
Abstract
Vasopressin (arginine vasopressin, AVP) plays a crucial role in maintaining body fluid homeostasis. Excessive release of vasopressin can lead to hyponatremia. Changes in cerebral circulation during vasopressin-induced chronic hyponatremia are not elucidated. The present study has been designed to investigate the effect of chronic vasopressin-induced hyponatremia on the regulation of the tone of the middle cerebral artery (MCA) of the rat. Chronic hyponatremia was induced in vivo with the help of vasopressin, released continuously from subcutaneously implanted ALZET mini-osmotic pumps, and a liquid diet. After 3.5 days of chronic hyponatremia, the plasma Na+ concentration decreased to 119 ± 3 mM. MCAs were isolated and placed in a MOPS-buffered saline solution containing 121 mM Na+. Chronic hyponatremia did not affect the response of the MCA to increased intravascular pressure, to the administration of acetylcholine (ACh) and nitric oxide (NO) donor (SNAP, S-nitroso-N-acetyl-DL-penicillamine), and to increased K+ concentration, but impaired the response of the MCA to increased extravascular H+ concentration. Disturbed response of the MCA to acidosis was associated neither with the impairment of KATP channels nor with the activation of vasopressin V1 receptor. Correction of hyponatremia did not restore the response of the MCA to acidosis. These results indicate that cerebral blood vessels do not fully adapt to prolonged vasopressin-induced hyponatremia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although hyposmotic hyponatremia is a relatively common clinical water-electrolyte balance disorder which may be asymptomatic, it may result in serious complications in patients with central nervous system (CNS) diseases [12, 27]. The first complication of hyponatremia is connected with osmotically driven brain edema [12] and the second complication is the dehydration of the brain as a consequence of too rapid correction of prolonged hyponatremia [29].
Hyposmotic brain edema triggers an adaptive mechanism, referred to as “regulatory volume decrease” (RVD). The initial step of this mechanism consists of the rapid extrusion of K+ and Cl− ions, and the obligatory efflux of water which is detectable within 3–24 h of the onset of hyponatremia [21]. The second step involves the slow loss of organic solutes, such as taurine, glycine, glutamine, myo-inositol, sorbitol, and creatine [24]. The decrease in organic osmolytes occurs within 2 days of hyponatremia [36].
The brain symptoms of hyponatremia depend on how fast the decrease in plasma sodium concentration occurs. The rapid onset of the disorder (within less than 48 h), referred to as acute hyponatremia, is associated with severe brain edema and a greater risk of death. When the onset of hyponatremia is slow (more than 48 h), the mechanism of RVD allows for the effective reduction of brain edema. As a result, symptoms of slowly developing chronic hyponatremia are milder or not diagnosed [7, 26].
Previous studies on the impact of chronic hyponatremia on the CNS focused on issues related to brain edema, such as analysis of brain water and electrolyte content [7, 34] or related to the development of brain demyelination after too rapid correction of hyponatremia [29].
According to the literature, chronic hyponatremia has a negative impact on the regulation of the cerebral blood vessels, despite the fully developed adaptive mechanism of RVD. Chronic hyponatremia induced by desmopressin in rabbits has been shown to impair cerebrovascular reactivity to hypercapnia and autoregulation to hypotension [23]. Furthermore, the functional impairment of cerebral microcirculation was reported in rats after 3.5 days of hyponatremia, particularly when low sodium concentration in the plasma was associated with an increased level of plasma vasopressin [16]. Vasopressin is known to play a crucial role in the development of hyponatremia in humans [7]. It acts through V2 receptors located at the distal and collecting tubules of the kidney causing increased reabsorption of water [15]. Vasopressin also has profound vascular effects acting through V1a receptors located in smooth muscle [15]. Recently, it has been shown that prolonged increase in vasopressin levels in plasma can lead to oxidative stress and upregulation of endothelin-1 (ET-1) in the cerebral blood vessels [11].
