative for CD10, MUM1, cyclin D1, and EBER. Whole-genome sequence demonstrated the presence of the following mutations: EZH2Y646N, IRF8Y23H, TNFRSF14, and UBR5, and complex chromosomal alterations. The patient was treated with 2 cycles of chemotherapy (cytarabine and methotrexate) and has been in complete remission for the last 3 years. PB-DLBCL is a disease with characteristic clinical presentation, and morphological and genetic features [55, 56]. It tends to affect younger patients; occurs more frequently in the femur, followed by the pelvis, vertebrae, and humerus; and has an excellent prognosis. Unlike most extranodal lymphomas, PB-DLBCL shows a centrocyte-like GCB-type GEP and a characteristic mutational profile similar to FL including alterations in B2M, EZH2, IRF8, and TNFRSF14 [55]. The distinctive clinicopathological features suggest that PB-DLBCL is an extranodal manifestation of DLBCL; however, the possibility of an extranodal t(14;18)-negative FL with predominantly large centrocytes, similar to primary cutaneous follicle center lymphoma, cannot be ruled out. Further studies are needed to characterize better this type of lymphomas.

Primary diffuse large B-cell lymphoma of the bone (A–B). Case LYWS-1231 courtesy of Y-Ch. Liu. A–B Imaging studies (MRI) of the left femur shows a lobulated mass extended from the mid diaphysis into the distal epiphysis including both the medial and lateral femoral condyles. B The bone biopsy showed a polymorphic infiltrate predominantly of large cells confirming the diagnosis of diffuse large B-cell lymphoma. C–L Diffuse large B-cell lymphoma with CCND1 rearrangement. Case LYWS-1380 courtesy of K.S. Kurz. C Lymph node biopsy with effaced architecture by a diffuse infiltrate of predominantly medium-sized to large blast cells. D Giemsa stain reveals that the tumor cells have basophilic cytoplasm and round to oval nuclei with open chromatin and prominent nucleolus. E Cyclin D1 is positive in the tumor cells. F In other areas of the lymph node the tumor cells were cyclin D1 negative. G CD5 stain is positive in the reactive T cells but negative in the tumor cells. H SOX11 remains negative. I MUM1 is positive in the majority of tumor cells whereas CD10 remained negative (not shown). J Ki-67 demonstrate a high proliferation rate. K–L Interphase FISH analysis using break apart probes for CCND1 and MYC reveals 1 fusion signal (yellow arrow), one red signal (red arrow), and one green signal (green arrow) indicative of CCND1 (K) and MYC (L) rearrangements
Overexpression of cyclin D1 in DLBCL has been associated mostly with gains of CCND1 gene copies [57, 58]. The demonstration of IGH::CCND1 rearrangement leading to cyclin D1 overexpression can occur in DLBCL and poses a diagnostic challenge with blastoid or pleomorphic mantle cell lymphoma [59,60,61]. Most cases have been reported associated with other chromosomal translocations including BCL6, BCL2, and MYC. The translocation has been reported mostly as secondary genetic event in the evolution of DLBCL and other lymphomas such as CLL [62] and FL [63]. Accordingly, the case LYWS-1380 presented by Katrin S. Kurz corresponded to a 58-year-old man, who presented in clinical stage IIA. Morphologically, the lymph node showed a diffuse infiltration by large cells that were CD20+, MUM1+, and BCL2+ (Fig. 8C–L). The tumor cells were negative for CD10, BCL6, CD5, and SOX11. The proliferation rate was 90% and MYC was positive in around 50% of the cells. Interestingly, cyclin D1 was strongly positive only in a part of the lymph node and the rest remained cyclin D1 negative. FISH analysis with BAP for CCND1 demonstrated a break only in those areas where cyclin D1 was positive indicating that the translocation was indeed a secondary genetic event in the evolution of this lymphoma. Moreover, MYC-R was also identified in the same area. The FISH analysis for BCL2 and BCL6 showed normal signals. Mutational analyses demonstrated several mutations (CARD11, CD79B, FOXO1, PIM1, SOCS1), supporting the diagnosis of DLBCL. The GEP performed by the panel confirmed the ABC-type signature. In general, cases with large cell morphology and cyclin D1 positivity but lack CD5 and SOX11 expression should raise the possibility of DLBCL over MCL. The expression of MUM1 or FOXP1 may be useful to achieve the correct diagnosis [59].
