
Abstract
Hypertension is one of the leading causes of premature death in humans and exhibits a complex aetiology including environmental and genetic factors. Mutations within the glucocorticoid receptor (GR) can cause glucocorticoid resistance, which is characterized by several clinical features like hypercortisolism, hypokalaemia, adrenal hyperplasia and hypertension. Altered glucocorticoid receptor signalling further affects sodium and potassium homeostasis as well as blood pressure regulation and cell proliferation and differentiation that influence organ development and function. In salt-sensitive hypertension, excessive renal salt transport and sympathetic nervous system stimulation may occur simultaneously, and, thus, both the mineralocorticoid receptor (MR) and the GR-signalling may be implicated or even act interdependently. This review focuses on identified GR mutations in human primary generalized glucocorticoid resistance (PGGR) patients and their related clinical phenotype with specific emphasis on adrenal gland hyperplasia and hypertension. We compare these findings to mouse and rat mutants harbouring genetically engineered mutations to further dissect the cause and/or the consequence of clinical features which are common or different.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypertension is a multifactorial disease and affects one-third of the human adult population [36]. It has been recently identified as the most prevalent cardiovascular comorbidity in patients infected with COVID-19 significantly increasing the risk of hospitalization and death [40, 53]. Glucocorticoid resistance is a condition characterized by generalized, partial target tissue resistance to this hormone [48]. Compensatory mechanisms lead to elevation in circulating adrenal steroids with mineralocorticoid and/or androgenic activity, and the clinical spectrum of this condition is broad, ranging from asymptomatic to severe cases of hyperandrogenism, fatigue and/or mineralocorticoid excess. The molecular basis of glucocorticoid resistance has been attributed to mutations in the GR gene impairing glucocorticoid signal transduction [2]. The role of the glucocorticoid receptor in salt-sensitive hypertension is less understood in human, and, therefore, animal models carrying GR mutations are useful to study the cause and the development of this disease.
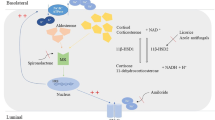
Glucocorticoids are ligands of the glucocorticoid receptor (GR), which is one of the most studied transcriptional regulatory factors (TRFs) involved in homeostasis and physiological regulation [76]. Glucocorticoids are small lipophilic signalling molecules produced by the adrenocortex under tight regulation of the hypothalamic–pituitary–adrenal gland (HPA) axis [63] (Fig. 1A). They present fundamental hormones playing a role in various biological functions as metabolism, inflammation processes or stress through the control of more than 1000 genes [20]. These hormones are released rhythmically with both a circadian and an ultradian (pulsatile) pattern [20]. Despite a precise regulation, glucocorticoids are sometimes secreted in excess or in an insufficient manner [57]. GR mutations leading to glucocorticoid insensitivity can affect glucocorticoid production through a dysregulation of the feedback loop (Fig. 1A) leading to a large spectrum of clinical phenotypes as adrenal hyperplasia and hypertension. Sustained glucocorticoid excess together with high salt intake might induce salt-sensitive hypertension in human, but it was not reported yet whether varying salt intake differentially affected subjects carrying GR mutations. Studies in animal models associated glucocorticoid excess with ACTH-induced steroidogenesis, increased deoxycorticosterone levels and/or increased glucocorticoid-mediated MR signalling, but also with abnormal renal hemodynamic response and failure of the distal nephron to adapt to high salt intake [26].

Pathogenesis of glucocorticoid resistance. A Consequences of GR mutations on the hypothalamic–pituitary axis causing hypercortisolism. ACTH adenocorticotropic hormone; CRH corticotropin-releasing hormone B Linear model of the human GR structure and localization of identified mutations. C Linear structure of rodent GRs carrying the mutant em2, em4 and β geo allele; NTD N-terminus domain, DBD DNA binding domain, HR hinge region, LBD ligand binding domain
In human, there are still questions that need to be addressed, (i) if and which kind of GR mutations are causative for the development of GR-mediated hypertension, and (ii) which are the main predisposing factors for this disease. Moreover, it was not clear until recently if some of these clinical features might be recapitulated in animal models to help dissect the underlying molecular mechanism. The aim of this review is to give an overview of GR pathological mutations found in humans and on studies of GR mutations done in experimental animals. We will discuss the prevalence of hypertension and adrenal hyperplasia in patients and animals carrying GR mutations and discuss possible mechanisms.
Primary structure of the glucocorticoid receptor
The primary structure of the human glucocorticoid receptor is known since 1985 [45], which is the date of its first reported cloning. The nuclear receptor subfamily 3 group C member 1 (NR3C1) gene, located on the short arm of chromosome 5 (5q31Y32) [14], encodes the GR. This gene is composed of nine exons (1 noncoding and 8 coding exons). Alternative splicing produces two main protein isoforms, namely hGRα and hGRβ leading further to eight receptor α and eight β isoforms [50]. The two isoforms are similar up to position 727, and hGRα is longer of 50 amino acids [52]. In 1990, Zong and colleagues [24] demonstrated that the promoter region (5′UTR-untranslated region) of human GR has a GC content of 72% but does not contain a TATA or CAAT box. The 184 nucleotides of exon 1 represent only the 5′UTR and are not coding for the protein. The major transcription start site is situated in exon 2, 134 bp upstream of the ATG initiation codon [23].
The 5′ part of the sequence encodes the N-terminus domain (NTD, 421 amino acids) of the receptor (Fig. 1). The main component of the NTD is the ligand-independent constitutive transcriptional activation function 1 (AF1), also named tau1 (Fig. 1B). AF1 is rich in acidic amino acids and necessary for GR transcriptional activation and for the interaction of GR with co-regulators, chromatin modulators and the basal transcription machinery. The NTD is followed by the DNA binding domain (DBD). A particularity of the DBD is the two highly conserved zinc fingers which are required for the DNA binding specificity of the receptor and the dimerization. These subdomains contain four cysteine residues coordinating a zinc ion, followed by an amphipathic helix and a peptide loop [76]. The loop of the second zinc finger is important for the dimerization via the distal box (D box) [76]. The DBD is the most conserved region among the nuclear hormone receptors, whereas the NTD is the most variable domain [34]. Between DBD and the ligand-binding domain (LBD), a small and flexible region is present from amino-acid 488 to amino-acid 527, the hinge region (HR) (Fig. 1B). Recent studies demonstrated its role in circadian transcriptional activity of the GR [32, 44]. At the C-terminus of the GR is the LBD encoded by exons 5 to 9. The LBD comprises three important regions, the transactivating domain 2 (Tau2), AF2 necessary for ligand-dependent interactions with co-regulators and an additional nuclear localization signal (NLS) (Fig. 1B). The hGR is virtually expressed in all tissues [69].