Therefore, we hypothesize that chronic hyponatremia associated with an increased plasma level of vasopressin has an adverse effect on the mechanisms regulating cerebral blood vessels, such as metabolic, myogenic, and endothelium-dependent regulation. To prove this hypothesis, the reactivity of pressurized MCAs, isolated from rat brains with chronic (3.5 days) vasopressin-induced hyponatremia, to increase of intravascular pressure, ACh, NO donor, extravascular increase of K+ and H+ ions, and extravascular increase of H+ ion concentration with KATP channels activator or specific antagonist of V1 vasopressin receptor in the background, was studied. In addition, the response of the MCA to acidosis was also studied 2 h after in vitro restoration of normonatremia.
Material and methods
Animals
Male Wistar rats (body weight 250–300 g, n = 65 animals) used in this study were supplied by the Animal House of the Mossakowski Medical Research Centre, Warsaw, Poland. All animal experiments were performed in accordance with the Law and Regulations on Animal Protection in Poland (Dz.U. 2015/266) and were approved by the Extramural First Committee for the Care and Use of Laboratory Animals for Experimental Procedures, National Medicines Institute in Warsaw.
Induction of chronic hyponatremia
Chronic hyponatremia was induced in vivo within 3.5 days using a vasopressin-filled mini-osmotic ALZET pump (Model 2002, Durect Corp., Cupertino, USA) and a rodent liquid diet (AIN-76, Bio-Serv, NJ, USA), according to Verbalis [34]. First, the rats were acclimated for 2 days to a rodent liquid diet administered in the amount of 75 ml and supplying 1.0 kcal/mL. During this time, they had free access to drinking water. Osmotic pumps filled with vasopressin were implanted subcutaneously on the neck under chloral hydrate anesthesia. The pump released vasopressin at a rate of 2.4 μg/24 h. Starting from the first day following implantation of the pump, the animals were fed the liquid diet AIN-76 containing 14% glucose in the amount of 50 mL/24 h. At this time, drinking water was withheld to prevent excessive water intake with escape from antidiuresis [35]. After 3.5 days of AVP infusion, the animals were decapitated under 5% isoflurane in 70% NO2/30% O2 anesthesia and the middle cerebral arteries were isolated as described below and blood samples were collected to measure the osmolality and concentration of Na+ in plasma.
Control rats were housed individually for the duration of the experiment with free access to drinking water and standard rat chow.
Isolated cerebral artery studies
After decapitation, the brains of the normonatremic control rats were removed from the cranium and placed in a cold (4 °C, pH = 7.4 adjusted by NaOH) physiological saline buffered with MOPS (3-(N-morpholino)propanesulfonic acid) (MOPS-PSS) containing 3.0 mM MOPS, 145.0 mM NaCl, 3.0 mM KCl, 2.5 mM CaCl2, 1.5 mM MgSO4, 1.21 mM NaH2PO4, 0.02 mM EDTA, 2.0 mM sodium pyruvate, 5.0 mM glucose, and 1% dialyzed bovine serum albumin (BSA). The brains of the rats with chronic hyponatremia were placed in a buffer identical in composition to the one described above, except for the NaCl concentration which was reduced to 121 mM. The osmolality of the solutions was 300 and 250 mOsm/kg H2O, respectively. Isolation and cannulation of the MCAs has been described in detail elsewhere [4]. In brief, after dissection from the brain, the MCAs were transferred to the organ chamber filled with cold MOPS buffer with 1% BSA, containing either 145 or 121 mM Na+. The vessels were cannulated and fixed on glass micropipettes. The organ chamber was placed on the stage of the inverted microscope (CKX41, Olympus, Germany) equipped with a video camera and a monitor for analysis of internal diameters of the arteries. The vessels were pressurized to 80 mmHg intravascular pressure and perfused with MOPS containing 1% BSA and 145 or 121 mM Na+ to maintain normonatremia or hyponatremia, respectively. Extraluminal fluid was replaced with MOPS without BSA, containing appropriate concentration of Na+, slowly heated to 37 °C and exchanged at a rate of 20 mL/min.
