
Abstract
Uric acid, the end product of purine metabolism in humans, is crucial because of its anti-oxidant activity and a causal relationship with hyperuricemia and gout. Several physiologically important urate transporters regulate this water-soluble metabolite in the human body; however, the existence of latent transporters has been suggested in the literature. We focused on the Escherichia coli urate transporter YgfU, a nucleobase-ascorbate transporter (NAT) family member, to address this issue. Only SLC23A proteins are members of the NAT family in humans. Based on the amino acid sequence similarity to YgfU, we hypothesized that SLC23A1, also known as sodium-dependent vitamin C transporter 1 (SVCT1), might be a urate transporter. First, we identified human SVCT1 and mouse Svct1 as sodium-dependent low-affinity/high-capacity urate transporters using mammalian cell-based transport assays. Next, using the CRISPR-Cas9 system followed by the crossing of mice, we generated Svct1 knockout mice lacking both urate transporter 1 and uricase. In the hyperuricemic mice model, serum urate levels were lower than controls, suggesting that Svct1 disruption could reduce serum urate. Given that Svct1 physiologically functions as a renal vitamin C re-absorber, it could also be involved in urate re-uptake from urine, though additional studies are required to obtain deeper insights into the underlying mechanisms. Our findings regarding the dual-substrate specificity of SVCT1 expand the understanding of urate handling systems and functional evolutionary changes in NAT family proteins.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The global increase in the incidence of gout [13, 25], the most prevalent form of inflammatory arthritis caused by hyperuricemia which is the sustained elevation of serum urate (the predominant form of uric acid under physiological conditions), has highlighted the significance of urate maintenance at optimal levels for a healthy life. The urate-degrading enzyme uricase (UOX) is genetically absent in humans [57], and uric acid is the final product of purine metabolism. Since urate cannot passively traverse cellular membranes, active transport plays a pivotal role in regulating urate handling in the human body, suggesting the physiological significance of urate transporter proteins. Indeed, other groups and we have identified physiologically important urate transporters of which functional changes influence serum urate levels, such as renal urate re-absorbers—urate transporter 1 (URAT1, also known as SLC22A12) [14] and glucose transporter 9 (GLUT9/SLC2A9) [8, 32, 52], and an intestinal and renal urate exporter—ATP-binding cassette transporter G2 (ABCG2) [21, 33, 56]. However, the previously described urate transporters do not thoroughly explain human urate handling systems. This suggests the presence of latent urate transporters in the human body. As a reference point for investigating such mechanisms, we focused on prokaryotic (non-mammalian) proteins exhibiting urate transport activity.
YgfU (NCBI accession; NP_417364.2), a member of the nucleobase-ascorbate transporter (NAT) family, was previously identified as a low-affinity and high-capacity urate transporter in Escherichia coli [37]. Given the functional similarities between homologous genes, we hypothesized that mammalian proteins homologous to YgfU might be involved in urate transport. Consistent with the fact that only SLC23A proteins are classified as members of the NAT family in humans [15], an in silico homology search conducted using the BLAST-P program (accessed in July 2013) revealed that SLC23A1 and SLC23A2 are the closest to YgfU in amino acid sequence, leading us to focus on them as a source for latent urate transporter(s).
SLC23A1 and SLC23A2 are sodium-dependent vitamin C transporters (SVCT1 and SVCT2, respectively) [50] involved in the cellular uptake of vitamin C (L-ascorbic acid) in various tissues/organs [6, 38, 42]. A previous study using Svct1 knockout (KO) mice suggested that SVCT1, which is expressed in the brush border of proximal tubules in the kidney [7, 26], is involved in the re-absorption of vitamin C from urine into the blood [11]. Despite its widespread distribution [54], SVCT2 is hardly expressed in the kidney [9, 26]. Svct2 KO mice exhibit low vitamin C levels in tissues and die soon after birth [39], demonstrating the physiological relevance of SVCT2. Despite the abundance of research on SVCTs, we could not find any information on whether SVCTs can transport urate or participate in urate handling in the human body. To address this concern, we focused on SVCT1/SLC23A1 and investigated its function as a urate transporter from molecular and physiological perspectives.
This study aimed to investigate the urate transport ability of human SVCT1 and mouse Svct1 using cell-based transport assays. Hyperuricemic Svct1 KO mice deficient in both Urat1 and Uox were generated using the CRISPR-Cas9 system. In addition, we performed functional assays to explore the effects of serum urate (SU)-affecting drugs and their active metabolites (a total of 28 compounds) on SVCT1-mediated urate and vitamin C transport. Our findings provide insights into the dual-substrate specificity of SVCT1 and its physiological implications.
Materials and methods
Materials
The critical materials and resources used in this study are summarized in Supplementary Table S1. pCAG-EGxxFP (Addgene plasmid # 50716), pCAG-EGxxFP-Cetn1 (Addgene plasmid # 50717), and pX330-Cetn1/1 (Addgene plasmid # 50718) were gifts from Masahito Ikawa [30]. Fuji Yakuhin (Saitama, Japan) kindly provided dotinurad under a material transfer agreement. The remaining chemicals were of analytical grade and commercially available. Detailed information on SU-affecting drugs and their active metabolites used in this study is summarized in Supplementary Table S2.
Plasmid construction
The full-length human SVCT1/SLC23A1 wild-type (WT) (NCBI accession; NM_005847.5) open reading frame (ORF) was inserted into the pEGFP-N1 vector (Clontech Laboratories, Palo Alto, CA, USA) using EcoRI and Sal I sites. A site-directed mutagenesis technique removed the original termination codon and achieved in-frame fusion with a downstream EGFP protein (i.e., SVCT1-EGFP expression). An expression vector with mouse Svct1/Slc23a1 WT (NCBI accession; NM_011397.4) was constructed similarly. Exon 6 was excluded from the Svct1-EGFP expression vector using the site-directed mutagenesis technique to express the Svct1 KO mutant.
The full-length human ORFs, URAT1/SLC22A12 WT (NCBI accession; NM_144585.3) in pEGFP-C1 [48] and human GLUT9a/SLC2A9a WT (NCBI accession, NM_020041.2) in pEGFP-N1 [44], were derived from our previous studies. All plasmid constructs were confirmed by sequencing using BigDye® Terminator v3.1 (Applied Biosystems, Foster City, CA, USA) on an Applied Biosystems® 3130 Genetic Analyzer (Applied Biosystems), according to the manufacturer’s guidelines. All plasmids used in the experiments were originated from the same batch.
Cell culture
Human embryonic kidney-derived HEK293 cells were maintained in Dulbecco’s Modified Eagle’s Medium (Nacalai Tesque, Kyoto, Japan) supplemented with 10% fetal bovine serum (Biowest, Nuaillé, France), 1% penicillin-streptomycin (Nacalai Tesque), 2 mM L-glutamine (Nacalai Tesque), and 1 × non-essential amino acid (Life Technologies, Tokyo, Japan) at 37°C in a humidified atmosphere of 5% (v/v) CO2 in air. In addition, HEK-derived 293A cells and Madin-Darby canine kidney II (MDCKII) cells were also maintained similarly.
For in vitro assays, each vector plasmid was transfected into HEK293 cells on 12-well cell culture plates 24 h after seeding (0.92 × 105 cells/cm2) using a forward transfection approach with polyethyleneimine “MAX” (PEI-MAX) (Polysciences, Warrington, PA, USA) as described previously [34]. For microscopic observation, HEK293 cells were seeded on collagen-coated glass-bottom dishes (Matsunami Glass, Tokyo, Japan). After 24 h of incubation, the medium was replaced with a fresh medium. Inhibitory tests with SU-affecting drugs and their active metabolites were performed on 293A cells.
For z-stack microscopic observation of polarized cells, MDCKII cells were seeded on collagen-coated glass-bottom dishes at a density of 1.31 × 105 cells/cm2 and transiently transfected with the respective plasmid vectors using PEI-MAX (2.0 μg plasmid/10 μL of PEI-MAX/dish) as described previously [35].
Animals
All animal experiments were conducted according to methods approved by the Institutional Animal Care and Use Committee of The University of Tokyo. All animals received humane care following the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health. The animals were maintained on a standard FR-1 diet (Funabashi Farm, Chiba, Japan) with ad libitum water, under 12-h light/dark cycles, as described previously [40].