Histologic transformation of small B-cell NHL to aggressive lymphoma is well recognized. However, transformation to an aggressive lymphoma with plasmablastic morphology and phenotype is rare [64]. Plasmablastic transformation has been reported in CLL, FL, and rarely LPL [64] [65]. Although these transformed tumors mimic primary PBL, they are not associated with immunodeficiency, and rarely have EBV infection or MYC alterations [64]. Furthermore, they display features indicating clonal evolution from a small B-cell lymphoma and not de novo PBL. Two cases submitted to the workshop (LYWS-1167 submitted by Silvia Tse Bunding, and LYWS-1405 submitted by Anu Peter) represented nice examples of cases that progressed/transformed to an aggressive lymphoma with PBL-like features. Case LYWS-1405 was a 68-year-old man with 8 years history of CLL, who received different treatments including ibrutinib in the last 3 years. He presented with a right groin mass that was biopsied and diagnosed as PBL with a MYC-R. Molecular studies demonstrated that the CLL and PBL were clonally related and shared the same TP53 mutation. Interestingly, recent reports have associated treatment of CLL with the BTK inhibitor ibrutinib and Richter’s transformation mimicking PBL [66,67,68]. Further studies are warranted to understand PBL-like transformation in CLL and in other lymphomas.
Conclusions
Because of new and improved techniques used for standard diagnosis and translational research, we have witnessed a growing list of genetic alterations, variably present in large B-cell lymphomas that are useful for diagnosis or to understand the biology and pathogenesis of certain diseases. Some of these alterations (IRF4-R and 11q aberration) became the defining genetic alteration of new disease entities (Box 1).
HG/LBCL-11q is defined by chromosome 11q-gains and telomeric loss and can occur in the setting of immunodeficiency including posttransplant and HIV+ patients but also in patients with A-T. The disease predominates in children and has a broad morphological spectrum and a Burkitt-like immunophenotype, but MYC expression is weak or negative, lacks MYC-R, and harbors a different mutational profile compared to BL. LMO2 is a useful marker but expressed only in 50% the cases. FISH analysis is recommended for the diagnosis, and in ambiguous cases, other more sophisticated chromosomal analysis is warranted. Whether concurrent TP53 mutations identified in adult patients represent clonal evolution or DLBCL with TP53 and 11q aberrations warrants further studies. Nonetheless, the presence of TP53 mutations conveyed a dismal prognosis in this cohort.
LBCL-IRF4 is mainly a disease of children and young adults but it also occurs in adults and elderly patients. In contrast to children and young adults, where the disease presents more often in the Waldeyer’s ring and in clinical stages I/II, in adults presents more often as nodal disease and in more advanced clinical stages. Nevertheless, the prognosis is excellent. Morphologically and genetically, the disease seems to be the same in children and adults. The aberrant expression of CD10, BCL6 and MUM1/IRF4 should prompt to perform IRF4 FISH analysis. In the cases submitted to the workshop, BCL6-R were rare when compared to other published series (7% vs 35%). In cases with FL3B, MUM1 should be performed, and if positive, IRF4 FISH is recommended. Cryptic IRF4-R by FISH occur in around 10% of the cases; however, in the correct context, breaks in IGH, IGK and IGL, as well as IRF4 mutations, support the diagnosis. The cases submitted to the workshop also raised important questions that need to be resolved in the future: (1) What is the prognostic significance of TP53 mutations? (2) Does localized disease with a follicular growth pattern needs systemic treatment, especially in children and young adults where a watch and wait strategy seems to be justified? (3) Do cases that have been reported with ABC-type GEP belong to the same disease?
IRF4-R can be observed in a variety of aggressive B-cell lymphomas in association with other chromosomal translocations. The IRF4-R might be the initial event but also a secondary event or acquired during transformation. These cases should not be diagnosed as LBCL-IRF4. More studies are warranted to characterize better these cases.
Finally, novel molecular groups were also discussed highlighting challenging features with some groups needing special attention such as PB-DLBCL, B-ALL with IGH::MYC rearrangement, DLBCL with CCND1-R, and PBL-like transformation.
