
Abstract
Autoantibodies against islet cell antigens are routinely used to identify subjects at increased risk of symptomatic type 1 diabetes, but their relation to the intra-islet pathogenetic process that leads to positivity for these markers is poorly understood. We screened 556 non-diabetic organ donors (3 months to 24 years) for five different autoantibodies and found positivity in 27 subjects, 25 single- and two double autoantibody-positive donors. Histopathological screening of pancreatic tissue samples showed lesion characteristic for recent-onset type 1 diabetes in the two organ donors with a high-risk profile, due to their positivity for multiple autoantibodies and HLA-inferred risk. Inflammatory infiltrates (insulitis) were found in a small fraction of islets (<5%) and consisted predominantly of CD3+CD8+ T-cells. Islets with insulitis were found in close proximity to islets devoid of insulin-positivity; such pseudo-atrophic islets were present in multiple small foci scattered throughout the pancreatic tissue or were found to be distributed with a lobular pattern. Relative beta cell area in both single and multiple autoantibody-positive donors was comparable to that in autoantibody-negative controls. In conclusion, in organ donors under age 25 years, insulitis and pseudo-atrophic islets were restricted to multiple autoantibody-positive individuals allegedly at high risk of developing symptomatic type 1 diabetes, in line with reports in older age groups. These observations may give further insight into the early pathogenetic events that may culminate in clinically overt disease.
Similar content being viewed by others


Introduction
Insulitis is an inflammatory lesion of the islets of Langerhans characteristic for patients with recent-onset type 1 diabetes. The infiltrate predominantly consists of CD8+ T-lymphocytes of which a fraction is thought to mediate beta cell specific cytotoxicity [1,2,3,4]. It is now widely accepted that clinical onset of type 1 diabetes is preceded by a usually long hidden disease process as judged by the appearance of circulating antibodies against beta cell antigens including islet cell cytoplasm (ICA) [5], insulin (IAA) [6], glutamic acid decarboxylase (GADA) [7], insulinoma-associated protein 2 (IA-2A) [8], and zinc transporter 8 (ZnT8A) [9], months to decades before diagnosis [10, 11]. These autoantibodies tend to develop sequentially rather than simultaneously [12], with IA-2A and ZnT8A usually appearing closer to clinical onset [13]. Both in children and in adults up to 40 years of age, presence of at least two molecularly defined autoantibodies confers 90% risk of developing clinical onset within 20 years, and is now termed asymptomatic type 1 diabetes [10, 11, 14]. However, the relationship between the development of autoantibodies and the development of histopathological hallmarks of type 1 diabetes during the natural history of asymptomatic disease is poorly understood. In a now classical model for the development of type 1 diabetes, autoantibodies are considered to be indicators for ongoing autoimmune beta cell destruction in genetically susceptible individuals, triggered, and promoted by as yet unidentified environmental factors [15, 16]. At clinical onset, functional beta cell mass has dropped to 10–40% of normal [17], but the timing and kinetics of its decline during the asymptomatic phase remain a matter of debate [15, 16]. The classical model remains to a large extent hypothetical since firm evidence supporting a causal link between insulitis and circulating autoantibodies is lacking. In previous histopathological studies, donor pancreata from autoantibody-positive non-diabetic organ donors—used as a model for at risk individuals [3, 4, 18,19,20,21,22]—showed no evidence of decreased relative beta cell area [4, 18, 20, 21]. Only four autoantibody-positive individuals in total were identified with insulitis according to international consensus criteria [4, 18], half of them in subjects >45 years of age, an age group that is substantially older than the group of 10–14 years in which the incidence of type 1 diabetes is at its highest level [1]. Insight into early histopathological changes in islet tissue in autoantibody-positive individuals under the age of 25 years is therefore limited and formed the rationale for studying a large cohort of non-diabetic organ donors below the age of 25 years. We compared histopathological changes in islet tissue from autoantibody-positive organ donors to a matched control group of autoantibody-negative organ donors from the same cohort.
Methods
Collection of pancreatic tissue
Human donor pancreases were obtained in the context of our islet transplantation program. A single tissue sample (~0.5 cm3) was taken from the pancreatic body of cold-preserved deceased donor organs provided by Eurotransplant Foundation (Leiden, The Netherlands), fixed in phosphate-buffered 4% formaldehyde or Bouin’s fixative, and embedded in paraffin. Some donor organs were eventually not processed for islet isolation, and in these cases, multiple sample blocks from the resected organ were available for study. Biopsies were taken as part of a quality control procedure that was approved by the local ethics committee (approval number BUN143201941720) and stored in a registered Biobank (Diabetes Biobank Brussels, approval number local ethics committee BUN143201524128). Between 1989 and 2019, 556 organs met the following inclusion criteria: donor age below 25 years, availability of minimal clinical data, a serum sample for islet cell autoantibody assays, and a formalin or Bouin fixed tissue sample. Diabetes was an exclusion criterion for organ procurement. Within the group of 556 organ donors, a total of 27 were found to be positive for one or more autoantibodies directed against islet cell antigens. Pancreas tissue from this subgroup was screened for histopathological changes as described below, and the results were compared to a control group of 27 autoantibody-negative organ donors obtained from the same cohort and matched for age, sex, and BMI. There is no overlap whatsoever between the pancreata from young donors with or without autoantibodies studied here (<25 years) and the pancreata from a previous report on older donors (≥25 years) [18].