Uox KO mice on a C57BL/6J genetic background were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) (Stock No: 002223, B6;129S7-Uoxtm1Bay/J) [58], Urat1-Uox double knockout (DKO) mice were obtained from our earlier investigations [17, 18, 55]. In addition, drinking water was supplemented with 180 mg/L allopurinol (FUJIFILM Wako Pure Chemical, Osaka, Japan) and 4 g/L Uralyt-U® (a standardized mixture of potassium citrate and sodium citrate,Nippon Chemiphar, Tokyo, Japan) to attenuate hyperuricemia phenotypes that cause severe renal impairment in mice with Uox−/− genetic background, as described previously [47].
Svct1-Urat1-Uox triple knockout (TKO) mice were generated using the CRISPR-Cas9 system [36], according to our previous studies [35, 47]. Briefly, single guide RNAs (sgRNAs) for the Svct1 gene disruption were designed using CRISPRdirect (https://crispr.dbcls.jp/, accessed in December 2015), each sgRNA was evaluated using an EGxxFP system [31]. For this purpose, HEK293 cells were transiently co-transfected with the evaluated sgRNA/pX330, genomic fragments containing sgRNA target sequence (approximately 320 bp)/pCAG-EGxxFP, and pCAG-mRFP. Based on the in vitro analyses (detailed in the “Results” section), we determined the target sequence for genome editing: 5′-ccagggtgcaatcatggtgtcca-3′ (detailed in the “Results” section). Next, the synthesized sgRNA (FASMAC, Kanagawa, Japan) and in vitro transcribed Cas9 mRNA were microinjected into mouse zygotes fertilized in vitro (WT ovum × Uox KO sperm), the microinjected embryos were transferred into recipient mice. We focused on a 19-bp deletion between intron 5 and exon 6 in the Svct1 gene found in Uox+/− F0 mice. After confirming the germ-line transmission of the deletion allele, the founder mouse was mated with WT mice (C57BL/6J, Japan SLC, Shizuoka, Japan) and Uox KO mice to generate Svct1 KO mice and Svct1-Uox DKO mice by the further crossing of mice, respectively. The latter mice were further mated with Urat1-Uox DKO mice to generate Svct1-Urat1-Uox TKO mice.
Genotyping of the 19-bp deletion was conducted during Svct1 KO allele isolation, using a TA cloning-based direct sequencing, as described previously [35]. After the generation of each mouse line, Svct1 genotypes were determined using specific PCR primer sets. Similar PCR-based methods were used to genotype Uox and Urat1 KO alleles. Information on each PCR primer is summarized in Supplementary Table S3.
Specimen collection
Spot urine samples were collected on a plastic wrap sheet and transferred to fresh tubes. Under anesthesia with isoflurane (FUJIFILM Wako Pure Chemical), blood was drawn from the jugular veins, and plasma or serum was obtained as described previously [35, 45]. All liquid specimens were stored at −80°C until further processing. Immediately following euthanasia, tissues were excised, weighed, and rapidly frozen in liquid nitrogen, and stored until subsequent analysis.
Preparation of protein lysates and immunoblotting
Whole-cell lysate (WCL) samples were prepared as described previously [35]. At 48 h post-plasmid transfection, HEK293 cells were washed twice with ice-cold potassium-free phosphate-buffered saline [PBS (−)] and lysed in ice-cold RIPA lysis buffer [50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 1% NP-40, 1 mM phenylmethylsulfonyl fluoride, and a Protease Inhibitor Cocktail for General Use (Nacalai Tesque)]. The solution was centrifuged at 15,000 × g at 4°C for 10 min, and the supernatant was collected in a separate tube. The protein concentration of the WCL was quantified using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Carlsbad, CA, USA) with bovine serum albumin (BSA) as a standard, according to the manufacturer’s protocol.
Protein extracts from mouse tissues were prepared in ice-cold RIPA lysis buffer using the methods in our previous study [45]. Immediately following euthanasia, tissues were excised, weighed, and then homogenized using a Physcotron homogenizer (Microtec, Chiba, Japan). Crude lysates were incubated at 4°C for 1 h with gentle rotation, and following the centrifugation at 15,000 × g at 4°C for 10 min, the resulting supernatant was collected in a fresh tube. The protein concentration was determined the Pierce™ BCA Protein Assay Kit.
Immunoblot analyses were performed with minor modifications from our previous study [35]. The loading samples were separated by SDS polyacrylamide gel electrophoresis and electroblotted on a PVDF membrane (Immobilon-P, Millipore, Bedford, MA, USA) at 15 V for 60 min. The membrane was blocked by incubation in Tris-buffered saline containing 0.05% Tween 20 and 5% skim milk (TBST-5%SM). Blots were probed with the specific antibodies diluted in TBST-5%SM (Supplementary Table S1), and an HRP-dependent luminescence was developed using ECL™ Prime Western Blotting Detection Reagent (GE Healthcare, Buckinghamshire, UK). Immunocomplexes were detected using a multi-imaging Analyzer Fusion Solo 4™ system (Vilber Lourmat, Eberhardzell, Germany).
Confocal microscopic observation
Confocal laser-scanning microscopy was conducted as described previously [35]. Post 48 h of plasmid transfection, cells were fixed with ice-cold methanol. After washing with PBS (–), the cells were treated with TO-PRO-3 iodide (Molecular Probes, Eugene, OR, USA) to visualize nuclei. After a subsequent wash with PBS (–), the cells were mounted on Fluorescence Mounting Medium (Agilent, Santa Clara, CA, USA). Fluorescence was detected using the FV10i Confocal Laser Scanning Microscope (Olympus, Tokyo, Japan) to analyze the localization of EGFP-fused transporter proteins.
Cell-based transport assay
We conducted cell-based urate uptake assays using human SVCT1- or mouse Svct1-expressing HEK293 cells 48 h after plasmid transfection with minor modifications from our previous studies [35, 47] to evaluate their latent urate transport activities. Krebs–Ringer buffer (133 mM NaCl, 4.93 mM KCl, 1.23 mM MgSO4, 0.85 mM CaCl2, 20 mM CAPS, 5 mM D-glucose, 5 mM L-glutamine, at pH 7.4 unless otherwise indicated, when using a Na+-free condition, NaCl was replaced with equimolar choline chloride) was employed as a transport buffer. After pre-incubation in the transport butter at 37°C for 15 min, the cells were incubated for 10 min or indicated periods in pre-warmed fresh transport buffer containing [8-14C]-uric acid (53 mCi/mmol; American Radiolabeled Chemicals, St. Louis, MO, USA) at 10 μM or specified concentrations. SVCT1-inhibiting effects of the target compounds, including vitamin C, were examined using Krebs–Ringer buffer either without (i.e., with only vehicle control) or with the individual compounds at the indicated concentrations. The cells were washed twice with ice-cold transport buffer and lysed with 500 μL of 0.2 M NaOH on ice. The lysates were neutralized with 100 μL of 1 M HCl. A liquid scintillator (Tri-Carb 3110TR; PerkinElmer) was used to measure the radioactivity of the lysates. Protein concentrations in the lysates were assessed using the Pierce™ BCA Protein Assay Kit.
The urate transport activity was calculated as incorporated clearance (μL/mg protein/min) = (incorporated level of urate [DPM/mg protein/min] / urate level in the incubation mixture [DPM/μL]). Subtracting the urate transport activity of mock cells from that of human SVCT1- and mouse Svct1-expressing cells determined their respective urate transport activities. The vitamin C transport activity was calculated similarly based on data obtained using [1-14C]-vitamin C (L-ascorbic acid) (7.3 mCi/mmol; PerkinElmer, Waltham, MA, USA) at 20 μM as a substrate.
Similar uptake assays were performed using URAT1-expressing HEK293 cells with a Cl−-free transport buffer [Cl−-free Hanks’ balanced salt solution: 125 mM Na-gluconate, 4.8 mM K-gluconate, 1.2 mM KH2PO4, 1.2 mM MgSO4, 1.3 mM Ca-gluconate, 25 mM HEPES, 5.6 mM D-glucose, at pH 7.4] according to our previous studies [34, 48]. In cell-based assays for GLUT9a, a high-potassium transport buffer (145.4 mM KCl, 0.8 mM MgSO4, 1.8 mM CaCl2, 25 mM HEPES, 25 mM Tris, 5 mM D-glucose, and pH 7.4) was employed.
Calculation of the kinetic parameters and half-maximal inhibitory concentration values
The Michaelis-Menten constant (Km) and maximal velocity (Vmax) of the SVCT1- or Svct1-mediated urate transport were determined by fitting the Michaelis–Menten model to experimental urate transport rates and concentrations using nonlinear regression curve fitting in GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA), as described previously [49].