Under such conditions, the vessels were allowed to equilibrate for 1 h to develop a myogenic tone, i.e., a contraction by about 30–40% of the diameter measured directly after pressurizing. Smooth muscle cell function was tested by increasing K+ ion concentration from 3 to 20 mM in an extraluminal bath.
The responses of the MCAs to increasing intravascular pressure, NO-dependent ACh, and NO donor SNAP, increased concentration of H+ and K+ ions, increased H+ ion concentration with KATP channels activator or with antagonist of V1 vasopressin receptor in the background were studied in separate arteries in the control and in the chronic hyponatremia group. In addition, the response of the MCA to increased H+ ion concentration was tested 2 h after the correction of chronic hyponatremia (i.e., return to the solution with Na+ concentration of 145 mM and osmolality of 300 mOsm/kg H2O).
In order to study the myogenic response of the MCA (constriction of the artery in response to increasing intravascular pressure), the intraluminal pressure was slowly lowered from 80 to 20 mmHg and then slowly raised from 40 to 100 mmHg, in 20 mmHg steps. The pressure was maintained for 10 min at each step before the diameter was measured. Next, the intravascular pressure was set again at 80 mmHg and the artery was allowed to equilibrate for 15 min in a calcium-free (1 mM EGTA) MOPS-buffered saline solution. The pressure steps were repeated according to the paradigm used prior to EGTA administration. This enabled the calculation of the diameter of MCA as a percentage of maximal dilation at each pressure step according to the formula DCa/D0Ca × 100, where DCa and D0Ca are the diameters measured in the presence and absence of Ca2+, respectively.
ACh was administered intraluminally in the following concentrations: 10−6 M, 10−5 M, and 10−4 M, and the diameter of the MCA was measured 15 min later. Only one concentration was studied in each experiment in order to avoid tachyphylaxis.
NO donor SNAP (10−5 M) was administered to the extravascular buffer, and the response of the MCA to this drug was assessed after 15 min.
Hyperkalemia was induced for 15 min by the increase of K+ ion concentration in the extravascular fluid from 3 to 20 mM.
Acidosis was induced for 15 min by adding 2.5% HCl to the extravascular buffer which resulted in a decrease of pH from 7.4 to 7.0. The effects of the activator of KATP channels (pinacidil, 10−5 M) or the antagonist of V1 vasopressin receptor ([Deamino-Pen1, O-Me-Tyr2, Arg8]-Vasopressin, 10−8 M) and increased H+ ion concentration with these compounds in the background were measured after 15 min of their extravascular administration.
Changes in the diameter of the MCAs in response to the tested compounds were calculated as a percentage of the diameter measured before the administration of the compound.
All reagents used in these experiments were purchased from Sigma-Aldrich.
Plasma analyses
During decapitation, blood was collected into heparinized Eppendorf tubes. Blood samples were centrifuged within 10 min (3000×g). Na+ concentration in plasma was measured by flame photometry (Jenway PFP7, Essex, UK). The osmolality of plasma was estimated by freezing-point depression using a semi-micro osmometer (Osmomat 030, Gonotec, Germany).
Statistics
Statistical analyses were performed using Statistica 10 software. The results are expressed as means ± S.E.M. The data were analyzed using one-way ANOVA followed by Tukey’s multiple comparison tests, or paired or unpaired Student’s t test, as appropriate. Differences with p < 0.05 were considered statistically significant.
Results
After 3.5 days of chronic hyponatremia, the body weight of the animals did not change. Not a single animal died due to chronic hyponatremia. Plasma Na+ concentration of hyponatremic rats decreased to 119 ± 3 mM (p < 0.001, n = 30), and plasma osmolality decreased to 252 ± 2 mOsm/kg H2O (p < 0.05, n = 6).