Autoantibodies and genetic risk markers
Serum samples were tested for the presence of ICA, IA-2A, GADA, and IAA [23], and when positive for at least one of those markers, also evaluated for ZnT8A [24]. ICA were assessed by indirect immunofluorescence and endpoint titers expressed as JDF units. IA-2A, GADA, IAA, and ZnT8A were measured by liquid-phase radiobinding assay and expressed as either percent tracer bound in hemolysis-free sera (IAA, ZnT8A) or WHO U/mL (IA-2A, GADA) [23, 24]. Cut-off values for autoantibody positivity were calculated as 99th percentile of autoantibody levels in 790 non-diabetic controls after exclusion of outlying values (≥12 JDF units for ICA, ≥1.4 WHO U/ml for IA-2A, ≥23 WHO U/ml for GADA, ≥0.6% tracer binding for IAA, and ≥1.28% (0–14 years) or ≥1.02% tracer binding (>14 years) for ZnT8A. The autoantibody assays were validated in successive international proficiency testing programs (DASP, IASP); all positive results were confirmed in a separate subsequent assay [13, 23]. Whole blood was haplotyped for DNA polymorphisms at the HLA-DQA1 and DQB1 gene loci and DQ-associated risk stratified as reported previously [25]. The presence of HLA class I alleles conferring susceptibility for type 1 diabetes independently of HLA class II inferred risk was deduced from information provided by Eurotransplant.
Screening for insulitis and pseudo-atrophic islets
Sections were processed and stained for CD45 and synaptophysin for the detection of insulitis and for insulin and glucagon to screen for pseudo-atrophic (insulin-deficient) islets as previously described [18] (see also Supplementary methods). We screened at least 1 cm2, a minimum of 100 islets, and an average of 280±43 (±SEM) islets per donor organ. Each section was analyzed by two observers, who were blinded as to the identity of the samples. When insulitis or pseudo-atrophic islets were found, the available blocks were sectioned completely for analysis of every twentieth section. The diagnosis of insulitis was made according to the JDRF-nPOD consensus criteria [26]. Briefly, insulitis is diagnosed when ≥3 islets with ≥15 CD45+ cells are found in an organ that also contains pseudo-atrophic islets. We considered clusters of ≥5 endocrine cells as islets and called them “insulitic” when the threshold of ≥15 CD45+ cells was exceeded and “pseudo-atrophic” when the islets were devoid of insulin immunoreactive cells.
Characterization of leucocytic infiltrates
Leucocytic infiltrates were immunophenotyped on paraffin sections using immunofluorescent staining for CD3, CD45, CD4, CD8, CD20, and CD68 as previously described [18] (see also Supplementary methods). Leucocytic infiltrates were examined with a fluorescence microscope Nikon Eclipse 80i (Nikon BeLux; Brussels; Belgium) equipped with an Orca AG camera (Hamamatsu; Herrsching am Ammersee, Germany) and imaging software NIS elements AR (Nikon BeLux). Negative controls included the omission of the primary antibody and positive controls used paraffin-embedded human tonsil.
Quantification of relative beta cell area and beta cell proliferation
For quantification of the relative beta cell area, paraffin sections were stained for insulin. The relative insulin-positive cell area was measured as previously published [18] (see also Supplementary methods).
Beta cell replication was quantified using double staining for insulin and Ki67 (Supplementary methods). A minimum of 1000 insulin-positive cells per donor were analyzed for Ki67 positivity.
Statistical analysis
Differences between groups were analyzed by an unpaired t test or Mann-Whitney test (two-tailed) for, respectively, normal distributions and non-normal distributions. Spearman correlation was used to examine the relationship between different parameters. All statistical tests were performed using GraphPad Prism 8 (GraphPad Software; San Diego; CA; USA) and considered significant at p<0.05.
Results
Screening for autoantibodies
The study population consisted of 556 organ donors <25 years with a median age of 19 years (range 3 months to 24 years) (Table 1). Serum samples were screened for the presence of autoantibodies resulting in a total of 27 (4.9%) positive donors. Their clinical characteristics did not differ significantly from that of the entire study population (Table 1). Twenty-five individuals were positive for a single autoantibody, with a predominance of GADA (Table S1). Two donors were positive for two autoantibody markers, one for ICA and GADA (DBB-3504), the other for IA-2A and ZnTA8 (DBB-A096) (Table S2).
From 22 of the 27 autoantibody-positive donors, a segmental pancreas was received, but from five individuals, the intact organ was available for study. The mean pancreatic weight of intact organs was 68.4±25.3g (±SD), which was not significantly different from that in ten controls matched for age, sex, and BMI (71.7±26.6g).
Beta cell area and replication
The relative beta cell area was measured in all 27 autoantibody-positive donors and an equal number of autoantibody-negative controls matched for age, sex, and BMI (Table 1). The mean relative beta cell area in autoantibody-positive donors was 1.38±0.53% (±SD), which was not significantly different from that in the matched controls (1.14±0.36%) (Fig. 1a). No significant difference could be found at the level of beta cell replication between the autoantibody-positive organ donors (median (IQR) 0.0% (0.0–0.1%)) and the matched controls (0.0% (0.0–0.3%)); some donors (both autoantibody-positive and -negative) showed high replication levels, up to 7% (Fig. 1b), corresponding to levels seen in earlier studies in donors characterized by a longer duration of stay in intensive care [52]. No correlation could be found between beta cell area and age, and between beta cell replication and age (data not shown).