NR3C1 gene mutations in humans
Since 1982, 38 NR3C1 gene mutations have been described in human (summarized in Fig. 1; Table 1). Among these mutations, four have been described separately in unrelated patients (G679S, R477S, L672P and R714Q). G679S is found both as homo- and heterozygous mutation in human. In 2001, Raef et al. [56] demonstrated that the homozygous G679S mutation of the GR-α gene is associated with severe cortisol resistance, whereas a heterozygous mutation can lead to subclinical cortisol resistance. The R477S mutation presents different clinical phenotypes (adrenal hyperplasia vs no adrenal hyperplasia; normotensive vs hypertensive). In total, 38 GR mutations are listed in Table 1, with their related clinical phenotype documented at the age of diagnosis as well as in follow-up studies. For example, the E198X mutation has been discovered in a 3-year-old patient without adrenal hyperplasia [64]. However, at the age of 12 years, this patient additionally presented with bilateral adrenal hyperplasia [64]. Overall, the age of diagnosed patients is ranging from 5 weeks to 70 years with an average of 32.8 ± 19 years even though, in families with GR mutations, the age of affected subjects is not always mentioned. Over the past years, the number of diagnosed patients carrying GR mutations is continuously increasing, and 16 hitherto unknown GR mutations were documented within the last 6 years. Overall, 26 new mutations were identified in women versus 16 in men (Table 1). It might be interesting to follow whether there is a generally higher incidence in women. The large majority are heterozygous (31 cases) in comparison to six homozygous GR mutations. Homozygous mutations might be underrepresented due to the severity of the clinical symptoms.
Molecular features of the NR3C1 gene mutations
In human, 62% of the NR3C1 gene mutations affect the LBD (Fig. 1), and, despite its size and its variability in the nuclear receptor family, only four mutations are localized within the N-terminus domain, with two of these situated within the AF1 domain (Q123T and E198X). Tatsi et al. [64] showed that E198X was a nonsense mutation resulting in a premature stop. The loss of 577 amino acids leads to degradation through the nonsense-mediated mRNA decay (NMD) mechanism. Q123T leads to glucocorticoid resistance and is characterized by a truncated non-functional protein [52]. The mutant D401H equally results in a truncated protein [9], whereas the P9R variant presents a missense mutation [35]. Two mutations (S551Y and T556I) are localized in the tau 2 domain of the LBD and I757V in the AF2 domain, respectively [73] (Fig. 1; Table 1). The homozygous S551Y mutation did not affect GR protein abundance but decreased its transcriptional activity, nuclear translocation and dimerization [36]. The T556I mutation did not affect GR protein abundance [78].
In vitro studies using transfections allowed to quantify the fluorescence of the different GR mutants in distinct subcellular compartments and to determine GR-dependent nuclear translocation [72]. The Δ612GR mutant GR protein could not be detected [65]. In truncated GR mutants Q123T, E198X and R469X, protein abundance is decreased [5, 52, 64]. Ligand binding is normal in D401H, V423A, R477H, R477S and Y478C mutations and decreased in S551Y, T556I, I559N, V571A, V575G, D641V, G679S, R714Q, H726R, V729I, I737L, I747M and L773VfsX25 mutations and lacking in the Δ612GR, L672P and F774SfsX24 mutations. Data are not reported for six mutations (H588LfsX5, L595V, P9R, R477C, I757V and I465SfsX22: Table 1). DNA binding is preserved in D401H, T556I, V575G, G679S, R714Q, H726R and I737L mutations, decreased in 2 mutations (V423A and Y478C; [59, 72]) and lacking in 3 mutations (R477H, R477S and L672P; [60, 72, 74]). Transactivation is only increased in the D401H mutation and lacks in the R477S, R491X, F774SfsX24, Δ612GR, Y660X and L672P mutations. Translocation of the GR-glucocorticoid complex is diminished in most of the GR mutations. Only the D401H mutation presents normal trafficking [9], and Δ612GR and L672P mutations depict no translocation of the GR-glucocorticoid complex [65, 72]. Overall, most described GR mutations exhibit a normal protein abundance, but decreased ligand binding, transactivation and translocation.
Clinical symptoms of NR3C1 mutation
Glucocorticoids are key hormones involved in multiple pathways and physiological processes as development, metabolism, or immunity [73]. Any GR mutation thus affects multiple pathways and might thus affect organ development and function leading to a systemic phenotype (Fig. 2). Patients carrying heterozygous or homozygous GR mutations present with hypercortisolism (100%), hypertension (51.3%), adrenal hyperplasia (38%), fatigue (28%), anxiety, hirsutism (25%) and hypokalaemia (21.6%: Fig. 2). Irregular menstruation cycle, infertility, obesity or low body weight, dyslipidaemia, diabetes, hyperandrogenism, polyuria, ambiguous genitalia, oligo/amenorrhea, hypoglycaemia, alopecia, acne and precocious pubarche, clitoromegaly and advanced bone age symptoms are more rare [52, 73] (Fig. 2). A few human studies additionally suggested impairment of sodium homeostasis although plasma Na+ levels were in the normal range [33, 36]. To further answer this question, more detailed and standardized phenotyping might be required in the future. More recently, Vitellius and Lombes concluded that there is no clear genotype–phenotype relationship between the severity of the disease and the degree of functional loss of the mutant GR and the age at presentation. On the other side, there was a tendency that young patients presented more severe clinical signs than adults [73]. The reason is, however, not known, and more case studies might be required. In the following part, we will further discuss the link between GR mutation, adrenal gland hyperplasia and hypertension.

Graphic representing percentages of all clinical features observed in patients carrying 38 GR mutations (see Table 1) causing glucocorticoid resistance
Is adrenal hyperplasia a symptom of GR resistance?