Box 1 • HG/LBCL-11q is defined by chromosome 11q-gains and telomeric loss and can occur in the setting of immunodeficiency including post-transplant and HIV+ patients but also in patients with ataxia-telangiectasia. • FISH analysis is recommended for the diagnosis of HG/LBCL-11q. • LMO2 is a useful diagnostic marker for HG/LBCL-11q but expressed only in 50% of the cases. Burkitt lymphoma is LMO2 negative. • LBCL-IRF4 is mainly a disease of children and young adults but it also occurs in adults and elderly patients. • LBCL-IRF4 in children and young adults affects predominantly the Waldeyer’s ring and presents in clinical stages I/II; in contrast, in adults, it is more often a nodal disease and presents in advanced clinical stages (III/IV). • LBCL-IRF4 is an indolent lymphoma with excellent prognosis. • Morphologically, LBCL-IRF4 can be purely follicular, follicular/diffuse, or diffuse and often shows aberrant expression of CD10, BCL6, and MUM1. CD5 expression is not rare, especially in children. • In adults, IRF4 rearrangements can be observed in a variety of aggressive B-cell lymphomas in association with other chromosomal alterations. These cases should not be diagnosed as LBCL-IRF4. • Other novel molecular groups are discussed. |
Data availability
Not applicable
References
Gascoyne RD, Campo E, Jaffe ES et al (2017) Diffuse large B-cell lymphoma, NOS. In: Swerdlow SH, Campo E, Harris NL et al (eds) WHO classification of tumours of haematopoetic and lymphoid tissues. International Agency for Research on Cancer, Lyon
Swerdlow SH, Campo E, Pileri SA et al (2016) The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127(20):2375–2390
Song JY, Dirnhofer S, Piris MA, Quintanilla-Martinez L, Pileri S, Campo E (2023) Diffuse large B-cell lymphomas, not otherwise specified, and emerging entities. Virchows Arch 482(1):179–192
Alizadeh AA, Eisen MB, Davis RE et al (2000) Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403(6769):503–511
Pedersen MO, Gang AO, Poulsen TS et al (2014) MYC translocation partner gene determines survival of patients with large B-cell lymphoma with MYC- or double-hit MYC/BCL2 translocations. Eur J Haematol 92(1):42–48
Copie-Bergman C, Cuilliere-Dartigues P, Baia M et al (2015) MYC-IG rearrangements are negative predictors of survival in DLBCL patients treated with immunochemotherapy: a GELA/LYSA study. Blood 126(22):2466–2474
Scott DW, King RL, Staiger AM et al (2018) High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood 131(18):2060–2064
Morin RD, Arthur SE, Hodson DJ (2022) Molecular profiling in diffuse large B-cell lymphoma: why so many types of subtypes? Br J Haematol 196(4):814–829
Chapuy B, Stewart C, Dunford AJ et al (2018) Molecular subtypes of diffuse large B-cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 24(5):679–690
Schmitz R, Wright GW, Huang DW et al (2018) Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med 378(15):1396–1407
de Leval L, Alizadeh AA, Bergsagel PL et al (2022) Genomic profiling for clinical decision making in lymphoid neoplasms. Blood 140(21):2193–2227
Bonzheim I, Sander P, Salmeron-Villalobos J et al (2022) The molecular hallmarks of primary and secondary vitreoretinal lymphoma. Blood Adv 6(5):1598–1607
Nayak L, Iwamoto FM, LaCasce A et al (2017) PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood 129(23):3071–3073
Hofscheier A, Ponciano A, Bonzheim I et al (2011) Geographic variation in the prevalence of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: a comparative analysis of a Mexican and a German population. Mod Pathol 24(8):1046–1054
Quintanilla-Martinez L, Swerdlow SH, Tousseyn T, Barrionuevo C, Nakamura S, Jaffe ES (2023) New concepts in EBV-associated B, T, and NK cell lymphoproliferative disorders. Virchows Arch 482(1):227–244
Cesarman E, Chadburn A, Rubinstein PG (2022) KSHV/HHV8-mediated hematologic diseases. Blood 139(7):1013–1025
Salaverria I, Philipp C, Oschlies I et al (2011) Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood 118(1):139–147