Relative beta cell area (a) and the percentage of replicating beta cells (b) in the pancreas of autoantibody-positive organ donors (n=27) and matched controls (n=27). The two donors with multiple autoantibodies and insulitis are indicated with a green dot (DBB-A096) and a red dot (DBB-3504). Results are expressed as mean (a) or median (b) with individual data points. No significant difference was found between both study groups using an unpaired Student t test (a) or an unpaired Mann-Whitney test (b) at the .05* level
Screening for insulitis
Pancreas biopsies from all 27 autoantibody-positive donors and an equal number of autoantibody-negative-matched controls were screened for the presence of insulitis as defined by the international consensus criteria. Sections were stained for synaptophysin and CD45, to detect and quantify the presence of leucocytic infiltrates. They were also stained for insulin and glucagon to detect insulin-deficient pseudo-atrophic islets. None of the 25 single autoantibody-positive donors, or the 27 controls, showed evidence of insulitis. However, the two donor organs with multiple autoantibodies were both diagnosed with insulitis and showed multiple insulitic islets together with focal areas rich in pseudo-atrophic islets.
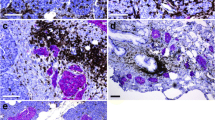
Donor DBB-3504 involved a 22-year-old male donor who died of polytrauma, 11 days after admission to the intensive care unit (ICU). He was tested positive for two autoantibodies (ICA, 400 JDF units; GADA, 29655 WHO units/ml) and had a protective HLA-DQA1/DQB1 genotype (02-02:01/03-03:01) and a HLA class I risk allele (A24) [27, 28]. The entire donor organ was received with a total weight of 53g and processed for islet isolation and transplantation. A single tissue sample (approx. 0.5 cm3) from the body of the gland was available for histopathological study. Low magnification images of tissue sections double-stained for insulin and glucagon showed both normal and pseudo-atrophic islets devoid of insulin-immunoreactivity, with a lobular distribution of the latter islet type. In total, 58.2% of all islets analyzed were categorized as pseudo-atrophic (Fig. 2a–b). Double staining for CD45 and synaptophysin showed insulitis in 3.8% of the islets, with the infiltrate being mainly (74%) present in the islet periphery (peri-insulitis) (Fig. 2c). The infiltrate consisted predominantly of CD3+CD8+-positive T-lymphocytes (76% of all CD3+ cells) (Fig. 2d). In addition, small numbers CD3+CD4+ T-lymphocytes and CD20+ B-lymphocytes were present. The infiltrates showed a CD20Lo insulitic profile, with a CD20/CD4 ratio <1.0 [29]. A more extensive characterization of the infiltrating cells was precluded by the small size of the infiltrates. All insulitic islets still contained insulin-positive beta cells. A beta cell replication rate of 0.6% was found in this donor, in addition to a relative beta cell area of 1.23% (Table 2).

Immunofluorescent staining of pancreas sections from a 22-year-old male organ donor positive for GADA and ICA autoantibodies (DBB-3504). Staining for insulin (green) and glucagon (red) reveals a lobular area (above the dotted line) (a) containing pseudo-atrophic islets devoid of insulin (b). Immunostaining for the leucocytic marker CD45 (brown) in combination with the pan-endocrine marker synaptophysin (red) showing peri-insulitis (c). Staining for CD3 (green), CD8 (red), and insulin (blue) shows an insulitic lesion consisting of a cluster of predominantly lymphocytic cells in the islet periphery (d)
Donor DBB-A096 involved a 17-year-old male donor who died of severe head trauma, 4 days after admission to ICU. He was tested positive for two autoantibodies (IA-2A, 7.3 WHO units/ml; ZnT8A, 1.6% tracer binding) and had a susceptible HLA-DQA1/DQB1 genotype (01-05:01/03:01-03:02) and a HLA class I risk allele (B15) [30]. The donor organ consisted of a segmental pancreas of unknown weight, divided into flash frozen samples of approximately 1 cm3 without positional information. All tissue samples were available for histopathological analysis. Low magnification tissue sections double-stained for insulin and glucagon showed >30 small focal areas of <2 mm diameter scattered throughout the parenchyma containing 3–5 pseudo-atrophic islets in addition to larger zones of a more lobular character (Fig. 3a–b). Overall, 7.7% of all islets were found to be of a pseudo-atrophic nature. Analysis of multiple biopsies showed insulitis in 0.2% of the islets, with the infiltrate being mainly present in the islet periphery (Fig. 3c). The infiltrates predominantly consisted of CD3+CD8+ T-lymphocytes (65% of all CD3+ cells) and CD3+CD4+ T-lymphocytes (9% of all CD3+ cells) (Fig. 3d). Infiltrates showed a CD20Lo insulitic profile with rare B-lymphocytes and a CD20/CD4 ratio <1.0 [29]. All insulitic islets contained insulin-positive beta cells. A diffuse CD68+ macrophage infiltration was present throughout the pancreatic tissue (data not shown). A beta cell replication rate of 2.8% was found in this donor (Fig. 3e), in addition to a relative beta cell area of 1.33% (Table 2).