In 2018, Vitellius et al. hypothesized that GR mutations might represent a novel genetic cause of adrenal hyperplasia [74]. In the French MUTA-GR study including 100 patients, they discovered 2 new mutations (R491X, Q501H) and 3 already described GR mutations (R469X, R477S and L672P). The prevalence of patients exhibiting adrenal hyperplasia and hypertension and/or presenting hypercortisolism is 5% thus encouraging further GR mutation screening to differentiate GR resistance from Cushing’s syndromes and to improve the follow-up of these patients. It is worthwhile mentioning that not all cases of PGGR are attributed to NR3C1gene mutations [46]. Among the 38 GR mutations identified in human, 14 (38%) are associated with adrenal gland hyperplasia (Table 1). Seven of these mutations are present in the LBD, 3 in the DBD (L672P, R477S and R469X), 2 in the HR (I465SfsX22 and R491X) and 2 in the NTD (P9R, E198X). The adrenal hyperplasia is significantly bilateral (11 cases versus 3 cases unilateral: Table 1). The relatively high percentage of adrenal hyperplasia strongly suggests the GR mutations being causative. Furthermore, a recent study done in 389 patients determining the genetic predisposition to primary bilateral macronodular adrenal hyperplasia (PBMAH) found a prevalence of 8.9% GR mutations [67].
Nine of these 14 GR mutations presented an elevated plasmatic level of the adrenocorticotropic hormone (ACTH) (Table 1), although also patients with normal ACTH levels were diagnosed (Table 1). Furthermore, patients carrying the Q501H mutation present a low level of ACTH despite adrenal hyperplasia. Thus, an increase of ACTH level might not be the only cause of adrenal gland hyperplasia in patients harbouring GR mutations. An autocrine positive regulatory feedback of glucocorticoid secretion was proposed that directly impacts NCI-H295R adrenocortical cell function [2]. Thus, following birth, the adrenal gland continues to mature undergoing rapid involution and differentiation into zona fasciculata (ZF) and zona glomerulosa mainly under the influence of ACTH and angiotensin II. At the age of 6 years, the zona reticularis will appear. Thus, GR may play a direct or indirect role in the adrenarche process. It could explain the adrenal gland hyperplasia diagnosed in a patient harbouring the E198X mutation only at the age of 12 and not at 3 years when the mutation has been found [64] (Figs. 1 and 3). Interestingly, 9 GR mutations out of 14 (64.3%) with adrenal hyperplasia are also associated with hypertension (Table 1), i.e. 47% of the GR mutations with hypertension are also associated with adrenal gland hyperplasia raising the question whether a direct link between hypertension and adrenal hyperplasia exists (Fig. 3). In several cases, an increase of the aldosterone level may explain hypertension. Furthermore, a high aldosterone level associated with an adrenal hyperplasia and hypokalaemia might be a sign of primary aldosteronism.

Hypothetical scheme of mechanisms implicated in the generation of salt -sensitive hypertension and adrenal hyperplasia in human and animal models
Are GR mutations causative of primary aldosteronism?
Primary aldosteronism (PA) is the first and most common cause of secondary hypertension [35] characterized by high blood pressure, adrenal hyperplasia, hyperaldosteronism, low renin concentration and often hypokalaemia [6, 77] (Fig. 3). Often underdiagnosed, PA is prevalent in 5–10% of hypertensive patients and reaches more than 20% prevalence in patients suffering of resistant hypertension [6, 77]. Genetic somatic alterations were identified in aldosterone-producing tumours and adrenocortical carcinoma and linked to aberrant intracellular calcium signalling and altered ATPase function leading to enhanced CYP11B2 expression and increased aldosterone production [62, 77]. Patients with the familial form 3 of hyperaldosteronism presented with massive adrenal hyperplasia, severe hypokalaemia and high concentrations of the hybrid steroids 18-oxocortisol and 18-hydroxycortisol in urine [13].
It is important to note that PA might also be associated with hypercortisolism or subclinical Cushing’s syndrome. Cortisol-producing tumours were associated with aberrant cAMP-protein kinase A signalling. Furthermore, germline mutations in the tumour suppressor gene armadillo repeat containing 5 (ARMC5) were the underlying cause for primary bilateral macronodular adrenal hyperplasia (PBMAH) (see for review [29]). Indeed, recent studies demonstrated that the prevalence of hypercortisolism in PA is ranging from 12.1 to 77.6% [25]. Furthermore, an impairment of the glucose metabolism in PA is associated with cortisol co-secretion and may lead to diabetes type II underlining hypercortisolism as common clinical feature for PA [19]. PA patients with abnormal activation of glucocorticoid receptor in keratinocytes were reported [4].
In summary, due to the heterogeneity of the data collected, we cannot conclude that GR mutations are causative for PA. There is certainly additional need for evaluation of the renin-aldosterone ratio and the plasma renin activity (PRA). To further understand the underlying mechanisms of glucocorticoid resistance, adrenal gland hyperplasia and hypertension, animal models are useful to further dissect the development of the symptoms under normal and challenging conditions. In the following section, we will discuss the findings in transgenic rodents exhibiting GR mutations (Table 2).