The urate and vitamin C transport activities were evaluated in the presence of several target compound concentrations to calculate their half-maximal inhibitory concentration (IC50) values against urate and vitamin C transport by SVCT1. The SVCT1-mediated transport activities were expressed as a percentage of the control (100%). Using the least-squares method with Excel 2019 (Microsoft, Redmond, WA, USA), fitting curves were obtained based on the calculated values according to the following formula as described previously [49]:
where Emax is the maximum effect, EC50 is the half-maximal effective concentration, C is the concentration of the test compound, and n is the sigmoid-fit factor. Finally, based on these results, the IC50 was calculated.
Vesicle transport assay using ABCG2-expressing plasma membrane vesicles
An in vitro vesicle transport assay [46] was conducted using ABCG2-expressing plasma membrane vesicles for the functional characterization of ABCG2. As described previously [34], the ABCG2-expressing plasma membrane vesicles and control vesicles were prepared from 293A cells infected with EGFP-ABCG2-expressing adenovirus and control (EGFP) adenovirus, respectively. Then, the transport assay was conducted using a rapid filtration technique [34]. Vesicles (0.5 mg/mL) were incubated at 37°C for 10 min in the presence of [8-14C]-urate (10 μM) or [1-14C]-vitamin C (500 μM), in a reaction mixture (10 mM Tris/HCl, 250 mM sucrose, 10 mM MgCl2, 10 mM creatine phosphate, 1 mg/mL creatine phosphokinase, pH 7.4, and 50 mM ATP or AMP as an ATP substitute). After washing by ice-cold stop buffer five times, radioactivity incorporated into the vesicles was detected using the liquid scintillator (Tri-Carb 3110TR). The transport activity was calculated as an incorporated clearance (mL/mg protein/min): (incorporated level of tested compound [DPM/mg protein/min] / tested compound level in the reaction mixture [DPM/mL]).
Experimental measurement of vitamin C by LC-PDA analysis
As described previously [23, 35], vitamin C concentrations in WT or Svct1 KO mice plasma were determined using liquid chromatography-photodiode array (LC-PDA) analysis with an ACQUITY UPLC® PDA Detector (Waters, Milford, MA, USA) coupled with an ACQUITY UPLC System (Waters). After deproteinization with equal volumes of 10% (w/v) metaphosphate solution containing 1 mM EDTA, each sample was centrifuged at 20,000 × g at 4°C for 10 min,the obtained supernatant was diluted with equal volumes of 25 mM phosphate buffer (pH 2.1) containing 60 μM acyclovir (FUJIFILM Wako Pure Chemical) as an internal control and then separated on a CAPCELL PAK ADME S3 column (maintained at 50°C; 3 μm, 2.1 × 100 mm; Osaka Soda, Osaka, Japan) with a gradient mobile phase (0–4 min: 2% B; 4–5 min: 2–98% B; 5–9 min: 98% B; 9–10 min: 98–2% B; 10–12 min: 2% B) of 25 mM phosphate buffer (pH 2.1) (A) and methanol (B) at a flow rate of 300 μL/min. Vitamin C and acyclovir were measured at 243 nm in the PDA spectrum. Calibration curves for the analyte were generated using a series of murine plasma spiked with L-ascorbic acid (the reduced form of vitamin C) standard solutions. Peaks were analyzed using MassLynx NT software v4.1 (Waters).
Experimental measurement of urate and creatinine by LC-MS/MS analysis
Serum and urinary urate and creatinine concentrations were determined using an LC-mass spectrometry (MS)/MS system comprising an ACQUITY UPLC® instrument coupled to a Xevo TQ-S triple-quadrupole MS/MS system (Waters), as described in previous investigations [19, 41], with some modifications. After adding acyclovir as an internal standard, specimens were purified by protein precipitation with four volumes of methanol, specimens were diluted in some cases. Each preprocessed sample was separated on an ACQUITY UPLC BEH C18 Column (maintained at 50°C; 1.7 μm, 2.1 × 150 mm; Waters) using gradient mobile phases: [0–3.5 min: 0% B; 3.5–5 min: 0–5% B; 5–5.5 min: 5–95% B; 5.5–10.5 min: 95% B; 10.5–11 min: 95–0% B; 11–15 min: 0% B] of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) at a flow rate of 300 μL/min for urate and [0–5 min: 10% D; 5–6 min: 10–90% D; 6–11 min: 90% D; 11–12 min: 90–10% D; 12–15 min: 10% D) of 5 mM ammonium acetate in water (C) and 0.1% formic acid in methanol (D) at a flow rate of 200 μL/min for creatinine. Ionization was performed using a heated electrospray ionization probe and analytes under optimized conditions for each target compound (Supplementary Table S4), and target compounds were monitored in multiple reactions monitoring mode. Calibration curves for the analyte were generated from a series of standard solutions of uric acid and creatinine. Peak analyses and quantification were conducted using MassLynx NT software version 4.1.
Calculation of fractional excretion of uric acid
Fractional excretion of uric acid (urate clearance/creatinine clearance ratio, FEUA) was calculated using the serum and urine concentrations of urate and creatinine mentioned above, using the following formula: FEUA = (urine urate concentration [μM]/serum urate concentration [μM]) / (urine creatinine concentration [μM]/serum creatinine concentration [μM]) × 100 [%].
Statistics
All statistical analyses were performed using Excel 2019 with Statcel4 add-in software (OMS publishing, Saitama, Japan). The number of biological replicates (n) and the different statistical tests conducted for each experiment are detailed in figure legends. The similarity of variance across multiple groups was compared using Bartlett’s test. When passing the test for homogeneity of variance, a parametric Tukey–Kramer multiple-comparison test for all pairwise comparisons or a parametric Williams’ multiple-comparison test for trend analysis was used. In the case of a single pair of quantitative data, after comparing the variances of a set of data using an F-test, an unpaired Student’s t-test was performed. P values less than 0.05 or 0.01 were considered statistically significant.
All experiments were monitored in a non-blinded fashion. Samples that experienced technical failure during processing were excluded from analyses. Each experiment used the minimum number of mice or samples required to obtain informative results and sufficient data for subsequent studies.
Results
Identification of human SVCT1 as a urate transporter
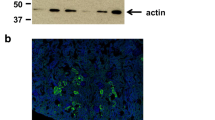
We evaluated the urate transport ability of SVCT1 using mammalian HEK293 cells transiently expressing SVCT1. Immunoblotting and confocal microscopy confirmed the expression (Fig. 1a) and plasma membrane localization (Fig. 1b) of EGFP-tagged SVCT1 in the cells. When incubated in Krebs–Ringer buffer mimicking blood ionic content, the SVCT1-expressing cells incorporated [1-14C]-vitamin C more efficiently than mock cells (Fig. 1c), demonstrating the functional expression of SVCT1. In a similar experimental condition, [8-14C]-urate transport into the SVCT1-expressing cells was also significantly greater than mock cells; however, Na+ exclusion from the Krebs–Ringer buffer blocked the SVCT1-mediated urate transport (Fig. 1d). These results indicated that SVCT1 is a sodium-dependent urate transporter. A high pH condition (the Krebs–Ringer buffer at pH 10) had minimal effect on the SVCT1-mediated urate transport activity (Fig. 1e).