Immunofluorescent staining of pancreas sections from a 17-year-old male organ donor positive for IA-2A and ZnT8 autoantibodies (DBB-A096). Staining for insulin (green) and glucagon (red) shows multiple small foci (a) or larger lobular areas of pseudo-atrophic islets (on the right of the dotted line) (b). Insulitic lesions were predominantly found at the islet periphery (c) and show small lymphocytic infiltrates composed of CD3+CD8+ T-lymphocytes (d). Insulin-containing islets show marked positivity for the replication marker Ki67 (e)
Discussion
Screening for five islet autoantibody types in sera from 556 pancreas donors aged under 25 years without symptomatic type 1 diabetes allowed to identify 25 organs from single marker-positive individuals and two from double positive ones. This substantially expands the limited number of previous single- or multiple autoantibody-positive (n=7 and n=3, respectively) organ donors under age 25 years and for whom the individual age was reported [3, 4, 19, 21]. This illustrates the considerable effort needed to study pancreas tissue from persons with apparent immune activation against islet cells or, in case of multiple antibody-positivity, who are allegedly in an asymptomatic disease stage [1, 4, 14, 18]. Histopathological alterations were discrete and restricted to both double positive individuals, hereby confirming previous observations [4, 18]. In the present study, histological analysis of the pancreata from both double positive donors showed discrete peri-insulitis in a small fraction of islets, as well as an intact relative beta cell area despite the presence of numerous pseudo-atrophic islets predominantly in a lobular area. A relatively high islet cell replication rate was also noted. An intriguing novel observation is the presence of >30 small focal areas of 3–5 pseudo-atrophic islets scattered throughout the parenchyma of an individual with alleged asymptomatic type 1 diabetes on the basis of positivity for IA-2A, ZnT8A, and the presence of HLA class I- and II-inferred susceptibility [10, 11, 14].
Strengths of the study include the procurement of donor organs, systematic screening for five autoantibody markers, and the use of international consensus criteria to define insulitis. The fact that for most donors only a single usually small tissue sample was received is a limitation. It can thus not be completely excluded that focal histopathological alterations may have been missed in some organs due to the non-random distribution of lesions throughout the organ. Combined screening for both insulitis and pseudo-atrophic islets, as performed in the present study, may decrease but not avoid such a risk. As molecularly defined autoantibodies, including GADA, may contribute to ICA reactivity, joint presence of ICA, and GADA and may not necessarily imply multiple autoantibody positivity [31]. However, high-titer ICA levels, such as present in donor DBB-3504, were associated with increased risk of progression to clinical onset [32]. Likewise in latent autoimmune diabetes in adult patients, the time to insulin-dependence was shorter in joint presence of ICA and GADA than in presence of GADA alone [33]. Part of the ICA positivity in in donor DBB-3504 may have been caused by an autoantibody specificity for which we did not test [13, 23, 34, 35].
When combining our findings with available histological and biological information on multiple autoantibody-positive organ donors (all ages) outside the context of gestational diabetes, polyendocrinopathy or exocrine co-morbidity, and for whom the exact age was reported, it is remarkable that so few histopathological hallmarks of type 1 diabetes were found in the pancreata from these individuals, who are allegedly in an asymptomatic disease stage (Table S3) [3, 4, 18, 20]. Discrete insulitis and foci of pseudo-atrophic islets were observed in 4/4 donors under age 25 years with two or more autoantibody markers and in 2/11 multiple marker-positive donors above that age (Table S3), in line with observations on a higher prevalence of insulitis with younger age at diagnosis in recent-onset type 1 diabetes [2, 36]. The composition of the islet infiltrates was similar in all cases, with a preponderance of CD8+ lymphocytes. The presence of HLA class I alleles reported to confer susceptibility independently of HLA class II-inferred risk [27, 28, 30, 37], and of a high-risk autoantibody profile (IA-2A or ZnT8A plus ≥1 other marker) [11, 13, 34] further support the probability of progressive subclinical disease in (many of) these donors, despite the presence of protective HLA class II haplotypes in some (Table S3); indeed, the latter plays no longer a major role once multiple antibodies have developed [11, 28, 38, 39].
In contrast, neither insulitis nor pseudo-atrophic islets were reported in pancreata from single autoantibody-positive organ donors screened for all antibody markers and reported individually or as a group [3, 4, 18,19,20,21,22]. Conceivably, some of the donors with low-level single autoantibody positivity may represent “statistical” positives due to the use of the 99th percentile of large numbers of healthy controls as cut-off in all assays [23, 24], conferring a risk up to 5% of a false-positive result when testing for five markers. Moreover, single autoantibody positivity is often a low-level transient phenomenon of uncertain significance [40]. Multiple autoantibody positivity, on the other hand, is associated with 90% 20-year risk of progression to clinical onset in children and adults [10, 11, 28].
We found no indication that autoantibody-positivity, be it for one or multiple specificities, was associated with a decreased relative beta cell area in the identified donor organs, in line with previous studies [4, 18, 20, 21, 41, 42]. Interestingly, even in presence of clear insulitic lesions and pseudo-atrophic islets devoid of beta cells, the relative beta cell area was not found to be significantly different from that in matched controls.
Of special interest is donor DBB-A096 with insulitis and multiple small foci of beta cell loss scattered throughout the gland. Such large numbers of small lesions, consisting of 3–5 pseudoatrophic islets, have not previously been described in recent-onset patients, although conversely, a lobular pattern of beta cell survival is relatively common in type 1 diabetes patients, especially those with older age at onset [43,44,45]. If the small focal lesions are indicative of a pre-diabetic state in this subject, it could be hypothesized that during disease progression, such focal lesions grow in number or size and finally fuse, leading to the characteristic presentation of a pancreatic gland at disease onset with predominantly pseudo-atrophic islets being present, but also with regions in which islets show no histopathological changes. The early disease processes leading to such (multi)focal lesions are unknown. Whether they represent focal points of (auto)immunity with the autoreactive cells migrating to nearby islets, possibly via the connective tissue septa, as observed in mouse models of the disease [46], or represent focal points of (possibly viral) infection spreading outwards [47], can only be speculated.