GR transgenic animal models and hypertension
The role of GR on salt sensitive hypertension, salt excretion and renal blood flow was first studied in GR haploinsufficient rodents. A lack of sensitivity to glucocorticoids was observed in mice carrying an impaired corticosteroid receptor function by inhibiting gene expression with antisense RNA. These mice showed increased plasma corticosterone as well as adrenocorticotropic hormone (ACTH) concentrations and slightly hyperplastic adrenal glands [54] (Table 2). Higher plasma corticosterone levels were also found in mice with loss of one functional GR allele (GR+/−) [16]. Further studies in these mice revealed the corticotropin-releasing hormone (CRH) as a major target for glucocorticoid feedback-control at the hypothalamic level [33]. Thereby, GR+/− mice were lacking the GR-dependent regulation of the hypothalamus-pituitary axis (HPA) established during foetal development [58]. In models of globally reduced GR expression, salt-sensitivity and sustained hypertension were observed most likely reflecting sustained mineralocorticoid receptor activation [26, 55]. Mice in which the GR was disrupted by gene-trap integration of a βgeo reporter gene (GRβgeo/+, [39]) displayed a hyperactive HPA with elevated plasma corticosterone and larger adrenal glands due to hyperplasia as well as hypertension due to the activation of the renin–angiotensin–aldosterone system (RAAS) [39]. A high salt diet induced furthermore an increase of corticosterone excretion and the BP in these mice. Similarly, rats with GR haploinsufficiency (GR+/em2, [68]) when kept on a standard diet showed significantly increased plasma aldosterone and corticosterone levels and an adrenocortex hyperplasia but a normal systolic blood pressure. Salt-sensitive hypertension was only provoked in combination with high salt intake in the transgenic rats [68]. During prolonged salt exposure, both transgenic and control rats progressively reduced their salt intake [68]. While, in the mouse, an adaptive failure of the renal vasculature and tubule resulting in transient sodium retention was documented, a dysregulation of the soluble epoxide hydroxylase enzyme (sEH) that regulates omega 3 and omega 6 non-polysaturated fatty acid metabolism was reported in rat, with a significant increase in less active metabolites [68]. Further studies are needed to elucidate whether this is a cause or consequence of salt-sensitive hypertension and/or this may affect the endothelial function.
To better understand the role of the vasculature in the generation and maintenance of glucocorticoid-mediated hypertension, vascular smooth muscle-specific (GRTie−Cre) knockout mice were analyzed [21]. These animals showed reduced vascular reactivity to dexamethasone, although their BP did not change significantly upon chronic oral administration of dexamethasone. Differences in circadian BP rhythm as well as higher baseline BP were observed suggesting that the vascular endothelial glucocorticoid receptor may function as a peripheral circadian clock [21]. Further studies are required to address the question whether glucocorticoid-mediated hypertension is primarily due to effects of glucocorticoids on the vasculature and not due to sodium retention and water absorption or both.
Persistent increased circulating glucocorticoids were proposed to contribute to nocturnal hypertension and induced a non-dipping blood pressure profile in mice with global reduction of GR abundance [28]. BP further increased by high salt diet in these mice, although systemic effects might not be excluded [26]. GR response elements (GREs) within the α-ENaC promoter region mediated glucocorticoid-induced transcription that might allow GR to contribute to the control of Na+ absorption [51]. Furthermore, GR activation dynamically regulated NCC phosphorylation and established the diurnal rhythm of NCC activity [27]. To further exclude systemic effects on BP by hypercortisolemia and understand the role of GR in combination with renal ion transporters in BP regulation, the implication of renal tubular GR was further analysed in mice lacking GR either along the nephron (Nr3c1Pax8/LC1 [8]) or only in the distal nephron [22]. Overall, Nr3c1Pax8/LC1 mice specifically lacking the renal tubular GR do not show alteration in sodium or potassium balance but present a transient sodium handling defect following diet switch to low sodium diet and an increased diastolic BP dipping upon switch to high salt diet. Analysis of Nr3c1Pax8/LC1 mice revealed that GR affected αENaC abundance and NCC activity in renal tubules [8]. Mice with a partial constitutive knockout in the distal nephron (GRloxP;Ksp−Cre+) exhibited mildly increased baseline BP, but similar hypertensive response to dexamethasone [22]. Interestingly, mice lacking the mineralocorticoid receptor (MR) along the nephron show increased GR protein abundance [8] and mice constitutively lacking the MR can partially be rescued with dexamethasone injections [3]. Mu and colleagues showed that dietary salt excess, coupled with β-adrenergic receptor stimulation, increased arterial BP via glucocorticoid receptors and WNK4 [42]. Salt loading increased blood pressure in isoproterenol-treated WT but not in distal nephron-specific glucocorticoid receptor knockout mice, suggesting that the GR is indispensable for βAR-mediated WNK4 downregulation and the development of salt-induced hypertension [18].
Glucocorticoid signalling regulates a wide range of systems in vertebrate organisms, and their responsiveness to glucocorticoids differs largely between individuals. Apart from the commonly used rodent models, the zebrafish has been proposed as an excellent in vivo model to study glucocorticoid resistance [61]. As a non-mammalian animal model, it has a GR β splice variant with dominant-negative activity and can thus be used to understand glucocorticoid action by GR β isoform overexpression or lacking glucocorticoid downregulation [61].
Conclusion
Glucocorticoid signalling stimulates several pathways concurrently and converges on common mechanisms. Although a relatively high number of human subjects suffering from glucocorticoid resistance and carrying NR3C1 mutations exhibit adrenal hyperplasia, this clinical feature may not be a prerequisite to develop hypertension. Nevertheless, not all patients with adrenal hyperplasia were analysed for GR mutations. Environmental factors as varying daily salt intake might play an additional important role here. In the contrary, there is a strong correlation between NR3C1 gene mutations, salt-sensitive hypertension and high salt intake. Increased adrenocorticotropic steroids are known to promote renal sodium reabsorption and implicate both GR and mineralocorticoid receptor (MR) pathways that contribute to the pathogenesis of hypertension. We thus cannot exclude compensatory mechanisms and/or glucocorticoid-mediated mineralocorticoid activation. To dissect the underlying mechanisms, animal models are required that can be challenged and, hence, the development of hypertension can be followed over time. The use of inhibitors as sEH enzyme inhibitors in models with acute kidney injury (AKI), diabetic nephrology (DN), chronic kidney diseases (CKD), hypertension and other renal dysfunctions may present a potential therapy although this needs further experimental studies. More complete and standardized phenotyping of human subjects as well as adapted animal models will further help to dissect the underlying molecular mechanisms of adrenal gland hyperplasia and salt-sensitive hypertension.