Identification and characterization of human SVCT1 as a urate transporter. All uptake assays were conducted using transiently SVCT1-expressing HEK293 cells 48 h after plasmid transfection in Krebs–Ringer buffer. Experimental conditions: pH 7.4 unless otherwise indicated, or pH 10 (h); [1-14C]-vitamin C in the transport buffer was 20 μM (c, j); incubation time and [8-14C]-urate in the transport buffer were 5 min and 10 μM, respectively, unless otherwise indicated. a–c Functional expression of SVCT1 in HEK293 cells 48 h after the transfection. (a) Immunoblot detection of SVCT1 protein in whole-cell lysates. (b) Intracellular localization of SVCT1 detected by confocal microscopy. Magnified images of representative cells denoted by yellow squares are demonstrated. Scale bars: 10 μm. (c) [1-14C]-Vitamin C transport activities into the cells. d–i SVCT1 as a urate transporter. (d) Sodium-dependency in SVCT1-mediated urate transport. (e) Insignificant effect of an alkaline pH condition (pH 10) on the urate transport activity of SVCT1. (f) Time-dependent [8-14C]-urate incorporation into SVCT1-expressing or mock (control) cells. (g) Time profile for SVCT1-mediated [8-14C]-urate uptake into HEK293 cells, which was calculated by subtracting the urate transport activity of mock cells from that of SVCT1-expressing cells. (h) Concentration dependence in SVCT1-mediated [8-14C]-urate transport. Regarding the estimated Michaelis-Menten constant (Km) and maximal velocity (Vmax), the 95% confidence interval values were provided in parentheses. (I) Concentration-dependent inhibition of SVCT1-mediated urate transport by vitamin C. IC50, the half-maximal inhibitory concentration. j Inhibitory effect of urate on SVCT1-mediated vitamin C transport at physiological concentrations. Values are shown as % of vehicle control (i, j). Data are expressed as the mean ± SD; where vertical bars are not displayed, the SD was contained within the limits of the symbol; n = 3. ††P < 0.01 (two-sided t-test; c, e); ns, not significantly different between groups; **P < 0.01 vs. the other groups (Tukey–Kramer multiple-comparison test; d); ##P < 0.01 vs. vehicle control (Williams’ multiple-comparison test; j)
Based on the results of time-course experiments (Fig. 1f, g), we investigated urate uptake at 5 min in subsequent analyses to determine the initial rates of SVCT1-mediated transport. Because uric acid is sparingly soluble at neutral pH, kinetic analyses were conducted at high pH to achieve high urate concentrations [up to 1.5 mM (approximately 25 mg/dL)] in the buffer during uptake experiments (Fig. 1h); the calculated Km and Vmax were 1511 μM and 265.3 pmol/mg protein/min, respectively, for SVCT1-mediated urate transport. Given this affinity indicator value, in terms of substrate self-inhibition, physiological levels of circulating urate [120–420 μM (approximately 2–7 mg/dL) in a healthy person] could have no significant effect on SVCT1-mediated urate transport. However, vitamin C inhibited urate transport activity in a concentration-dependent manner, with an IC50 value of 95.06 μM (Fig. 1i), suggesting that physiological levels of circulating vitamin C (50–90 μM in a healthy person with ≥100 mg of daily vitamin C intake [27]) would affect SVCT1-mediated urate transport. Moreover, 500 μM urate decreased SVCT1-mediated vitamin C transport to approximately 40% of the vehicle control (Fig. 1j). These findings indicate a potential interaction between urate and vitamin C mediated by SVCT1 in humans.
Considering the dual functionality of SVCT1 as a vitamin C and urate transporter (Fig. 2a), we investigated vitamin C transport activity in three well-characterized physiologically important urate transporters: URAT1 (Fig. 2b), GLUT9 (Fig. 2c), and ABCG2 (Fig. 2d). Even under appropriate experimental conditions in which urate transport activities were detected, neither URAT1, GLUT9, nor ABCG2 could transport vitamin C.

Investigation of vitamin C transport activities in physiologically important urate transporters. Regarding SVCT1 (a), URAT1 (b), GLUT9a (c), and ABCG2 (d), [8-14C]-urate or [1-14C]-vitamin C transport activities were investigated. SVCT1 exhibited a dual substrate specificity, whereas URTA1, GLUT9a, and ABCG2 lacked vitamin C transport activities. Cell-based uptake assays were conducted in Krebs–Ringer buffer (a), Cl−-free Hanks’ balanced salt solution (b), or high-potassium transport buffer (c); incubation time for uptake was 5 min. In vitro transport assay was carried out using ABCG2-expressing plasma membrane vesicles; incubation time for uptake was 10 min (d). Data are expressed as the mean ± SD; n = 3. ††P < 0.01; ns, not significantly different between groups (two-sided t-test)
Identification of mouse Svct1 as a urate transporter
We investigated the urate transport activity of mouse Svct1 in vitro (Fig. 3) prior to the generation of Svct1 KO mice. Functional expression of Svct1 in HEK293 cells was confirmed by immunoblotting (Fig. 3a), confocal microscopy (Fig. 3b), and vitamin C transport assay (Fig. 3c). Our findings confirmed that Svct1 is a sodium-dependent urate transporter of which urate transport activity was maintained even at high pH (Fig. 3d). Since the time profile of Svct1-mediated urate transport (Fig. 3e) was quite similar to SVCT1 (Fig. 1g), kinetic analyses for Svct1 (Fig. 3f) were conducted under identical experimental conditions. Svct1-mediated urate transport had a Km of 646.9 μM and a Vmax of 430.1 pmol/mg protein/min, suggesting no significant difference in molecular characteristics between human SVCT1 and mouse Svct1. Vitamin C also inhibited Svct1-mediated urate transport (similar to SVCT1) with an IC50 value of 129.7 μM (Fig. 3g). In addition, 500 μM urate decreased the Svct1-mediated vitamin C transport to approximately 60% of the vehicle control (Fig. 3h), indicating that Svct1 might be more resistant to the inhibitory effect of urate on vitamin C transport activity than SVCT1.

Identification and characterization of mouse Svct1 as a urate transporter. All uptake assays were conducted using transiently Svct1-expressing HEK293 cells 48 h after plasmid transfection in Krebs–Ringer buffer. Experimental conditions: pH 7.4 unless otherwise indicated, or pH 10 (f); [1-14C]-vitamin C in the transport buffer was 20 μM (c, h); incubation time and [8-14C]-urate in the transport buffer were 5 min and 10 μM, respectively, unless otherwise indicated. a–c Functional expression of Svct1 in HEK293 cells 48 h after the transfection. (a) Immunoblot detection of Svct1 protein in whole-cell lysates. (b) Intracellular localization of Svct1 detected by confocal microscopy. Magnified images of representative cells denoted by yellow squares are demonstrated. Scale bar: 10 μm. (c) [1-14C]-Vitamin C transport activities into the cells. d–g Svct1 as a urate transporter. (d) Sodium-dependency in Svct1-mediated urate transport, which was insignificantly affected by an alkaline pH condition (pH 10). (e) Time profile for Svct1-mediated [8-14C]-urate uptake into HEK293 cells. (f) Concentration dependence in Svct1-mediated [8-14C]-urate transport. Regarding the estimated Michaelis-Menten constant (Km) and maximal velocity (Vmax), the 95% confidence interval values were provided in parentheses. (g) Concentration-dependent inhibition of Svct1-mediated urate transport by vitamin C. IC50, the half-maximal inhibitory concentration. h Mild inhibitory effect of urate on Svct1-mediated vitamin C transport at physiological concentrations. Values are shown as % of vehicle control (g, h). Data are expressed as the mean ± SD; where vertical bars are not displayed, the SD was contained within the limits of the symbol; n = 6 (f), 3 (the others). ††P < 0.01; ns, not significantly different between groups (two-sided t-test; c, d); **P < 0.01 vs. the other groups for pH 7.4 (Tukey–Kramer multiple-comparison test; d); ##P < 0.01 vs. vehicle control (Williams’ multiple-comparison test; h)
Generation and analyses of Svct1 KO mice with a genetic background of Uox and Urat1 deficiency
Next, we generated Svct1 KO mice to examine the involvement of Svct1 in urate handling (Fig. 4). Contrary to humans, uric acid is further metabolized by Uox in WT mice; thus, the Uox KO genetic background was used in this study to address urate levels in vivo. As described in the “Materials and methods” section, genome editing was conducted using the CRISPR-Cas9 system with a functional gRNA targeting the exon 6 of Svct1 (Fig. 4a), resulting in a 19-bp deletion including exon/intron boundary sequences located on the front of exon 6 (Fig. 4b, c). Given that this type of mutation leads to single exon skipping [3], the deletion must have resulted in c.487–688del (p.V163Lfs*37). This frameshift mutation disrupted the vitamin C transport activity of Svct1 (Fig. 4d). Moreover, the loss-of-function was confirmed in vivo in terms of vitamin C handling (Fig. 4e, f) and Svct1 protein expression (Fig. 4g). Indeed, Svct1 KO mice exhibited lower vitamin C levels in plasma (Fig. 4e) and higher values of the urine/plasma vitamin C concentration ratio (Fig. 4f) than WT mice. Immunoblotting successfully detected the renal Svct1 expression, which was absent in Svct1 KO mice (Fig. 4g, Supplementary Fig. S1). These results were consistent with a previous report showing that Svct1 is involved in the re-uptake of vitamin C from urine into the blood by the kidneys [11]. Using MDCKII cells, a polarized renal cell line, we confirmed the apical localization of both human SVCT1 and mouse Svct1 (Supplementary Fig. S2).