In the absence of longitudinal studies on islet pathology in high-risk individuals, it cannot be excluded that the observed histopathological changes in multiple autoantibody-positive donor pancreata do not represent true pre-diabetic lesions. This is, however, unlikely since multiple autoantibody-positivity is rarely transient, and confers a high probability of progression to clinically overt disease [11, 28, 38, 39]. The seemingly preserved beta cell mass in the identified multiple autoantibody-positive donors should not be taken as argument against a high probability of later disease progression. Indeed, data by us and others [48, 49] indicate that the functional beta cell mass, as assessed by (stimulated) C-peptide release, only starts to sharply decline within 2 years from diagnosis. The age range of the multiple autoantibody-positive donors in Table S3 indicates an underrepresentation of children in all series of donor pancreata and may point to a selection bias towards slow progressors [50]; hence, the observed histopathological alterations may not be fully representative for asymptomatic childhood-onset type 1 diabetes. The described lesions may also reflect remnants of a low-level immune process during which islets are still able to compensate for any immune-mediated loss as indicated by the presence of islet cell replication in some of these donors. We can also not fully discount the possibility that the relatively minor level of insulitis that is observed is a consequence rather than a cause of the pathogenetic events taking place.
The finding of relatively high levels of replication in the islets of Langerhans of two donors with insulitis is of interest, especially because of the virtual absence of replication in most adult organ donors [51]. However, the interpretation of these observations and the increased CD68 macrophage infiltration in one donor is complicated by studies showing that organ donors with an increased duration of stay in intensive care show evidence of tissue repair, including increased numbers of M2 macrophages, increased vascular density, and increased replication of all pancreatic cell types [52]. As both donors in the present study are characterized by a relatively long duration of stay in intensive care, it cannot be excluded that the increase in beta cell replication found in these two donors is caused by a repair process and is not related to the presumed autoimmune process itself. The percentage of Ki67-positive beta cells was not correlated with relative insulin-positive area, both in our autoantibody-positive and in autoantibody-negative donors (data not shown). This does, however, not exclude compensatory beta cell regeneration via, e.g., transdifferentiation.
In conclusion, evidence for early type 1 diabetes-related histopathological lesions in the pancreas of donors under age 25 years appears restricted to organs from multiple autoantibody-positive individuals at high risk of developing symptomatic disease, in line with observations in older age groups [13]. Donors with single autoantibody-positivity presented no histopathological evidence of immune-mediated beta cell destruction. Single autoantibody-positivity may thus not be a sufficient indicator of islet lesions and not a suitable parameter to select patients for clinical intervention studies. More stringent inclusion criteria are necessary to identify individuals at high risk for the development of type 1 diabetes, such as those proposed in a recent successful immune intervention study where inclusion was limited to relatives of patients with type 1 diabetes with multiple autoantibodies and evidence of dysglycemia [53]. A better insight into the relationship between autoantibody-positivity, insulitis, and beta cell destruction will help in devising better therapies aimed at preventing or curing the disease. However, the fact that none of the donors with multiple autoantibodies and insulitis reported in the present study showed evidence of a decreased relative beta cell area indicates that the relationship between T-cell infiltration and beta cell destruction is more complex than proposed in the classic models of the disease. Alternative models have recently been proposed in which both islet autoimmunity and beta cell dysfunction are suggested to play equally essential roles [54]. Histopathological studies of multiple autoantibody positive donors may help to characterize such pathogenetic pathways, investigating to what extent islet autoimmunity is accompanied by evidence of beta cell dysfunction and stress.
Availability of data and material
The detailed datasets generated during and/or analyzed during the current study are available from the corresponding author upon request.
Abbreviations
- GADA:
-
Glutamic acid decarboxylase autoantibodies
- IA-2A:
-
Insulinoma-associated protein 2 autoantibodies
- IAA:
-
Insulin autoantibodies
- ICA:
-
Islet cell cytoplasm autoantibodies
- ZnT8A:
-
Zinc transporter 8 autoantibodies
References
DiMeglio LA, Evans-Molina C, Oram RA (2018) Type 1 diabetes. Lancet 391(10138):2449–2462. https://doi.org/10.1016/S0140-6736(18)31320-5
Gepts W (1965) Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14(10):619–633. https://doi.org/10.2337/diab.14.10.619
Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TWH, Atkinson MA, Roep BO, von Herrath MG (2012) Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med 209(1):51–60. https://doi.org/10.1084/jem.20111187
Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, Atkinson MA (2016) Insulitis and β-cell mass in the natural history of type 1 diabetes. Diabetes 65(3):719–731. https://doi.org/10.2337/db15-0779
Bottazzo G, Florin-Christensen A, Doniach D (1974) Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 304(7892):1279–1283. https://doi.org/10.1016/S0140-6736(74)90140-8
Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK, Paquette TL (1983) Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science 222(4630):1337–1339. https://doi.org/10.1126/science.6362005
Baekkeskov S, Aanstoot H-J, Christgau S et al (1990) Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature 347(6289):151–156. https://doi.org/10.1038/347151a0
Lan MS, Wasserfall C, Maclaren NK, Notkins AL (1996) IA-2, a transmembrane protein of the protein tyrosine phosphatase family, is a major autoantigen in insulin-dependent diabetes mellitus. Proc Natl Acad Sci 93(13):6367–6370. https://doi.org/10.1073/pnas.93.13.6367
Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, Rewers M, Eisenbarth GS, Jensen J, Davidson HW, Hutton JC (2007) The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci 104(43):17040–17045. https://doi.org/10.1073/pnas.0705894104
Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, Winkler C, Ilonen J, Veijola R, Knip M, Bonifacio E, Eisenbarth GS (2013) Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 309(23):2473–2479. https://doi.org/10.1001/jama.2013.6285
Gorus FK, Balti EV, Messaaoui A, Demeester S, van Dalem A, Costa O, Dorchy H, Mathieu C, van Gaal L, Keymeulen B, Pipeleers DG, Weets I (2017) Twenty-year progression rate to clinical onset according to autoantibody profile, age, and HLA-DQ genotype in a registry-based group of children and adults with a first-degree relative with type 1 diabetes. Diabetes Care 40(8):1065–1072. https://doi.org/10.2337/dc16-2228
Yu L, Rewers M, Gianani R, Kawasaki E, Zhang Y, Verge C, Chase P, Klingensmith G, Erlich H, Norris J, Eisenbarth GS (1996) Antiislet autoantibodies usually develop sequentially rather than simultaneously. J Clin Endocrinol Metab 81(12):4264–4267. https://doi.org/10.1210/jcem.81.12.8954025
De Grijse J, Asanghanwa M, Nouthe B et al (2010) Predictive power of screening for antibodies against insulinoma-associated protein 2 beta (IA-2β) and zinc transporter-8 to select first-degree relatives of type 1 diabetic patients with risk of rapid progression to clinical onset of the disease: implicat. Diabetologia 53(3):517–524. https://doi.org/10.1007/s00125-009-1618-y
Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, Greenbaum CJ, Herold KC, Krischer JP, Lernmark Å, Ratner RE, Rewers MJ, Schatz DA, Skyler JS, Sosenko JM, Ziegler AG (2015) Staging presymptomatic type 1 diabetes: a scientific statement of jdrf, the endocrine society, and the American diabetes association. Diabetes Care 38(10):1964–1974. https://doi.org/10.2337/dc15-1419
Eisenbarth GS (1986) Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 314(21):1360–1368. https://doi.org/10.1056/NEJM198605223142106
Van Belle TL, Coppieters KT, Von Herrath MG (2011) Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev 91(1):79–118. https://doi.org/10.1152/physrev.00003.2010
Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, Schandene L, Crenier L, de Block C, Seigneurin JM, de Pauw P, Pierard D, Weets I, Rebello P, Bird P, Berrie E, Frewin M, Waldmann H, Bach JF, Pipeleers D, Chatenoud L (2005) Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 352(25):2598–2608. https://doi.org/10.1056/NEJMoa043980
in’t Veld P, Lievens D, De Grijse J et al (2007) Screening for insulitis in adult autoantibody-positive organ donors. Diabetes 56(9):2400–2404. https://doi.org/10.2337/db07-0416
Wagner R, McNally JM, Bonifacio E et al (1994) Lack of immunohistological changes in the islets of nondiabetic, autoimmune, polyendocrine patients with beta-selective GAD-specific islet cell antibodies. Diabetes 43(7):851–856. https://doi.org/10.2337/diab.43.7.851
Gianani R, Putnam A, Still T, Yu L, Miao D, Gill RG, Beilke J, Supon P, Valentine A, Iveson A, Dunn S, Eisenbarth GS, Hutton J, Gottlieb P, Wiseman A (2006) Initial results of screening of nondiabetic organ donors for expression of islet autoantibodies. J Clin Endocrinol Metab 91(5):1855–1861. https://doi.org/10.1210/jc.2005-1171
Oikarinen M, Tauriainen S, Honkanen T, Vuori K, Karhunen P, Vasama-Nolvi C, Oikarinen S, Verbeke C, Blair GE, Rantala I, Ilonen J, Simell O, Knip M, Hyöty H (2008) Analysis of pancreas tissue in a child positive for islet cell antibodies. Diabetologia 51(10):1796–1802. https://doi.org/10.1007/s00125-008-1107-8
Wiberg A, Granstam A, Ingvast S, Härkönen T, Knip M, Korsgren O, Skog O (2015) Characterization of human organ donors testing positive for type 1 diabetes-associated autoantibodies. Clin Exp Immunol 182(3):278–288. https://doi.org/10.1111/cei.12698