References
Al Argan R, Saskin A, Yang JW, D’Agostino MD, Rivera J (2018) Glucocorticoid resistance syndrome caused by a novel NR3C1 point mutation. Endocr J 65:1139–1146. https://doi.org/10.1507/endocrj.EJ18-0135
Asser L, Hescot S, Viengchareun S, Delemer B, Trabado S, Lombès M (2014) Autocrine positive regulatory feedback of glucocorticoid secretion: glucocorticoid receptor directly impacts H295R human adrenocortical cell function. Mol Cell Endocrinol 395:1–9. https://doi.org/10.1016/j.mce.2014.07.012
Berger S, Bleich M, Schmid W, Greger R, Schütz G (2000) Mineralocorticoid receptor knockout mice: lessons on Na+ metabolism. Kidney Int 57:1295–1298. https://doi.org/10.1046/j.1523-1755.2000.00965.x
Boix J, Bigas J, Sevilla LM, Iacobone M, Citton M, Torresan F, Caroccia B, Rossi GP, Pérez P (2017) Primary aldosteronism patients show skin alterations and abnormal activation of glucocorticoid receptor in keratinocytes. Sci Rep 7:15806. https://doi.org/10.1038/s41598-017-16216-5
Bouligand J, Delemer B, Hecart A-C, Meduri G, Viengchareun S, Amazit L, Trabado S, Fève B, Guiochon-Mantel A, Young J, Lombès M (2010) Familial glucocorticoid receptor haploinsufficiency by non-sense mediated mRNA decay, adrenal hyperplasia and apparent mineralocorticoid excess. PLoS ONE 5:e13563. https://doi.org/10.1371/journal.pone.0013563
Boulkroun S, Fernandes-Rosa FL, Zennaro M-C (2020) Old and new genes in primary aldosteronism. Best Pract Res Clin Endocrinol Metab 34:101375. https://doi.org/10.1016/j.beem.2020.101375
Cannavò S, Benvenga S, Messina E, Moleti M, Ferraù F (2019) Comment to glucocorticoid resistance syndrome caused by a novel NR3C1 point mutation’ by Al Argan et al. Endocr J 66:657. https://doi.org/10.1507/endocrj.EJ19-0122
Canonica J, Frateschi S, Boscardin E, Ebering A, Sergi C, Jäger Y, Peyrollaz T, Mérillat A-M, Maillard M, Klusonova P, Odermatt A, Koesters R, Debonneville A, Staub O, Verouti SN, Hummler E (2019) Lack of renal tubular glucocorticoid receptor decreases the thiazide-sensitive Na+/Cl– cotransporter NCC and transiently affects sodium handling. Front Physiol 10:989. https://doi.org/10.3389/fphys.2019.00989
Charmandari E, Ichijo T, Jubiz W, Baid S, Zachman K, Chrousos GP, Kino T (2008) A novel point mutation in the amino terminal domain of the human glucocorticoid receptor (hGR) gene enhancing hGR-mediated gene expression. J Clin Endocrinol Metab 93:4963–4968. https://doi.org/10.1210/jc.2008-0892
Charmandari E, Kino T, Ichijo T, Jubiz W, Mejia L, Zachman K, Chrousos GP (2007) A novel point mutation in helix 11 of the ligand-binding domain of the human glucocorticoid receptor gene causing generalized glucocorticoid resistance. J Clin Endocrinol Metab 92:3986–3990. https://doi.org/10.1210/jc.2006-2830
Charmandari E, Kino T, Souvatzoglou E, Vottero A, Bhattacharyya N, Chrousos GP (2004) Natural glucocorticoid receptor mutants causing generalized glucocorticoid resistance: molecular genotype, genetic transmission, and clinical phenotype. J Clin Endocrinol Metab 89:1939–1949. https://doi.org/10.1210/jc.2003-030450
Charmandari E, Raji A, Kino T, Ichijo T, Tiulpakov A, Zachman K, Chrousos GP (2005) A novel point mutation in the ligand-binding domain (LBD) of the human glucocorticoid receptor (hGR) causing generalized glucocorticoid resistance: the importance of the C terminus of hGR LBD in conferring transactivational activity. J Clin Endocrinol Metab 90:3696–3705. https://doi.org/10.1210/jc.2004-1920
Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, Lolis E, Wisgerhof MV, Geller DS, Mane S, Hellman P, Westin G, Åkerström G, Wang W, Carling T, Lifton RP (2011) K + channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331:768–772. https://doi.org/10.1126/science.1198785
Chrousos GP (1993) Syndromes of glucocorticoid resistance. Ann Intern Med 119:1113. https://doi.org/10.7326/0003-4819-119-11-199312010-00009
Chrousos GP, Vingerhoeds A, Brandon D, Eil C, Pugeat M, DeVroede M, Loriaux DL, Lipsett MB (1982) Primary cortisol resistance in man. A glucocorticoid receptor-mediated disease. J Clin Invest 69:1261–1269. https://doi.org/10.1172/JCI110565
Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, Fantuzzi G, Hummler E, Unsicker K, Schütz G (1995) Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev 9:1608–1621. https://doi.org/10.1101/gad.9.13.1608
Donner KM, Hiltunen TP, Jänne OA, Sane T, Kontula K (2013) Generalized glucocorticoid resistance caused by a novel two-nucleotide deletion in the hormone-binding domain of the glucocorticoid receptor gene NR3C1. Eur J Endocrinol 168:K9–K18. https://doi.org/10.1530/EJE-12-0532
Ellison DH, Brooks VL (2011) Renal nerves, WNK4, glucocorticoids, and salt transport. Cell Metab 13:619–620. https://doi.org/10.1016/j.cmet.2011.05.007
Gerards J, Heinrich DA, Adolf C, Meisinger C, Rathmann W, Sturm L, Nirschl N, Bidlingmaier M, Beuschlein F, Thorand B, Peters A, Reincke M, Roden M, Quinkler M (2019) Impaired glucose metabolism in primary aldosteronism is associated with cortisol cosecretion. J Clin Endocrinol Metab 104:3192–3202. https://doi.org/10.1210/jc.2019-00299
Gjerstad JK, Lightman SL, Spiga F (2018) Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 21:403–416. https://doi.org/10.1080/10253890.2018.1470238