Generation and validation of Svct1 knockout mice. a An EGxxFP-based in vitro evaluation of sgRNAs for Svct1 knockout. Control, previously validated sgRNA sequence targeting Centrin 1 as a positive control for the EGxxFP system; Svct1-P, sgRNA sequence targeting Svct1 performed the best in this study (5′-ccagggtgcaatcatggtgtcca-3′); Svct1-N, a representative sgRNA sequence targeting Svct1 performed poorly in this study (5′-catgaggtcgtggattcagcagg-3′). b Schematic illustration of the location of 19-bp deletion in the Svct1 gene. The 19-bp deletion was found at the boundary of intron 5 and exon 6 (Ex6); this deletion is theoretically expected to result in exon 6 skipping, resulting in the frameshift variant p.V163Lfs*37. WT, wild-type; KO, knockout; ORF, open reading frame. c Representative results of PCR-based genotyping for each knockout allele in Svct1 KO mice. NC, non-template control; L, 100-bp ladder marker. d Functional validation of Svct1 KO variant. The KO variant, Svct1 p.V163Lfs*37, transiently expressed in HEK293 cells was ineffective as a vitamin C transporter in Krebs–Ringer buffer (pH 7.4) containing 20 μM [1-14C]-vitamin C. Data are expressed as the mean ± SD, n = 3. e, f Vitamin C levels in plasma (e) and calculated urine-to-plasma vitamin C concentration ratios (f) of Svct1 single KO mice. Data are expressed as the mean ± SEM, n = 5. ††P < 0.01 (two-sided t-test; d–f). g Immunoblot detection of Svct1 in each murine tissue using the anti-Svct1 antibody. Whole cell lysate (WCL) samples prepared from Svct1-expressing and mock HEK293 cells 48 h after transfection were used as positive and negative controls, respectively. A strong signal for Svct1 was detected in the kidney of WT mice but not in Svct1 KO mice, indicating the successful knockout of Svct1 in this study; a side-by-side comparison is shown in Supplementary Fig. S1. Na+/K+ ATPase, a marker for plasma membrane components
The physiological function of Svct1 as a vitamin C re-absorber expressed on the apical membrane of renal epithelial cells prompted us to hypothesize that Svct1 might also serve as a renal urate re-absorber. Given that in healthy conditions, Urat1 is the most influential apical mechanism in the kidney, we knocked out Urat1 from Svct1-Uox DKO mice to sharpen the potency of Svct1, followed by in vivo analyses of the resulting Svct1-Urat1-Uox TKO mice (Fig. 5). The TKO mice exhibited no significant differences in body weight (Fig. 5a), or serum creatinine (a renal function marker) concentrations (Fig. 5b) from control Urat1-Uox DKO mice, demonstrating that Svct1 deficiency did not induce severe renal dysfunction. Serum urate concentrations in the TKO mice were significantly lower than in the control DKO mice (Fig. 5c), indicating the physiological impact of Svct1 on urate handling. Although FEUA values were not significantly different between the two lines (Fig. 5d), an increasing trend (13.7% in TKO; 11.8% in DKO) was consistent with Svct1 deficiency reducing serum urate.

Effects of Svct1 knockout on urate handling in mice deficient in Urat1 and Uox. Body weight (a), serum creatinine (b), serum urate (c), and fractional renal urate excretion (FEUA, urate clearance/creatinine clearance ratio) (d) in Svct1-Urat1-Uox triple knockout (TKO) mice and control [Urat1-Uox double knockout (DKO)] mice. Serum urate levels in TKO mice were significantly lower than DKO mice. Data are expressed as the mean ± SEM, n = 14 (DKO) and 10 (TKO). †P < 0.05; ns, not significantly different between groups (two-sided t-test)
Investigation of inhibitory effects of serum urate-affecting drugs on SVCT1-mediated urate and vitamin C transport
Some SU-affecting drugs have been previously reported to inhibit physiologically important urate transporters with clinical implications via off-target interactions, including febuxostat–ABCG2 and losartan–organic anion transporter 10 (OAT10) [34, 49]. However, there was no information on their effects on SVCT1. Therefore, we investigated the effects of SU-affecting drugs and their active metabolites (a total of 28 compounds listed in Supplementary Table S2) on SVCT1 function as a urate and vitamin C transporter (Fig. 6, Supplementary Fig. S3).

Effects of serum urate-affecting drugs and their active metabolites on the urate and vitamin C transport activities of SVCT1. Forty-eight h after transfection, SVCT1-expressing 293A cells were subjected to a cell-based urate or vitamin C (VC) transport assay. The transport activities were measured for 2.5 min in the presence of each compound at the indicated concentrations. BBR, benzbromarone; 6-OH BBR, 6-hydroxybenzbromarone; DU, dotinurad; IC50, the half-maximal inhibitory concentration. a The effects of each compound [experimentally maximum concentrations (refer to Supplementary Table S2)] on the urate transport activities of SVCT1. Comparison of the effects on the urate and VC transport activities of SVCT1 was shown in Supplementary Fig. S3. b–g Concentration-dependent inhibition of SVCT1-mediated urate (b, d, f) and VC (c, e, g) transport by BBR, 6-OH BBR, and DU. Values are shown as % of vehicle control (0.1% dimethyl sulfoxide); data are expressed as the mean ± SEM; n = 4
An in vitro screening was conducted at maximum drug concentrations (3–1000 μM; Supplementary Table S2). Although some tested drugs caused a moderate enhancement of the urate transport activities of SVCT1 (Fig. 6a), inhibitory effects on SVCT1-mediated urate and vitamin C were comparable in most cases (Supplementary Fig. S3). Benzbromarone, 6-hydroxybenzbromarone, and dotinurad reduced urate transport activities of SVCT1 to <50% of the vehicle controls (Fig. 6a). The IC50 values were then calculated by measuring urate and vitamin C transport activities at different concentrations of each compound (Fig. 6b–g). The calculated IC50 values against urate and vitamin C transport activities of SVCT1 for benzbromarone, 6-hydroxybenzbromarone, and dotinurad were 9.83 μM and 8.39 μM, 9.97 μM and 2.86 μM, and 59.57 μM and 11.76 μM, respectively.
Next, the IC50 values of each compound were compared to its estimated maximum unbound concentration in human plasma (fuCmax) (Supplementary Table S5). We calculated the ratio of fuCmax/IC50 as an indicator, whose high value (≥1) suggested a possible inhibition of SVCT1 by each compound in clinical settings. The calculated values were much lower than 1 for benzbromarone and dotinurad. Despite the lack of 6-hydroxybenzbromarone pharmacokinetic data in humans, the pharmacokinetic parameters of 6-hydroxybenzbromarone and benzbromarone were similar in a prior investigation on monkeys [43]. Our results suggested that neither benzbromarone, 6-hydroxybenzbromarone, nor dotinurad, in circulation impaired SVCT1 function.
Discussion
In this study, we performed in vitro cell-based assays (Figs. 1 and 3) followed by in vivo approaches using hyperuricemic mice harboring the genetic background of Svct1 KO (Figs. 4 and 5) to investigate SVCT1 as a candidate for mammalian urate transporter based on its similarity to YgfU, a prokaryote urate transporter. Given the sodium-dependence of SVCT1-mediated transport and the higher sodium ion concentrations in external fluids relative to the cytosol, SVCT1 must be involved in cellular urate uptake under physiological conditions. Also, we found that urate and vitamin C could compete for SVCT1-mediated transport (Fig. 1i, j). Since both urate and vitamin C serve significant roles as anti-oxidants in the body [1], comprehending their potential interactions in maintaining stable levels may contribute to a more extensive understanding of biological homeostasis regulation.
Our findings suggest that SVCT1 is not specific to vitamin C. These results contradict a widely accepted notion regarding mammalian NATs, which were considered highly specific for vitamin C because SVCTs did not recognize nucleobases as substrates [15, 24, 50]. Traditionally, NAT proteins have been divided into three groups based on their substrate specificities: the first is exclusive to bacteria and specific for uracil,the second is present in bacteria, fungi, and plants and specific for nucleobases and oxidized purines, such as xanthine and/or uric acid (urate); the third is found in vertebrates and specific for vitamin C (L-ascorbate) [15]. Consequently, microbial and plant NATs are nucleobase transporters but not vitamin C transporter, whereas vertebrate NATs have the opposite substrate specificity. All mammalian NATs, except for rat Slc23a4, the first sodium-dependent nucleobase transporter (SNBT1), failed to display substrate selectivity for nucleobases such as uracil and xanthine [50, 54, 59]. However, rSnbt1/Slc23a4 was not involved in vitamin C transport [59], and orthologues of rat Snbt1/Slc23a4 are pseudogenes in almost all primates, including humans [24]. Thus, evidence obtained herein that SVCT1 recognizes both vitamin C and urate (an oxidized purine) as substrates suggests that primate SVCTs could have retained some functional aspects of their ancestral proteins that must have been nucleobase transporters.