Liu Q, Salaverria I, Pittaluga S et al (2013) Follicular lymphomas in children and young adults: a comparison of the pediatric variant with usual follicular lymphoma. Am J Surg Pathol 37(3):333–343
Quintanilla-Martinez L, Sander B, Chan JK et al (2016) Indolent lymphomas in the pediatric population: follicular lymphoma, IRF4/MUM1+ lymphoma, nodal marginal zone lymphoma and chronic lymphocytic leukemia. Virchows Arch 468(2):141–157
Salaverria I, Martin-Guerrero I, Wagener R et al (2014) A recurrent 11q aberration pattern characterizes a subset of MYC-negative high-grade B-cell lymphomas resembling Burkitt lymphoma. Blood 123(8):1187–1198
Gonzalez-Farre B, Ramis-Zaldivar JE, Salmeron-Villalobos J et al (2019) Burkitt-like lymphoma with 11q aberration: a germinal center-derived lymphoma genetically unrelated to Burkitt lymphoma. Haematologica 104(9):1822–1829
Wagener R, Seufert J, Raimondi F et al (2019) The mutational landscape of Burkitt-like lymphoma with 11q aberration is distinct from that of Burkitt lymphoma. Blood 133(9):962–966
Leoncini L, Campo E, Stein H, Harris NL, Jaffe ES, Kluin PM (2017) Burkitt-like lymphoma with 11q aberration. In: Swerdlow SH, Campo E, Harris NL et al (eds) WHO classification of Tumours of heamtopoetic and lymphoid tissue. International Agency for Research on Cancer, Lyon, p 334
Au-Yeung RKH, Arias Padilla L, Zimmermann M et al (2020) Experience with provisional WHO-entities large B-cell lymphoma with IRF4-rearrangement and Burkitt-like lymphoma with 11q aberration in paediatric patients of the NHL-BFM group. Br J Haematol 190(5):753–763
Campo E, Jaffe ES, Cook JR et al (2022) The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood 140(11):1229–1253
Alaggio R, Amador C, Anagnostopoulos I et al (2022) The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 36(7):1720–1748
Pastorczak A, Attarbaschi A, Bomken S et al (2022) Consensus recommendations for the clinical management of hematological malignancies in patients with DNA double stranded break disorders. Cancers (Basel) 14(8)
Ferreiro JF, Morscio J, Dierickx D et al (2015) Post-transplant molecularly defined Burkitt lymphomas are frequently MYC-negative and characterized by the 11q-gain/loss pattern. Haematologica 100(7):e275–e279
Baptista MJ, Tapia G, Munoz-Marmol AM et al (2022) Genetic and phenotypic characterisation of HIV-associated aggressive B-cell non-Hodgkin lymphomas, which do not occur specifically in this population: diagnostic and prognostic implications. Histopathology 81(6):826–840
Kim JA, Kim HY, Kim SJ, Kim HJ, Kim SH (2021) A case of Burkitt-like lymphoma with 11q aberration with HIV infection in East Asia and literature review. Ann Lab Med 41(6):593–597
Wang J, Ma L, Guo J, Xi Y, Xu E (2021) Burkitt-like lymphoma with 11q aberration in a patient with AIDS and a patient without AIDS: two cases reports and literature review. Open Med (Wars) 16(1):428–434
Horn H, Kalmbach S, Wagener R et al (2021) A diagnostic approach to the identification of Burkitt-like lymphoma with 11q aberration in aggressive B-cell lymphomas. Am J Surg Pathol 45(3):356–364
Gagnon MF, Pearce KE, Greipp PT et al (2021) MYC break-apart FISH probe set reveals frequent unbalanced patterns of uncertain significance when evaluating aggressive B-cell lymphoma. Blood Cancer J 11(11):184
Chong LC, Ben-Neriah S, Slack GW et al (2018) High-resolution architecture and partner genes of MYC rearrangements in lymphoma with DLBCL morphology. Blood Adv 2(20):2755–2765
van Os NJ, Roeleveld N, Weemaes CM et al (2016) Health risks for ataxia-telangiectasia mutated heterozygotes: a systematic review, meta-analysis and evidence-based guideline. Clin Genet 90(2):105–117
Hall MJ, Bernhisel R, Hughes E et al (2021) Germline pathogenic variants in the ataxia telangiectasia mutated (ATM) gene are associated with high and moderate risks for multiple cancers. Cancer Prev Res (Phila) 14(4):433–440