Goodwin JE, Zhang J, Gonzalez D, Albinsson S, Geller DS (2011) Knockout of the vascular endothelial glucocorticoid receptor abrogates dexamethasone-induced hypertension. J Hypertens 29:1347–1356. https://doi.org/10.1097/HJH.0b013e328347da54
Goodwin JE, Zhang J, Velazquez H, Geller DS (2010) The glucocorticoid receptor in the distal nephron is not necessary for the development or maintenance of dexamethasone-induced hypertension. Biochem Biophys Res Commun 394:266–271. https://doi.org/10.1016/j.bbrc.2010.02.123
Govindan MV, Pothier F, Leclerc S, Palaniswami R, Xie B (1991) Human glucocorticoid receptor gene promotor—homologous down regulation. J Steroid Biochem Mol Biol 40:317–323. https://doi.org/10.1016/0960-0760(91)90197-D
Zong J, Ashraf J, Thompson EB (1990) The promoter and first, untranslated exon of the human glucocorticoid receptor element sites. Mol Cell Biol 10:5580–5585. https://doi.org/10.1128/mcb.10.10.5580-5585.1990
Inoue K, Nishikawa T (2018) The spectrum of subclinical primary aldosteronism and incident hypertension. Ann Intern Med 168:754. https://doi.org/10.7326/L18-0024
Ivy JR, Evans LC, Moorhouse R, Richardson RV, Al-Dujaili EAS, Flatman PW, Kenyon CJ, Chapman KE, Bailey MA (2018) Renal and blood pressure response to a high-salt diet in mice with reduced global expression of the glucocorticoid receptor. Front Physiol 9:848. https://doi.org/10.3389/fphys.2018.00848
Ivy JR, Jones NK, Costello HM, Mansley MK, Peltz TS, Flatman PW, Bailey MA (2019) Glucocorticoid receptor activation stimulates the sodium-chloride cotransporter and influences the diurnal rhythm of its phosphorylation. Am J Physiol-Ren Physiol 317:F1536–F1548. https://doi.org/10.1152/ajprenal.00372.2019
Ivy JR, Oosthuyzen W, Peltz TS, Howarth AR, Hunter RW, Dhaun N, Al-Dujaili EAS, Webb DJ, Dear JW, Flatman PW, Bailey MA (2016) Glucocorticoids induce nondipping blood pressure by activating the thiazide-sensitive cotransporter. Hypertension 67:1029–1037. https://doi.org/10.1161/HYPERTENSIONAHA.115.06977
Kamilaris CDC, Hannah-Shmouni F, Stratakis CA (2020) Adrenocortical tumorigenesis: lessons from genetics. Best Pract Res Clin Endocrinol Metab 34:101428. https://doi.org/10.1016/j.beem.2020.101428
Karl M, Lamberts SW, Detera-Wadleigh SD, Encio IJ, Stratakis CA, Hurley DM, Accili D, Chrousos GP (1993) Familial glucocorticoid resistance caused by a splice site deletion in the human glucocorticoid receptor gene. J Clin Endocrinol Metab 76:683–689. https://doi.org/10.1210/jcem.76.3.8445027
Karl M, Lamberts SW, Koper JW, Katz DA, Huizenga NE, Kino T, Haddad BR, Hughes MR, Chrousos GP (1996) Cushing’s disease preceded by generalized glucocorticoid resistance: clinical consequences of a novel, dominant-negative glucocorticoid receptor mutation. Proc Assoc Am Physicians 108:296–307
Kino T, Chrousos GP (2011) Acetylation-mediated epigenetic regulation of glucocorticoid receptor activity: circadian rhythm-associated alterations of glucocorticoid actions in target tissues. Mol Cell Endocrinol 336:23–30. https://doi.org/10.1016/j.mce.2010.12.001
Kretz O, Reichardt HM, Schütz G, Bock R (1999) Corticotropin-releasing hormone expression is the major target for glucocorticoid feedback-control at the hypothalamic level. Brain Res 818:488–491. https://doi.org/10.1016/S0006-8993(98)01277-3
Kumar R, Thompson EB (2005) Gene regulation by the glucocorticoid receptor: structure: function relationship. J Steroid Biochem Mol Biol 94:383–394. https://doi.org/10.1016/j.jsbmb.2004.12.046
Lin L, Wu X, Hou Y, Zheng F, Xu R (2019) A novel mutation in the glucocorticoid receptor gene causing resistant hypertension. Am J Hypertens 32:1126–1128. https://doi.org/10.1093/ajh/hpz137
Ma L, Tan X, Li J, Long Y, Xiao Z, De J, Ren Y, Tian H, Chen T, a. o. (2020) A novel glucocorticoid receptor mutation in primary generalized glucocorticoid resistance disease. Endocr Pract 26:651–659. https://doi.org/10.4158/EP-2019-0475
McMahon SK, Pretorius CJ, Ungerer JPJ, Salmon NJ, Conwell LS, Pearen MA, Batch JA (2010) Neonatal complete generalized glucocorticoid resistance and growth hormone deficiency caused by a novel homozygous mutation in helix 12 of the ligand binding domain of the glucocorticoid receptor gene (NR3C1). J Clin Endocrinol Metab 95:297–302. https://doi.org/10.1210/jc.2009-1003
Mendonca BB, Leite MV, de Castro M, Kino T, Elias LLK, Bachega TAS, Arnhold IJP, Chrousos GP, Latronico AC (2002) Female pseudohermaphroditism caused by a novel homozygous missense mutation of the GR gene. J Clin Endocrinol Metab 87:1805–1809. https://doi.org/10.1210/jcem.87.4.8379
Michailidou Z, Carter RN, Marshall E, Sutherland HG, Brownstein DG, Owen E, Cockett K, Kelly V, Ramage L, Al-Dujaili EAS, Ross M, Maraki I, Newton K, Holmes MC, Seckl JR, Morton NM, Kenyon CJ, Chapman KE (2008) Glucocorticoid receptor haploinsufficiency causes hypertension and attenuates hypothalamic-pituitary-adrenal axis and blood pressure adaptions to high-fat diet. FASEB J 22:3896–3907. https://doi.org/10.1096/fj.08-111914
Mills KT, Stefanescu A, He J (2020) The global epidemiology of hypertension. Nat Rev Nephrol 16:223–237. https://doi.org/10.1038/s41581-019-0244-2
Molnár Á, Patócs A, Likó I, Nyírő G, Rácz K, Tóth M, Sármán B (2018) An unexpected, mild phenotype of glucocorticoid resistance associated with glucocorticoid receptor gene mutation case report and review of the literature. BMC Med Genet 19:37. https://doi.org/10.1186/s12881-018-0552-6
Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F, Marumo T, Yatomi Y, Geller DS, Tanaka H, Fujita T (2011) Epigenetic modulation of the renal β-adrenergic–WNK4 pathway in salt-sensitive hypertension. Nat Med 17:573–580. https://doi.org/10.1038/nm.2337
Nader N, Bachrach BE, Hurt DE, Gajula S, Pittman A, Lescher R, Kino T (2010) A novel point mutation in helix 10 of the human glucocorticoid receptor causes generalized glucocorticoid resistance by disrupting the structure of the ligand-binding domain. J Clin Endocrinol Metab 95:2281–2285. https://doi.org/10.1210/jc.2009-2463
Nader N, Chrousos GP, Kino T (2009) Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: potential physiological implications. FASEB J 23:1572–1583. https://doi.org/10.1096/fj.08-117697
Nawata H, Sekiya K, Higuchi K, Kato K-I, Ibayashi H (1987) Decreased deoxyribonucleic acid binding of glucocorticoid-receptor complex in cultured skin fibroblasts from a patient with the glucocorticoid resistance syndrome *. J Clin Endocrinol Metab 65:219–226. https://doi.org/10.1210/jcem-65-2-219
Nicolaides NC, Chrousos GP (2018) Bilateral adrenal incidentalomas and NR3C1 mutations causing glucocorticoid resistance: is there an association? Eur J Endocrinol 179:C1–C4. https://doi.org/10.1530/EJE-18-0471
Nicolaides NC, Geer EB, Vlachakis D, Roberts ML, Psarra A-MG, Moutsatsou P, Sertedaki A, Kossida S, Charmandari E (2015) A novel mutation of the hGR gene causing Chrousos syndrome. Eur J Clin Invest 45:782–791. https://doi.org/10.1111/eci.12470
Nicolaides NC, Kino T, Chrousos G, Charmandari E (2000) Primary generalized glucocorticoid resistance or chrousos syndrome. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrère B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP (eds) Endotext. MDText.com, Inc., South Dartmouth (MA)
Nicolaides NC, Roberts ML, Kino T, Braatvedt G, Hurt DE, Katsantoni E, Sertedaki A, Chrousos GP, Charmandari E (2014) A novel point mutation of the human glucocorticoid receptor gene causes primary generalized glucocorticoid resistance through impaired interaction with the LXXLL motif of the p160 coactivators: dissociation of the transactivating and transreppressive activities. J Clin Endocrinol Metab 99:E902–E907. https://doi.org/10.1210/jc.2013-3005
Oakley RH, Cidlowski JA (2011) Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem 286:3177–3184. https://doi.org/10.1074/jbc.R110.179325
Otulakowski G, Rafii B, Bremner HR, O’Brodovich H (1999) Structure and hormone responsiveness of the gene encoding the alpha-subunit of the rat amiloride-sensitive epithelial sodium channel. Am J Respir Cell Mol Biol 20:1028–1040. https://doi.org/10.1165/ajrcmb.20.5.3382
Paragliola RM, Costella A, Corsello A, Urbani A, Concolino P (2020) A novel pathogenic variant in the N-terminal domain of the glucocorticoid receptor, causing glucocorticoid resistance. Mol Diagn Ther 24:473–485. https://doi.org/10.1007/s40291-020-00480-9
Peng M, He J, Xue Y, Yang X, Liu S, Gong Z (2021) Role of hypertension on the severity of COVID-19: a review. J Cardiovasc Pharmacol 78:e648–e655. https://doi.org/10.1097/FJC.0000000000001116
Pepin M-C, Pothier F, Barden N (1992) Impaired type II glucocorticoid-receptor function in mice bearing antisense RNA transgene. Nature 355:725–728. https://doi.org/10.1038/355725a0
Ponce de León V, Mérillat A-M, Tesson L, Anegón I, Hummler E (2014) Generation of TALEN-mediated GRdim knock-in rats by homologous recombination. PLoS ONE 9:e88146. https://doi.org/10.1371/journal.pone.0088146
Raef H, Baitei EY, Zou M, Shi Y (2008) Genotype–phenotype correlation in a family with primary cortisol resistance: possible modulating effect of the ER22/23EK polymorphism. Eur J Endocrinol 158:577–582. https://doi.org/10.1530/EJE-07-0629
Raff H, Sharma ST, Nieman LK (2014) Physiological basis for the etiology, diagnosis, and treatment of adrenal disorders: cushing’s syndrome, adrenal insufficiency, and congenital adrenal hyperplasia. In: Terjung R (ed) Comprehensive Physiology, 1st edn. Wiley, pp 739–769
Reichardt HM, Schütz G (1996) Feedback control of glucocorticoid production is established during fetal development. Mol Med Camb Mass 2:735–744
Roberts ML, Kino T, Nicolaides NC, Hurt DE, Katsantoni E, Sertedaki A, Komianou F, Kassiou K, Chrousos GP, Charmandari E (2013) A novel point mutation in the DNA-binding domain (DBD) of the human glucocorticoid receptor causes primary generalized glucocorticoid resistance by disrupting the hydrophobic structure of its DBD. J Clin Endocrinol Metab 98:E790–E795. https://doi.org/10.1210/jc.2012-3549
Ruiz M, Lind U, Gåfvels M, Eggertsen G, Carlstedt-Duke J, Nilsson L, Holtmann M, Stierna P, Wikström A-C, Werner S (2001) Characterization of two novel mutations in the glucocorticoid receptor gene in patients with primary cortisol resistance: Two novel mutations glucocorticoid receptor gene mutations. Clin Endocrinol (Oxf) 55:363–371. https://doi.org/10.1046/j.1365-2265.2001.01323.x
Schoonheim PJ, Chatzopoulou A, Schaaf MJM (2010) The zebrafish as an in vivo model system for glucocorticoid resistance. Steroids 75:918–925. https://doi.org/10.1016/j.steroids.2010.05.010
Seccia TM, Caroccia B, Gomez-Sanchez EP, Vanderriele P-E, Gomez-Sanchez CE (1979) Rossi GP (2017) Review of markers of zona glomerulosa and aldosterone-producing adenoma cells. Hypertens Dallas Tex 70:867–874. https://doi.org/10.1161/HYPERTENSIONAHA.117.09991