YgfU, a homolog of SVCT1 in Escherichia coli, functions as a urate transporter [37], indicating that specific ancestral urate transporters acquired vitamin C transport abilities during evolution. Inhibitory effects of vitamin C on UapA (a well-studied fungus NAT)-mediated xanthine transport [24] suggest a potential molecular interaction between nucleobase-specific NAT(s) and vitamin C under very low-affinity recognition. Thus, primate SVCTs might have evolved from ancestral nucleobase transporters, retaining a portion of their original molecular properties as nucleobase transporters while also acquiring vitamin C transport abilities. Considering a hypothesis that this functional acquisition might have resulted from the evolutional optimization of the sub-function of ancestral proteins [24], such a shift in substrate specificity of NATs (nucleobases–vitamin C) may reflect an evolutionary alternation in the significance of these compounds for each species.
The Km values of SVCT1 for urate [1511 μM, human SVCT1 (Fig. 1h); 647 μM, mouse Svct1 (Fig. 3f)] were higher than those for vitamin C established in previous studies (65–237 μM, human SVCT1 [38], 29 μM, rat Svct1 [50]. SVCT1, therefore, has a strong affinity for vitamin C than urate. The results of this study imply that SVCT1 could have a greater physiological significance as a vitamin C transporter than as a urate transporter, as genetic disruption of Svct1 had a greater impact on vitamin C levels in the blood rather than urate (Figs. 4e and 5c). Nevertheless, our current study deepens the understanding of urate handling systems.
The physiological roles of SVCT1 should be discussed, accompanied by some limitations and perspectives of this study. Further studies are required to understand the mechanism by which SVCT1 regulates serum urate levels, a crucial aspect of urate management. We hypothesized that Svct1 might serve as a renal urate re-absorber based on its high expression in the kidney (Fig. 4g) and function as a renal vitamin C re-absorber [11]. Given the effects of genetic dysfunction on FEUA in humans, URAT1 has the greatest contribution on the net amount of renal urate re-uptake from the luminal side of the proximal tubule among renal urate re-absorbers (including unidentified transporters) [12, 20]. Hence, this study employed the genetic background of Urat1 KO to emphasize the potential function. As expected, serum urate levels in Svct1-Urat1-Uox TKO mice were lower than in control Urat1-Uox DKO mice (Fig. 5c), while there was no statistical difference in the values of FEUA (an indicator for renal urate clearance) between the mice groups (Fig. 5d). To bridge this gap, we can envision a several possibilities as follows.
Since the effects of Svct1-knockout on urate handling was not sufficiently large (but significant in serum urate), sample size used in this study could have not allow us to detect a statistically significant difference in FEUA (calculated by four variables—serum urate concentration and the other three parameters; details are in the “Materials and methods” section). Based on this assumption, our results suggest that SVCT1 could have a role as the third member of renal urate re-absorbers expressed on the apical side of renal proximal tubular cells following URAT1 and OAT10 (a recently identified physiologically important machinery) [4, 16, 49]. Due to their different urate Km values—371 μM, URAT1 [14], 558 μM, OAT10 [49], 1511 μM, SVCT1 (Fig. 1h), when the high-affinity/low-capacity transporter (URAT1) is saturated or dysfunctional, the other low-affinity/high-capacity transporters might compensate. As we have previously proposed with regard to OAT10 [49], this notion for the presence of compensation/back up system(s) is consistent with clinical features of renal hypouricemia which is a pathological condition characterized by low serum urate (≤ 120 μM) and increased renal urate elimination (decreased renal urate reabsorption)—in renal hypouricemia type 2 related to the genetic dysfunction of GLUT9 (a basal machinery involved in urate secretion from renal tubular cells into the blood), typical values of FEUA are extremely high (≥ 100%), in renal hypouricemia type 1 related to that of URAT1, such FEUA values are high (tens of %) but relatively lower than those in type 2 [22]. This difference in FEUA would reflect the contribution of apical machineries for urate re-uptake other than URAT, however, all the remained cannot be explained by OAT10 alone, given the effect of its dysfunction on renal urate handling revealed by a clinico-genetic analysis [49]. Accordingly, SVCT1 could be a component of the systems that have not been entirely identified yet.
Moreover, further supportive information is available. Renal proximal tubules are divided into S1–S3 segments that reportedly have various roles (reabsorption, secretion, and post-secretory reabsorption, respectively) in renal urate handling [29]. Recent transcriptome analysis of murine renal tubule segments [9] revealed that all the three urate transporters were highly expressed in the S2 and S3 segments (Supplementary Fig. S4), however, contrary to Oat10, Urat1 and Svct1 were expressed also in the S1 segment. Although the segmental distribution of each protein in mouse and human kidneys needs to be validated, such expression profiles can strengthen the notion for the backup systems. In addition, previous studies reported that SVCT1-mediated vitamin C transport activity was significantly lower at low (acidic) pH conditions [50, 54], a similar and comparable effect (reduction in relative transport activity) was found in SVCT1-mediated urate transport activity (Supplementary Fig. S5). Contrary to SVCT1, cellular function of OAT10 was significantly higher at such condition [49]. Given these molecular properties in pH sensitivity, physiological contributions of SVCT1 and OAT10 on urate handling might be altered according to pH in primary urine. Thus, addressing what differences in the impact on urate handling exist between SVCT1 and OAT10 will be a future issue.
The latent involvement of non-renal Svct1 may provide an alternate explanation for the TKO mice phenotype. A previous study using Svct1 KO mice showed that accumulation levels of an orally-administered vitamin C analog (6-bromo-6-deoxy-L-ascorbic acid, an SVCT1 substrate [10] in intestinal mucosa were comparable between WT and Svct1 KO mice, suggesting that Svct1 could only have a limited effect on the intestinal vitamin C absorption [11]. The contribution of Svct1-mediated intestinal urate absorption to serum urate levels is unlikely in this context, given Svct1 expression was not detected elsewhere other than the kidney by immunoblotting (Fig. 4g).
Additionally, although vitamin C intestinal absorption has been characterized as an SVCT1-mediated mechanism in recent literature [28, 51], there is no evidence regarding the molecular machinery involved in this process in humans. This speculation seemed plausible at the onset of SVCT1 research due to the expression of SVCT1 mRNA in the intestine [54] and the apical localization of transfected SVCT1 protein in polarized Caco-2 cells (a human colon carcinoma-derived cell line) [5]. However, Svct1 KO mice were still capable of absorbing vitamin C from the diet [11], and a non-functional splice variant of SVCT1 (which cannot been distinguished with a functional variant by northern blotting) was expressed in normal human enterocytes [53], therefore, the role of SVCT1 in mediating the intestinal vitamin C absorption should be carefully interpreted. Our findings on the chemical inhibition of SVCT1-mediated vitamin C transport (Fig. 6) may be relevant pharmacologically. A pharmacokinetic model of gastrointestinal absorption [2] suggests that maximal clinical dosages of benzbromarone (50 mg) and dotinurad (4 mg) can result in maximum concentrations in the intestinal lumen of 379 μM and 35.9 μM, respectively, which are sufficient for substantial inhibition of SVCT1.
In conclusion, we established that SVCT1 functions as a sodium-dependent urate transporter, of which genetic disruption decreased serum urate levels in a hyperuricemic mouse model. Although further studies are needed to unravel the physiological mechanisms underlying the phenotype, our findings contribute to a better understanding of urate handling systems. Furthermore, given the dual-substrate specificity of SVCT1, this NAT family transporter could be a key molecule for elucidating a latent interaction between urate and vitamin C in the human body.
Data availability
Data are available from the corresponding author upon reasonable request. All data relevant to the study are included in the article or uploaded as online Supplementary Information.