Pittaluga S, Harris NL, Siebert R, Salaverria I (2017) Large B-cell lymphoma with IRF4 rearrangement. In: Swerdlow SH, Campo E, Harris NL et al (eds) WHO classification of tumours of haematopoietic and lymphoid tissues. International Agency for Research on Cancer, Lyon, France, pp 280–281
Laurent C, Cook JR, Yoshino T, Quintanilla-Martinez L, Jaffe ES (2023) Follicular lymphoma and marginal zone lymphoma: how many diseases? Virchows Arch 482(1):149–162
Ramis-Zaldivar JE, Gonzalez-Farre B, Balague O et al (2020) Distinct molecular profile of IRF4-rearranged large B-cell lymphoma. Blood 135(4):274–286
Frauenfeld L, Castrejon-de-Anta N, Ramis-Zaldivar JE et al (2022) Diffuse large B-cell lymphomas in adults with aberrant coexpression of CD10, BCL6, and MUM1 are enriched in IRF4 rearrangements. Blood Adv 6(7):2361–2372
Klapper W, Kreuz M, Kohler CW et al (2012) Patient age at diagnosis is associated with the molecular characteristics of diffuse large B-cell lymphoma. Blood 119(8):1882–1887
Burkhardt B, Zimmermann M, Oschlies I et al (2005) The impact of age and gender on biology, clinical features and treatment outcome of non-Hodgkin lymphoma in childhood and adolescence. Br J Haematol 131(1):39–49
Berg HE, Peterson JF, Lee HE, McPhail ED (2023) Large B-cell lymphoma with IRF4 gene rearrangements: Differences in clinicopathologic, immunophenotypic and cytogenetic features between pediatric and adult patients. Hum Pathol 131:108–115
Woessmann W, Quintanilla-Martinez L (2019) Rare mature B-cell lymphomas in children and adolescents. Hematol Oncol 37(Suppl 1):53–61
Chisholm KM, Mohlman J, Liew M et al (2019) IRF4 translocation status in pediatric follicular and diffuse large B-cell lymphoma patients enrolled in Children's Oncology Group trials. Pediatr Blood Cancer 66(8):e27770
Salaverria I, Martin-Guerrero I, Burkhardt B et al (2013) High resolution copy number analysis of IRF4 translocation-positive diffuse large B-cell and follicular lymphomas. Genes Chromosomes Cancer 52(2):150–155
Streich S, Frauenfeld L, Otto F et al (2023) Prevalence of IRF4 rearrangement in large B-cell lymphomas of the Waldeyer's ring in adults. Virchows Arch 482(3):551–560
Siddiqi A, Madhusudhana S, Glazyrin A (2018) 'Blastoid' variant of Burkitt lymphoma with additional partial 1q tetrasomy. Mol Clin Oncol 8(5):637–639
Roug AS, Wendtland P, Bendix K, Kjeldsen E (2014) Supernumerary isochromosome 1, idic(1)(p12), leading to tetrasomy 1q in Burkitt lymphoma. Cytogenet Genome Res. 142(1):7–13
Sato Y, Kurosawa H, Fukushima K, Okuya M, Arisaka O (2016) Burkitt-type acute lymphoblastic leukemia with precursor B-cell immunophenotype and partial tetrasomy of 1q: a case report. Medicine (Baltimore) 95(10):e2904
Wagener R, Lopez C, Kleinheinz K et al (2018) IG-MYC (+) neoplasms with precursor B-cell phenotype are molecularly distinct from Burkitt lymphomas. Blood 132(21):2280–2285
Li Y, Gupta G, Molofsky A et al (2018) B lymphoblastic leukemia/lymphoma with Burkitt-like morphology and IGH/MYC rearrangement: report of 3 cases in adult patients. Am J Surg Pathol 42(2):269–276
Kuppers R, Dalla-Favera R (2001) Mechanisms of chromosomal translocations in B-cell lymphomas. Oncogene 20(40):5580–5594
Varano G, Raffel S, Sormani M et al (2017) The B-cell receptor controls fitness of MYC-driven lymphoma cells via GSK3beta inhibition. Nature 546(7657):302–306
de Groen RAL, van Eijk R, Bohringer S et al (2021) Frequent mutated B2M, EZH2, IRF8, and TNFRSF14 in primary bone diffuse large B-cell lymphoma reflect a GCB phenotype. Blood Adv 5(19):3760–3775
Li X, Xu-Monette ZY, Yi S et al (2017) Primary bone lymphoma exhibits a favorable prognosis and distinct gene expression signatures resembling diffuse large B-cell lymphoma derived from centrocytes in the germinal center. Am J Surg Pathol 41(10):1309–1321
Vela-Chavez T, Adam P, Kremer M et al (2011) Cyclin D1 positive diffuse large B-cell lymphoma is a post-germinal center-type lymphoma without alterations in the CCND1 gene locus. Leuk Lymphoma 52(3):458–466
Chen BJ, Ruminy P, Roth CG et al (2019) Cyclin D1-positive mediastinal large B-cell lymphoma with copy number gains of CCND1 Gene: a study of 3 cases with nonmediastinal disease. Am J Surg Pathol 43(1):110–120