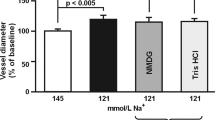
The diameter of the MCAs isolated from rats with chronic hyponatremia decreased from 255 ± 3 μm to 173 ± 4 μm (p < 0.001, n = 31) after 1-h equilibration on 80 mmHg intravascular pressure, in a buffer containing 121 mM Na+. This response was comparable to that observed in control normonatremic vessels. Extravascular increase in K+ concentration from 3 to 20 mM led to a comparable statistically significant dilation of the MCA in normonatremia (39 ± 3%, p < 0.001) and in hyponatremia (38 ± 5%, p < 0.001), (Fig. 1). Correction of hyponatremia (return to the buffer containing 145 mM Na+, 300 mOsm/kg H2O) caused an increase in MCA diameter by 19 ± 6%, (from 167 ± 11 μm to 197 ± 11 μm, p < 0.05, n = 5).

Response of the MCA to the extravascular increase in K+ concentration from 3 to 20 mM in normonatremia (n = 31) and in vasopressin-induced chronic hyponatremia (n = 35). MCA diameter is expressed as a percentage of baseline diameter. Data are expressed as means ± S.E.M
Pressure-diameter relationship of MCA
Increases in intravascular pressure from 40 to 100 mmHg resulted in a comparable constriction of isolated rat MCA regardless of the level of natremia. In the absence of Ca2+ in MOPS-PSS (1 mM EGTA), step elevations in pressure elicited increases in the diameter of the MCA in normonatremic and hyponatremic vessels. The ratio of the diameters recorded in the presence and absence of Ca2+ in MOPS-PSS indicates that chronic hyponatremia does not affect the myogenic response of MCA (Fig. 2).

MCA pressure-diameter relationships in normonatremia (n = 5) and in vasopressin-induced chronic hyponatremia (n = 5). Diameter of the MCA is expressed as a percentage of the maximal dilation measured at each pressure in Ca2+-free solution (EGTA 1 mM). Data are expressed as means ± S.E.M
Response of MCA to ACh and SNAP
Intravascular administration of endothelium-dependent vasodilator ACh (10−6 M, 10−5 M, 10−4 M) elicited a statistically significant and comparable relaxation of the MCA in normonatremia and in hyponatremia (Fig. 3). SNAP (10−5 M), an exogenous NO donor increased the MCA diameter by 45 ± 4%, p < 0.001 in normonatremia and by 32 ± 4%, p < 0.05 in hyponatremia. There were no significant differences between the groups (Fig. 4).

Response of the MCA to intravascular administration of endothelium-dependent vasodilator ACh (10−6 M, 10−5 M, 10−4 M) in normonatremia (n = 14) and in vasopressin-induced chronic hyponatremia (n = 14). Diameter of the MCA is expressed as a percentage of baseline diameter. Only one concentration was studied in each experiment. Data are expressed as means ± S.E.M

Response of the MCA to extravascular administration of NO donor SNAP (10−5 M) in normonatremia (n = 6), and in vasopressin-induced chronic hyponatremia (n = 6). Diameter of the MCA is expressed as a percentage of baseline diameter. Data are expressed as means ± S.E.M
Response of MCA to acidosis
A reduction of extravascular pH from 7.4 to 7.0 elicited a statistically significant increase in the MCA diameter by 19 ± 2% (p < 0.001) in normonatremia. In contrast, in hyponatremia, a reduction of pH to 7.0 did not cause statistically significant dilation of MCA (3 ± 2%, NS), (Fig. 5). Correction of hyponatremia (return to the buffer with normal Na+ concentration and osmolality) did not improve the response of the MCA to acidosis (4 ± 2%, NS).