Abbreviations
- ABCG2:
-
ATP-binding cassette transporter G2
- BSA:
-
Bovine serum albumin
- DMSO:
-
Dimethyl sulfoxide
- DKO:
-
Double knockout
- FEUA :
-
Fractional excretion of uric acid
- GLUT:
-
Glucose transporter
- IC50 :
-
Half-maximal inhibitory concentration
- HEK:
-
Human embryonic kidney
- MDCKII:
-
Madin-Darby canine kidney II
- V max :
-
Maximal velocity
- fuCmax :
-
Maximum unbound concentration in human plasma
- K m :
-
Michaelis-Menten constant
- NAT:
-
Nucleobase-ascorbate transporter
- ORF:
-
Open reading frame
- OAT10:
-
Organic anion transporter 10
- SU-affecting drug:
-
Serum urate-affecting drug
- sgRNA:
-
Single guide RNA
- SVCT:
-
Sodium-dependent vitamin C transporter
- TKO:
-
Triple knockout
- URAT1:
-
Urate transporter 1
- UOX:
-
Uricase
- WT:
-
Wild-type
References
Ames BN, Cathcart R, Schwiers E, Hochstein P (1981) Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A 78(11):6858–6862. https://doi.org/10.1073/pnas.78.11.6858
Ando H, Hisaka A, Suzuki H (2015) A new physiologically based pharmacokinetic model for the prediction of gastrointestinal drug absorption: translocation model. Drug Metab Dispos 43(4):590–602. https://doi.org/10.1124/dmd.114.060038
Anna A, Monika G (2018) Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet 59(3):253–268. https://doi.org/10.1007/s13353-018-0444-7
Bahn A, Hagos Y, Reuter S, Balen D, Brzica H, Krick W, Burckhardt BC, Sabolic I, Burckhardt G (2008) Identification of a new urate and high affinity nicotinate transporter, hOAT10 (SLC22A13). J Biol Chem 283(24):16332–16341. https://doi.org/10.1074/jbc.M800737200
Boyer JC, Campbell CE, Sigurdson WJ, Kuo SM (2005) Polarized localization of vitamin C transporters, SVCT1 and SVCT2, in epithelial cells. Biochem Biophys Res Commun 334(1):150–156. https://doi.org/10.1016/j.bbrc.2005.06.069
Burzle M, Suzuki Y, Ackermann D, Miyazaki H, Maeda N, Clemencon B, Burrier R, Hediger MA (2013) The sodium-dependent ascorbic acid transporter family SLC23. Mol Aspects Med 34(2–3):436–454. https://doi.org/10.1016/j.mam.2012.12.002
Castro T, Low M, Salazar K et al (2008) Differential distribution of the sodium-vitamin C cotransporter-1 along the proximal tubule of the mouse and human kidney. Kidney Int 74(10):1278–1286. https://doi.org/10.1038/ki.2008.329
Caulfield MJ, Munroe PB, O’Neill D et al (2008) SLC2A9 is a high-capacity urate transporter in humans. PLoS Med 5(10):e197. https://doi.org/10.1371/journal.pmed.0050197
Chen L, Chou CL, Knepper MA (2021) A comprehensive map of mRNAs and their isoforms across all 14 renal tubule segments of mouse. J Am Soc Nephrol 32(4):897–912. https://doi.org/10.1681/ASN.2020101406
Corpe CP, Lee JH, Kwon O, Eck P, Narayanan J, Kirk KL, Levine M (2005) 6-Bromo-6-deoxy-L-ascorbic acid: an ascorbate analog specific for Na+-dependent vitamin C transporter but not glucose transporter pathways. J Biol Chem 280(7):5211–5220. https://doi.org/10.1074/jbc.M412925200
Corpe CP, Tu H, Eck P et al (2010) Vitamin C transporter Slc23a1 links renal reabsorption, vitamin C tissue accumulation, and perinatal survival in mice. J Clin Invest 120(4):1069–1083. https://doi.org/10.1172/JCI39191
Dalbeth N, Choi HK, Joosten LAB, Khanna PP, Matsuo H, Perez-Ruiz F, Stamp LK (2019) Gout. Nat Rev Dis Primers 5(1):69. https://doi.org/10.1038/s41572-019-0115-y
Dehlin M, Jacobsson L, Roddy E (2020) Global epidemiology of gout: prevalence, incidence, treatment patterns and risk factors. Nat Rev Rheumatol 16(7):380–390. https://doi.org/10.1038/s41584-020-0441-1
Enomoto A, Kimura H, Chairoungdua A et al (2002) Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 417(6887):447–452. https://doi.org/10.1038/nature742
Gournas C, Papageorgiou I, Diallinas G (2008) The nucleobase-ascorbate transporter (NAT) family: genomics, evolution, structure-function relationships and physiological role. Mol Biosyst 4(5):404–416. https://doi.org/10.1039/b719777b
Higashino T, Morimoto K, Nakaoka H et al (2020) Dysfunctional missense variant of OAT10/SLC22A13 decreases gout risk and serum uric acid levels. Ann Rheum Dis 79(1):164–166. https://doi.org/10.1136/annrheumdis-2019-216044
Hosoyamada M, Takiue Y, Morisaki H et al (2010) Establishment and analysis of SLC22A12 (URAT1) knockout mouse. Nucleosides Nucleotides Nucleic Acids 29(4–6):314–320. https://doi.org/10.1080/15257771003738634
Hosoyamada M, Tsurumi Y, Hirano H, Tomioka NH, Sekine Y, Morisaki T, Uchida S (2016) Urat1-Uox double knockout mice are experimental animal models of renal hypouricemia and exercise-induced acute kidney injury. Nucleosides Nucleotides Nucleic Acids 35(10–12):543–549. https://doi.org/10.1080/15257770.2016.1143559
Hou H, Xiong W, Zhang X, Song D, Tang G, Hu Q (2012) LC-MS-MS Measurements of urinary creatinine and the application of creatinine normalization technique on cotinine in smokers’ 24 hour urine. J Anal Methods Chem 2012:245415. https://doi.org/10.1155/2012/245415
Hyndman D, Liu S, Miner JN (2016) Urate handling in the human body. Curr Rheumatol Rep 18(6):34. https://doi.org/10.1007/s11926-016-0587-7
Ichida K, Matsuo H, Takada T et al (2012) Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat Commun 3:764. https://doi.org/10.1038/ncomms1756
Kawamura Y, Nakayama A, Shimizu S et al (2021) A proposal for practical diagnosis of renal hypouricemia: evidenced from genetic studies of nonfunctional variants of URAT1/SLC22A12 among 30,685 Japanese Individuals. Biomedicines 9(8):1012. https://doi.org/10.3390/biomedicines9081012
Kondo Y, Inai Y, Sato Y et al (2006) Senescence marker protein 30 functions as gluconolactonase in L-ascorbic acid biosynthesis, and its knockout mice are prone to scurvy. Proc Natl Acad Sci U S A 103(15):5723–5728. https://doi.org/10.1073/pnas.0511225103
Kourkoulou A, Pittis AA, Diallinas G (2018) Evolution of substrate specificity in the nucleobase-ascorbate transporter (NAT) protein family. Microb Cell 5(6):280–292. https://doi.org/10.15698/mic2018.06.636
Kuo CF, Grainge MJ, Zhang W, Doherty M (2015) Global epidemiology of gout: prevalence, incidence and risk factors. Nat Rev Rheumatol 11(11):649–662. https://doi.org/10.1038/nrrheum.2015.91
Lee JH, Oh CS, Mun GH, Kim JH, Chung YH, Hwang YI, Shin DH, Lee WJ (2006) Immunohistochemical localization of sodium-dependent L-ascorbic acid transporter 1 protein in rat kidney. Histochem Cell Biol 126(4):491–494. https://doi.org/10.1007/s00418-006-0186-1
Levine M, Conry-Cantilena C, Wang Y et al (1996) Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A 93(8):3704–3709. https://doi.org/10.1073/pnas.93.8.3704
Lykkesfeldt J, Tveden-Nyborg P (2019) The pharmacokinetics of vitamin C. Nutrients 11(10):2412. https://doi.org/10.3390/nu11102412
Maesaka JK, Fishbane S (1998) Regulation of renal urate excretion: a critical review. Am J Kidney Dis 32(6):917–933. https://doi.org/10.1016/s0272-6386(98)70067-8
Mashiko D, Fujihara Y, Satouh Y, Miyata H, Isotani A, Ikawa M (2013) Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci Rep 3:3355. https://doi.org/10.1038/srep03355
Mashiko D, Young SA, Muto M et al (2014) Feasibility for a large scale mouse mutagenesis by injecting CRISPR/Cas plasmid into zygotes. Dev Growth Differ 56(1):122–129. https://doi.org/10.1111/dgd.12113
Matsuo H, Chiba T, Nagamori S et al (2008) Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet 83(6):744–751. https://doi.org/10.1016/j.ajhg.2008.11.001
Matsuo H, Takada T, Ichida K et al (2009) Common defects of ABCG2, a high-capacity urate exporter, cause gout: a function-based genetic analysis in a Japanese population. Sci Transl Med 1(5):5ra11. https://doi.org/10.1126/scitranslmed.3000237
Miyata H, Takada T, Toyoda Y, Matsuo H, Ichida K, Suzuki H (2016) Identification of febuxostat as a new strong ABCG2 inhibitor: potential applications and risks in clinical situations. Front Pharmacol 7:518. https://doi.org/10.3389/fphar.2016.00518
Miyata H, Toyoda Y, Takada T, Hiragi T, Kubota Y, Shigesawa R, Koyama R, Ikegaya Y, Suzuki H (2022) Identification of an exporter that regulates vitamin C supply from blood to the brain. iScience 25(1):103642. https://doi.org/10.1016/j.isci.2021.103642