Winnock F, Christie MR, Batstra MR, Aanstoot HJ, Weets I, Decochez K, Jopart P, Nicolaij D, Gorus FK, Belgian Diabetes Registry (2001) Autoantibodies to a 38-kDa glycosylated islet cell membrane-associated antigen in (pre)type 1 diabetes: association with IA-2 and islet cell autoantibodies. Diabetes Care 24(7):1181–1186. https://doi.org/10.2337/diacare.24.7.1181
Vermeulen I, Weets I, Asanghanwa M, Ruige J, van Gaal L, Mathieu C, Keymeulen B, Lampasona V, Wenzlau JM, Hutton JC, Pipeleers DG, Gorus FK, The Belgian Diabetes Registry (2011) Contribution of antibodies against IA-2β and zinc transporter 8 to classification of diabetes diagnosed under 40 years of age. Diabetes Care 34(8):1760–1765. https://doi.org/10.2337/dc10-2268
Van der Auwera BJ, Schuit FC, Weets I, Ivens A, Van Autreve JE, Gorus FK (2002) Relative and absolute HLA-DQA1-DQB1 linked risk for developing type I diabetes before 40 years of age in the Belgian population: implications for future prevention studies. Hum Immunol 63(1):40–50. https://doi.org/10.1016/S0198-8859(01)00362-7
Campbell-Thompson ML, Atkinson MA, Butler AE, Chapman NM, Frisk G, Gianani R, Giepmans BN, von Herrath MG, Hyöty H, Kay TW, Korsgren O, Morgan NG, Powers AC, Pugliese A, Richardson SJ, Rowe PA, Tracy S, in’t Veld PA (2013) The diagnosis of insulitis in human type 1 diabetes. Diabetologia 56(11):2541–2543. https://doi.org/10.1007/s00125-013-3043-5
Lipponen K, Gombos Z, Kiviniemi M, Siljander H, Lempainen J, Hermann R, Veijola R, Simell O, Knip M, Ilonen J (2010) Effect of HLA class I and class II alleles on progression from autoantibody positivity to overt type 1 diabetes in children with risk-associated class II genotypes. Diabetes 59(12):3253–3256. https://doi.org/10.2337/db10-0167
Balke EM, Balti EV, Van Der Auwera B et al (2018) Accelerated progression to type 1 diabetes in the presence of HLA-A∗24 and-B∗18is restricted to multiple islet autoantibody-positive individuals with distinct HLA-DQ and autoantibody risk profiles. Diabetes Care 41(5):1076–1083. https://doi.org/10.2337/dc17-2462
Leete P, Willcox A, Krogvold L, Dahl-Jørgensen K, Foulis AK, Richardson SJ, Morgan NG (2016) Differential insulitic profiles determine the extent of β-cell destruction and the age at onset of type 1 diabetes. Diabetes 65(5):1362–1369. https://doi.org/10.2337/db15-1615
Windsor L, Puschendorf M, Allcock R, Scott A, Sayer D, Kucharzak R, Gut I, McCann V, Davis E, Witt C, Christiansen F, Price P (2005) Does a central MHC gene in linkage disequilibrium with HLA-DRB1*0401 affect susceptibility to type 1 diabetes? Genes Immun 6(4):298–304. https://doi.org/10.1038/sj.gene.6364210
Korhonen S, Knip MM, Kulmala P, Savola K, Åkerblom HK, Knip M (2002) Autoantibodies to GAD, IA-2 and insulin in ICA-positive first-degree relatives of children with type 1 diabetes: a comparison between parents and siblings. Diabetes Metab Res Rev 18(1):43–48. https://doi.org/10.1002/dmrr.258
Tarn AC, Dean BM, Schwarz G, Thomas JM, Ingram D, Franco Bottazzo G, Gale EAM (1988) Predicting insulin-dependent diabetes. Lancet 331(8590):845–850. https://doi.org/10.1016/S0140-6736(88)91601-7
Turner R, Stratton I, Horton V, Manley S, Zimmet P, Mackay IR, Shattock M, Bottazzo GF, Holman R (1997) UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes. Lancet 350(9087):1288–1293. https://doi.org/10.1016/S0140-6736(97)03062-6
Achenbach P, Warncke K, Reiter J, Naserke HE, Williams AJK, Bingley PJ, Bonifacio E, Ziegler AG (2004) Stratification of type 1 diabetes risk on the basis of islet autoantibody characteristics. Diabetes 53(2):384–392. https://doi.org/10.2337/diabetes.53.2.384
McLaughlin KA, Richardson CC, Ravishankar A et al (2016) Identification of Tetraspanin-7 as a target of autoantibodies in type 1 diabetes. Diabetes 65(6):1690–1698. https://doi.org/10.2337/db15-1058
Pipeleers D, Ling Z (1992) Pancreatic beta cells in insulin-dependent diabetes. Diabetes Metab Rev 8(3):209–227. https://doi.org/10.1002/dmr.5610080303
Noble JA, Valdes AM (2011) Genetics of the HLA region in the prediction of type 1 diabetes. Curr Diab Rep 11(6):533–542. https://doi.org/10.1007/s11892-011-0223-x
Ilonen J, Kiviniemi M, Lempainen J, Simell O, Toppari J, Veijola R, Knip M, The Finnish Pediatric Diabetes Register (2016) Genetic susceptibility to type 1 diabetes in childhood - estimation of HLA class II associated disease risk and class II effect in various phases of islet autoimmunity. Pediatr Diabetes 17:8–16. https://doi.org/10.1111/pedi.12327
Krischer JP, Liu X, Lernmark Å, Hagopian WA, Rewers MJ, She JX, Toppari J, Ziegler AG, Akolkar B, TEDDY Study Group (2017) The influence of type 1 diabetes genetic susceptibility regions, age, sex, and family history on the progression from multiple autoantibodies to type 1 diabetes: a TEDDY study report. Diabetes 66(12):3122–3129. https://doi.org/10.2337/db17-0261
Kukko M, Kimpimäki T, Korhonen S et al (2005) Dynamics of diabetes-associated autoantibodies in young children with human leukocyte antigen-conferred risk of type 1 diabetes recruited from the general population. J Clin Endocrinol Metab 90(5):2712–2717. https://doi.org/10.1210/jc.2004-1371
Diedisheim M, Mallone R, Boitard C, Larger E (2016) β-cell mass in nondiabetic autoantibody-positive subjects: an analysis based on the network for pancreatic organ donors database. J Clin Endocrinol Metab 101(4):1390–1397. https://doi.org/10.1210/jc.2015-3756