Juskevicius D, Ruiz C, Dirnhofer S, Tzankov A (2014) Clinical, morphologic, phenotypic, and genetic evidence of cyclin D1-positive diffuse large B-cell lymphomas with CYCLIN D1 gene rearrangements. Am J Surg Pathol 38(5):719–727
Al-Kawaaz M, Mathew S, Liu Y et al (2015) Cyclin D1-positive diffuse large B-cell lymphoma with IGH-CCND1 translocation and BCL6 rearrangement: a report of two cases. Am J Clin Pathol 143(2):288–299
Cheng J, Hashem MA, Barabe F et al (2021) CCND1 genomic rearrangement as a secondary event in high grade B-cell lymphoma. Hemasphere 5(1):e505
Schliemann I, Oschlies I, Nagel I, Maria Murga Penas E, Siebert R, Sander B (2016) The t(11;14)(q13;q32)/CCND1-IGH translocation is a recurrent secondary genetic aberration in relapsed chronic lymphocytic leukemia. Leuk Lymphoma 57(11):2672–2676
Koduru PR, Chen W, Garcia R, Fuda F (2015) Acquisition of a t(11;14)(q13;q32) in clonal evolution in a follicular lymphoma with a t(14;18)(q32;q21) and t(3;22)(q27;q11.2). Cancer Genet 208(6):303–309
Martinez D, Valera A, Perez NS et al (2013) Plasmablastic transformation of low-grade B-cell lymphomas: report on 6 cases. Am J Surg Pathol 37(2):272–281
de Leval L, Copie-Bergman C, Rosenwald A et al (2017) B-cell lymphomas with discordance between pathological features and clinical behavior. Virchows Arch 471(4):439–451
Aqil B, Kaur A, Ramos J et al (2023) Richter transformation to aggressive plasmablastic neoplasm related to selection of a BTK-mutated clone in a patient with CLL/SLL treated by ibrutinib. Leuk Lymphoma 64(1):242–245
Marvyin K, Tjonnfjord EB, Breland UM, Tjonnfjord GE (2020) Transformation to plasmablastic lymphoma in CLL upon ibrutinib treatment. BMJ Case Rep 13(9)
Kittai AS, Huang Y, Beckwith KA et al (2023) Patient characteristics that predict Richter's transformation in patients with chronic lymphocytic leukemia treated with ibrutinib. Am J Hematol 98(1):56–65
Acknowledgements
The authors thank Vanessa Borgmann and Barbara Mankel from the Institute of Pathology, University of Tübingen, Germany, for performing the mutational analyses and additional FISH analyses in the cases submitted to the workshop. We want to thank also Tim-Colin Schade PhD student at the University of Tübingen for conducting the gene expression profiling. We want to thank HTG Molecular diagnostics Inc., for donating the HTG EdgeSeq DLBCL Cell of Origin assay. The authors also want to thank Drs Virginia Mancini, Margherita Vannucchi, Gioia Di Stefano, Domenico Ferrara, and Stefano Lazzi from University of Siena for performing and evaluating additional immunostainings, FISH, and mutational analyses, and for their invaluable help in organizing the workshop. The authors also thank the workshop participants for their case submissions and for allow us to use the case images in this review.
Funding
Open Access funding enabled and organized by Projekt DEAL. S-B.N is supported by the National Medical Research Council Senior Investigator Clinician Scientist Award (MOH-001104). LQ-M is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research foundation) under Germany’s Excellence Strategy-EXC2180-390900677.
Author information
Authors and Affiliations
Contributions
L.Q-M, C.L, and L.L reviewed all the cases, designed the analysis of the cases, and wrote the manuscript. L.dL, A.W, A.Z, S.D, S.O, L.S, FC, and S-B.N helped to write the manuscript. All authors read and agree with the content of this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 44 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Quintanilla-Martinez, L., Laurent, C., Soma, L. et al. Emerging entities: high-grade/large B-cell lymphoma with 11q aberration, large B-cell lymphoma with IRF4 rearrangement, and new molecular subgroups in large B-cell lymphomas. A report of the 2022 EA4HP/SH lymphoma workshop. Virchows Arch 483, 281–298 (2023). https://doi.org/10.1007/s00428-023-03590-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-023-03590-x