Response of the MCA to the reduction of pH from 7.4 to 7.0 in normonatremia (n = 14) and in vasopressin-induced chronic hyponatremia (n = 14). Neither the KATP channel activator, pinacidil (10−5 M), nor the antagonist of V1 vasopressin receptor, [Deamino-Pen1, O-Me-Tyr2, Arg8]-Vasopressin (10−8 M), was able to restore the response to acidosis in hyponatremia (*p < 0.05, between the groups). Diameter of the MCA is expressed as a percentage of baseline diameter. Data are expressed as means ± S.E.M
Effect of KATP channels activator on the response of MCA to acidosis
Pinacidil (10−5 M), a specific activator of the KATP channel, led to a comparable relaxation of the MCA in normonatremia and in chronic hyponatremia, by 29 ± 6%, p < 0.05 and 29 ± 4%, p < 0.05, respectively. Presence of pinacidil did not affect the response to acidosis either in normonatremia or in hyponatremia (Fig. 5). Combined application of pinacidil and acidosis caused relaxation of MCA in normonatremia by 15 ± 6%, p < 0.05, which was not significantly different from the response observed without pinacidil. The deprived response of the MCA to acidosis in hyponatremia was not improved by pinacidil, indicating that the KATP channel is not impaired in chronic vasopressin-induced hyponatremia (Fig. 5).
Effect of V1 vasopressin receptor antagonist on the response of MCA to acidosis
[Deamino-Pen1, O-Me-Tyr2, Arg8]-Vasopressin (10−8 M), a selective antagonist of V1 vasopressin receptor neither changed the basal MCA diameter nor the response of the vessel to acidosis in normonatremia. The MCA diameter increased during acidosis combined with V1 receptor antagonist by 21 ± 6%, p < 0.05, which was not significantly different from the response to acidosis observed without an antagonist. In contrast, administration of the V1 receptor antagonist in chronic hyponatremia resulted in a statistically significant dilation of the MCA by 19 ± 2%, p < 0.05. However, in an acidotic environment with V1 receptor antagonist in the background, there was no further vasorelaxation, i.e., blocking of V1 receptor did not improve the response of the MCA to acidosis in chronic hyponatremia (Fig. 5).
Discussion
The main finding of this study is that cerebral blood vessels do not fully adapt to prolonged vasopressin-induced hyponatremia. Although autoregulation, the response to hyperkalemia, and NO-dependent dilation were well-preserved, the response to acidosis was abolished. Disturbed response of the MCA to acidosis was associated neither with the impairment of KATP channels nor with the activation of vasopressin V1 receptor. In this study, prolonged hyponatremia was induced according to Verbalis [34], using mini-osmotic pumps releasing vasopressin at the rate of 2.4 μg/24 h and a rodent liquid diet formula with 14% dextrose addition. Such an infusion rate of AVP produced an increase in the vasopressin concentration in the plasma of the rats to about 17 pg/mL [34], which is a concentration comparable to that observed in patients with the SIADH [37]. Under experimental conditions, chronic hyponatremia can be induced by either vasopressin or by a peptidergic analogue of vasopressin—desmopressin (dDAVP) [34]. In our study, vasopressin was chosen due to its clinical implementation in the development of hyponatremia in patients with central nervous system disorders [27]. Vasopressin is known to affect cerebral circulation in a complex manner. It has been reported to constrict small cerebral arterioles and dilate large cerebral arteries in vitro [30]. Studies performed in vivo revealed that intracarotid administration of vasopressin elevates cerebral blood flow (CBF), and this effect is associated with an increase in cerebral oxygen consumption. This vasopressin-induced increase of CBF was mediated by V2-like receptors but not by V1 receptors [17]. Recently, Faraco et al. reported that prolonged increase in vasopressin levels in plasma cause oxidative stress and upregulation of ET-1 in the cerebral blood vessels [11].
We did not observe mortality in rats with chronic hyponatremia, which is consistent with the results published by Verbalis [34]. In contrast, some studies in hyponatremia induced by the administration of vasopressin have reported mortality rates up to 30% in male rats and up to 60% in female rats [7, 13]. However, in these quoted studies, chronic hyponatremia was induced with subcutaneous AVP at higher doses and intraperitoneal 140 mM dextrose in water was given two to five times over 3 to 4 days.