Straub RH, Cutolo M (2016) Glucocorticoids and chronic inflammation. Rheumatol 55:ii6–ii14. https://doi.org/10.1093/rheumatology/kew348
Tatsi C, Xekouki P, Nioti O, Bachrach B, Belyavskaya E, Lyssikatos C, Stratakis CA (2019) A novel mutation in the glucocorticoid receptor gene as a cause of severe glucocorticoid resistance complicated by hypertensive encephalopathy. J Hypertens 37:1475–1481. https://doi.org/10.1097/HJH.0000000000002048
Trebble P, Matthews L, Blaikley J, Wayte AWO, Black GCM, Wilton A, Ray DW (2010) Familial glucocorticoid resistance caused by a novel frameshift glucocorticoid receptor mutation. J Clin Endocrinol Metab 95:E490–E499. https://doi.org/10.1210/jc.2010-0705
Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23:99–103. https://doi.org/10.1038/12703
Vaczlavik A, Vaduva P, Bouys L, Neou M, Septier A, Heurtier V, Libe R, Kroiss M, Borson-Chazot F, Chanson P, Vanthyghem M-C, Tabarin A, Assie G, Stratakis C, Fragoso M, Ragazzon B, Bertherat J (2019) Genetic predisposition to primary bilateral macronodular adrenal hyperplasia (PBMAH): next generation sequencing ARMC5, NR3C1 (glucocorticoid receptor) and PDE11A4 (phosphodiesterase 11) in 389 patients. Endocr Abstr. https://doi.org/10.1530/endoabs.63.OC10.3
Vanderriele P-E, Wang Q, Mérillat A-M, Ino F, Aeschlimann G, Ehret X, Ancin Del Olmo D, Ponce de León V, Scholl UI, Winter DV, Odermatt A, Hummler E, Verouti SN (2021) Salt-sensitive hypertension in GR+/− rats is accompanied with dysregulation in adrenal soluble epoxide hydrolase and polyunsaturated fatty acid pathways. Int J Mol Sci 22:13218. https://doi.org/10.3390/ijms222413218
Vandevyver S, Dejager L, Libert C (2014) Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr Rev 35:671–693. https://doi.org/10.1210/er.2014-1010
Velayos T, Grau G, Rica I, Pérez-Nanclares G, Gaztambide S (2016) Glucocorticoid resistance syndrome caused by two novel mutations in the NR3C1 gene. Endocrinol Nutr 63:369–371. https://doi.org/10.1016/j.endonu.2016.03.010
Vitellius G, Delemer B, Caron P, Chabre O, Bouligand J, Pussard E, Trabado S, Lombes M (2019) Impaired 11β-hydroxysteroid dehydrogenase type 2 in glucocorticoid-resistant patients. J Clin Endocrinol Metab 104:5205–5216. https://doi.org/10.1210/jc.2019-00800
Vitellius G, Fagart J, Delemer B, Amazit L, Ramos N, Bouligand J, Le Billan F, Castinetti F, Guiochon-Mantel A, Trabado S, Lombès M (2016) Three novel heterozygous point mutations of NR3C1 causing glucocorticoid resistance: human mutation. Hum Mutat 37:794–803. https://doi.org/10.1002/humu.23008
Vitellius G, Lombes M (2020) Genetics in endocrinology: glucocorticoid resistance syndrome. Eur J Endocrinol 182:R15–R27. https://doi.org/10.1530/EJE-19-0811
Vitellius G, Trabado S, Hoeffel C, Bouligand J, Bennet A, Castinetti F, Decoudier B, Guiochon-Mantel A, Lombes M, Delemer B, Amiot-Chapoutot F, Ancelle D, Bertoin F, Brue T, Caron P, Borson-Chazot F, Christin-Maitre S, Chabre O, Dessailloud R, Estour B, Grulet H, Illouz F, Jeandidier N, Kerlan V, Klein M, Penfornis A, Pierre P, Tabarin A, Touraine P, Vantyghem MC, Young J (2018) Significant prevalence of NR3C1 mutations in incidentally discovered bilateral adrenal hyperplasia: results of the French MUTA-GR Study. Eur J Endocrinol 178:411–423. https://doi.org/10.1530/EJE-17-1071
Vottero A, Kino T, Combe H, Lecomte P, Chrousos GP (2002) A novel, C-terminal dominant negative mutation of the GR causes familial glucocorticoid resistance through abnormal interactions with p160 steroid receptor coactivators. J Clin Endocrinol Metab 87:2658–2667. https://doi.org/10.1210/jcem.87.6.8520
Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR (2017) Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol 18:159–174. https://doi.org/10.1038/nrm.2016.152
Zennaro M-C, Boulkroun S, Fernandes-Rosa FL (2020) Pathogenesis and treatment of primary aldosteronism. Nat Rev Endocrinol 16:578–589. https://doi.org/10.1038/s41574-020-0382-4
Zhu H, Dai Y, Wang O, Li M, Lu L, Zhao W, Xing X, Pan H, Li N, Gong F (2011) Generalized glucocorticoid resistance accompanied with an adrenocortical adenoma and caused by a novel point mutation of human glucocorticoid receptor gene. Chin Med J (Engl) 124:551–555
Funding
Open access funding provided by University of Lausanne This work was supported by the Swiss National Foundation Grants FNRS 31003A-182478/1 and 31003A-163347 (to E.H.) and the National Center of Competence in Research (NCCR) “Kidney.CH, kidney control of homeostasis” Lausanne, Switzerland (to E.H.).
Author information
Authors and Affiliations
Contributions
All the authors contributed to the study conception and design. Edith Hummler had the idea for the article, Sophia Verouti and Paul-Emmanuel Vanderriele performed the literature search and data analysis and wrote the manuscript and Edith Hummler critically revised the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue on Kidney Control of Homeostasis in Pflügers Archiv—European Journal of Physiology.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verouti, S., Hummler, E. & Vanderriele, PE. Role of glucocorticoid receptor mutations in hypertension and adrenal gland hyperplasia. Pflugers Arch - Eur J Physiol 474, 829–840 (2022). https://doi.org/10.1007/s00424-022-02715-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-022-02715-6