Rodriguez-Calvo T, Zapardiel-Gonzalo J, Amirian N, Castillo E, Lajevardi Y, Krogvold L, Dahl-Jørgensen K, von Herrath MG (2017) Increase in pancreatic proinsulin and preservation of β-cell mass in autoantibody-positive donors prior to type 1 diabetes onset. Diabetes 66(5):1334–1345. https://doi.org/10.2337/db16-1343
Gepts W, Lecompte PM (1981) The pancreatic islets in diabetes. Am J Med 70(1):105–115. https://doi.org/10.1016/0002-9343(81)90417-4
Löhr M, Klöppel G (1987) Residual insulin positivity and pancreatic atrophy in relation to duration of chronic type 1 (insulin-dependent) diabetes mellitus and microangiopathy. Diabetologia 30(10):757–762. https://doi.org/10.1007/BF00275740
Keenan HA, Sun JK, Levine J, Doria A, Aiello LP, Eisenbarth G, Bonner-Weir S, King GL (2010) Residual insulin production and pancreatic beta-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes 59:2846–2853
Coppieters K, Amirian N, von Herrath M (2012) Intravital imaging of CTLs killing islet cells in diabetic mice. J Clin Invest 122(1):119–131. https://doi.org/10.1172/JCI59285
Krogvold L, Edwin B, Buanes T, Frisk G, Skog O, Anagandula M, Korsgren O, Undlien D, Eike MC, Richardson SJ, Leete P, Morgan NG, Oikarinen S, Oikarinen M, Laiho JE, Hyöty H, Ludvigsson J, Hanssen KF, Dahl-Jørgensen K (2015) Detection of a low-grade enteroviral infection in the islets of Langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes 64(5):1682–1687. https://doi.org/10.2337/db14-1370
Vandemeulebroucke E, Keymeulen B, Decochez K et al (2010) Hyperglycaemic clamp test for diabetes risk assessment in IA-2-antibody-positive relatives of type 1 diabetic patients. Diabetologia 53(1):36–44. https://doi.org/10.1007/s00125-009-1569-3
Ferrannini E, Mari A, Nofrate V, Sosenko JM, Skyler JS (2010) Progression to diabetes in relatives of type 1 diabetic patients: mechanisms and mode of onset. Diabetes 59(3):679–685. https://doi.org/10.2337/db09-1378
Long AE, Wilson IV, Becker DJ, Libman IM, Arena VC, Wong FS, Steck AK, Rewers MJ, Yu L, Achenbach P, Casas R, Ludvigsson J, Williams AJK, Gillespie KM (2018) Characteristics of slow progression to diabetes in multiple islet autoantibody-positive individuals from five longitudinal cohorts: the SNAIL study. Diabetologia 61(6):1484–1490. https://doi.org/10.1007/s00125-018-4591-5
in’t Veld P, De Munck N, Van Belle K et al (2010) β-Cell replication is increased in donor organs from young patients after prolonged life support. Diabetes 59(7):1702–1708. https://doi.org/10.2337/db09-1698
Smeets S, Stangé G, Leuckx G, Roelants L, Cools W, de Paep DL, Ling Z, de Leu N, in’t Veld P (2020) Evidence of tissue repair in human donor pancreas after prolonged duration of stay in intensive care. Diabetes 69(3):401–412. https://doi.org/10.2337/db19-0529
Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio L, Dufort MJ, Gitelman SE, Gottlieb PA, Krischer JP, Linsley PS, Marks JB, Moore W, Moran A, Rodriguez H, Russell WE, Schatz D, Skyler JS, Tsalikian E, Wherrett DK, Ziegler AG, Greenbaum CJ, Type 1 Diabetes TrialNet Study Group (2019) An anti-CD3 antibody, replizumab, in relatives at risk for type 1 diabetes. N Engl J Med 381(7):603–613. https://doi.org/10.1056/NEJMoa1902226
Roep BO, Thomaidou S, van Tienhoven R, Zaldumbide A (2020) Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nat Rev Endocrinol. https://doi.org/10.1038/s41574-020-00443-4
Acknowledgements
We thank Nicole Buelens, Sabrina D’Haese, Veerle De Punt, Cindy Raemdonck, Sylvie Duys, Patrick Goubert, Chris Groven, and Ling Jin for expert technical assistance. Crystalline humulinTM was a gift from Dr. H. Schmitt, E. Lilly Co; cDNA for GAD65 a gift from Dr. Å. Lernmark; and cDNA for IA-2ic a gift from Dr. M. Christie. We gratefully acknowledge Willem Staels and Nico De Leu for their suggestions and help during the preparation of this manuscript.
Funding
This study was supported by grants of the Vrije Universiteit Brussel (onderzoekszwaartepunt ref. SRP55), Innoviris (2013-PFS-EH-16), the European Commission (H2020 681070), the Juvenile Diabetes Research Foundation (2-SRA-2019-708-S-B), and the Flemish Government (IWT130138).
Author information
Authors and Affiliations
Contributions
S.S., F.G., and P.V. wrote the manuscript and researched data. S.S., D.L.D.P., G.S., K.V., B.V.D.A., and Z.L. researched data and contributed to discussion. B.K. and I.W. reviewed/edited the manuscript and contributed to the discussion. P.V. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
Ethics approval
The study was approved by the local ethics committee with number B.U.N. 143201941720.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Peter in’t Veld and Frans Gorus shared last authorship
Supplementary Information
ESM 1
(DOCX 52 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Smeets, S., De Paep, D.L., Stangé, G. et al. Insulitis in the pancreas of non-diabetic organ donors under age 25 years with multiple circulating autoantibodies against islet cell antigens. Virchows Arch 479, 295–304 (2021). https://doi.org/10.1007/s00428-021-03055-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-021-03055-z