Nakao H, Harada T, Nakao K, Kiyonari H, Inoue K, Furuta Y, Aiba A (2016) A possible aid in targeted insertion of large DNA elements by CRISPR/Cas in mouse zygotes. Genesis 54(2):65–77. https://doi.org/10.1002/dvg.22914
Papakostas K, Frillingos S (2012) Substrate selectivity of YgfU, a uric acid transporter from Escherichia coli. J Biol Chem 287(19):15684–15695. https://doi.org/10.1074/jbc.M112.355818
Savini I, Rossi A, Pierro C, Avigliano L, Catani MV (2008) SVCT1 and SVCT2: key proteins for vitamin C uptake. Amino Acids 34(3):347–355. https://doi.org/10.1007/s00726-007-0555-7
Sotiriou S, Gispert S, Cheng J et al (2002) Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat Med 8(5):514–517. https://doi.org/10.1038/nm0502-514
Takada T, Yamanashi Y, Konishi K, Yamamoto T, Toyoda Y, Masuo Y, Yamamoto H, Suzuki H (2015) NPC1L1 is a key regulator of intestinal vitamin K absorption and a modulator of warfarin therapy. Sci Transl Med 7(275):27ra5223. https://doi.org/10.1126/scitranslmed.3010329
Takada T, Yamamoto T, Matsuo H et al (2018) Identification of ABCG2 as an exporter of uremic toxin indoxyl sulfate in mice and as a crucial factor influencing CKD progression. Sci Rep 8(1):11147. https://doi.org/10.1038/s41598-018-29208-w
Takanaga H, Mackenzie B, Hediger MA (2004) Sodium-dependent ascorbic acid transporter family SLC23. Pflugers Arch 447(5):677–682. https://doi.org/10.1007/s00424-003-1104-1
Taniguchi T, Ashizawa N, Matsumoto K et al (2019) Pharmacological evaluation of dotinurad, a selective urate reabsorption inhibitor. J Pharmacol Exp Ther 371(1):162–170. https://doi.org/10.1124/jpet.119.259341
Toyoda Y, Takada T, Miyata H, Ishikawa T, Suzuki H (2016) Regulation of the axillary osmidrosis-associated ABCC11 protein stability by N-linked glycosylation: effect of glucose condition. PLoS One 11(6):e0157172. https://doi.org/10.1371/journal.pone.0157172
Toyoda Y, Takada T, Umezawa M, Tomura F, Yamanashi Y, Takeda K, Suzuki H (2019) Identification of hepatic NPC1L1 as an NAFLD risk factor evidenced by ezetimibe-mediated steatosis prevention and recovery. FASEB Bioadv 1(5):283–295. https://doi.org/10.1096/fba.2018-00044
Toyoda Y, Takada T, Suzuki H (2019) Inhibitors of human ABCG2: from technical background to recent updates with clinical implications. Front Pharmacol 10:208. https://doi.org/10.3389/fphar.2019.00208
Toyoda Y, Takada T, Miyata H et al (2020) Identification of GLUT12/SLC2A12 as a urate transporter that regulates the blood urate level in hyperuricemia model mice. Proc Natl Acad Sci U S A 117(31):18175–18177. https://doi.org/10.1073/pnas.2006958117
Toyoda Y, Takada T, Saito H, Hirata H, Ota-Kontani A, Kobayashi N, Tsuchiya Y, Suzuki H (2020) Inhibitory effect of Citrus flavonoids on the in vitro transport activity of human urate transporter 1 (URAT1/SLC22A12), a renal re-absorber of urate. NPJ Sci Food 4:3. https://doi.org/10.1038/s41538-020-0063-7
Toyoda Y, Kawamura Y, Nakayama A et al (2022) OAT10/SLC22A13 acts as a renal urate re-absorber: clinico-genetic and functional analyses with pharmacological impacts. Front Pharmacol 13:842717. https://doi.org/10.3389/fphar.2022.842717
Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang Y, Brubaker RF, Hediger MA (1999) A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 399(6731):70–75. https://doi.org/10.1038/19986
Tveden-Nyborg P (2021) Vitamin C deficiency in the young brain-findings from experimental animal models. Nutrients 13(5):1685. https://doi.org/10.3390/nu13051685
Vitart V, Rudan I, Hayward C et al (2008) SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet 40(4):437–442. https://doi.org/10.1038/ng.106
Wang H, Dutta B, Huang W, Devoe LD, Leibach FH, Ganapathy V, Prasad PD (1999) Human Na(+)-dependent vitamin C transporter 1 (hSVCT1): primary structure, functional characteristics and evidence for a non-functional splice variant. Biochim Biophys Acta 1461(1):1–9. https://doi.org/10.1016/s0005-2736(99)00182-0
Wang Y, Mackenzie B, Tsukaguchi H, Weremowicz S, Morton CC, Hediger MA (2000) Human vitamin C (L-ascorbic acid) transporter SVCT1. Biochem Biophys Res Commun 267(2):488–494. https://doi.org/10.1006/bbrc.1999.1929
Watanabe T, Tomioka NH, Watanabe S, Suzuki Y, Tsuchiya M, Hosoyamada M (2016) The mechanism of false in vitro elevation of uric acid level in mouse blood. Biol Pharm Bull 39(7):1081–1084. https://doi.org/10.1248/bpb.b15-01046
Woodward OM, Kottgen A, Coresh J, Boerwinkle E, Guggino WB, Kottgen M (2009) Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc Natl Acad Sci U S A 106(25):10338–10342. https://doi.org/10.1073/pnas.0901249106
Wu XW, Muzny DM, Lee CC, Caskey CT (1992) Two independent mutational events in the loss of urate oxidase during hominoid evolution. J Mol Evol 34(1):78–84. https://doi.org/10.1007/BF00163854
Wu X, Wakamiya M, Vaishnav S, Geske R, Montgomery C Jr, Jones P, Bradley A, Caskey CT (1994) Hyperuricemia and urate nephropathy in urate oxidase-deficient mice. Proc Natl Acad Sci U S A 91(2):742–746
Yamamoto S, Inoue K, Murata T, Kamigaso S, Yasujima T, Maeda JY, Yoshida Y, Ohta KY, Yuasa H (2010) Identification and functional characterization of the first nucleobase transporter in mammals: implication in the species difference in the intestinal absorption mechanism of nucleobases and their analogs between higher primates and other mammals. J Biol Chem 285(9):6522–6531. https://doi.org/10.1074/jbc.M109.032961
Acknowledgements
The authors thank Drs. Ryoko Kudo and Motoki Goto for their technical support with developmental engineering. We also appreciate Fuji Yakuhin (Saitama, Japan) for providing dotinurad. YT is an Excellent Young Researcher in the MEXT Leading Initiative for Excellent Young Researchers. HMi was a Japan Society for the Promotion of Science (JSPS) Research Fellow; KM is also a JSPS Research Fellow.
Funding
Open access funding was provided by The University of Tokyo. This study was supported by the JSPS KAKENHI (Grant Numbers [21H03350 to YT], [16J01063 and 21K15293 to HMi], and [16H01808, 20H00568, 22K19369, and 22KK0152 to TT]). YT received a research grant from “The Nakajima Foundation”; TT received research grants from “Takeda Science Foundation,” “Gout and uric acid foundation of Japan,” and “The Nakatomi Foundation.”
Author information
Authors and Affiliations
Contributions
YT, HMi, and TT contributed to the study conception. YT, HMi, NU, and KM validated the study. YT and HMi carried out the formal analysis. YT, HMi, NU, KM, and RS performed investigations. YT, HMi, NU, KM, RS, HK, KN, NHT, HMa, KI, MH, and AA acquired resources for the study. YT and HMi performed data curation. YT prepared the original draft. YT and TT reviewed and edited the manuscript. YT and HMi performed visualization. HS and TT supervised the study. YT and TT were project administrators. YT, HMi, and TT acquired funding. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All animal experiments were performed according to methods approved by the Institutional Animal Care and Use Committee of The University of Tokyo. All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health.
Consent to participate
N/A.
Competing interests
Financial interests: KI and TT received fees for advisory boards and speaking fees from Mochida Pharmaceutical and Fuji Yakuhin. Non-financial interests: dotinurad was kindly provided by Fuji Yakuhin to TT under a material transfer agreement. All the companies had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation. The remaining authors declare they have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Toyoda, Y., Miyata, H., Uchida, N. et al. Vitamin C transporter SVCT1 serves a physiological role as a urate importer: functional analyses and in vivo investigations. Pflugers Arch - Eur J Physiol 475, 489–504 (2023). https://doi.org/10.1007/s00424-023-02792-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-023-02792-1