Our previous studies indicate that acute hyponatremia, which was induced in vitro without the presence of vasopressin, did not affect autoregulation, but blunted the response of the MCA to ACh, NO donor, and acidosis [4, 5]. Acute hyponatremia also decreased the sensitivity of the BKCa channels in vascular smooth muscle cells to agonists [4], which would explain the impaired response of the MCA to the abovementioned vasodilators. The results of this study do not speak in favor of the impairment of the BKCa channels in the MCA during vasopressin-induced chronic hyponatremia as the response of this vessel to ACh and NO donor is unchanged. In support of this, it has been demonstrated that vasopressin had no direct effect on the KCa channels [32]. Interestingly, there are data in the literature demonstrating that vasopressin inhibits another type of K+ channel involved in the response of the MCA to acidosis, the KATP channel. Wakatsuki et al. have shown, using the patch-clamp technique, that vasopressin directly blocked the KATP channels in smooth muscle cells isolated from the porcine coronary artery [32]. Another study performed on smooth muscle cells isolated from the mesenteric rat artery also confirmed that vasopressin produced a concentration-dependent inhibition of pinacidil-activated currents [28]. However, in the present study, activator of KATP channels did not restore the response of the MCA to acidosis in chronic hyponatremia, indicating that disturbed response to acidosis was not associated with the decreased sensitivity of these channels to their agonists. It should be stressed, however, that our previous studies [4] as well as the studies of Lindauer et al. [18] indicated that the response of the MCA to acidosis mainly depends on BKCa channels, at least, in acute hyponatremia and in physiological conditions.
Since chronic hyponatremia was induced in our study with the help of vasopressin, we decided to test the possible impact of the activation of V1 vasopressin receptors to the disturbed response of the MCA to acidosis. Although vasopressin was absent in the buffer during reactivity testing of the MCAs, the vessels were exposed to this peptide for 3.5 days, which might have left a long-term imprint on them. Indeed, antagonist of V1 vasopressin receptor evoked dilation of the MCA in ex vivo chronic hyponatremia, while it did not change vessel diameter in ex vivo normonatremia. It is difficult to interpret the result obtained in hyponatremia, although, we see two putative explanations. The first one is that during chronic hyponatremia, there is a change in the receptor-peptide interaction resulting in a slower dissociation of vasopressin from the receptor in hyponatremic buffer without vasopressin. If this is the case, in our experimental set-up in which the extravascular buffer circulates in a closed circuit, vasopressin may appear and exert vasoconstriction acting on smooth muscle V1 receptors. This explanation has to be verified in a separate study. Another possibility is that the antagonist used in this experiment is a partial agonist of V1 receptors and activates NO synthase in the endothelium resulting in the endothelium-dependent vasodilation [30]. The latter explanation is less likely, as it has been shown by Manning et al. that [Deamino-Pen1, O-Me-Tyr2, Arg8]-Vasopressin has no evident vasopressor agonism, i.e., does not activate V1 receptor [20].
The present study also showed that combined application of an antagonist of V1 vasopressin receptor and acidosis did not improve the response of the MCA to a decrease of pH in chronic hyponatremia, indicating that disturbed response of the MCA to acidosis is not associated with vasopressin. Similar results were obtained in studies by Nelson et al., who noted that cerebrovascular reactivity to hypercapnic acidosis was impaired in a model of chronic hyponatremia induced not with vasoactive vasopressin but with desmopressin [23]. Thus, Nelson’s and our present results suggest that the impairment of the response of cerebral blood vessels to hypercapnic (Nelson et al.) and normocapnic (present study) acidosis in chronic hyponatremia is related to a prolonged decrease of plasma osmolality and a decreased of sodium concentration. Another study demonstrated that vasopressin does not affect the response of the MCA to hypercapnic or normocapnic acidosis in normonatremia [25]. Thus, disturbed response of the MCA did not depend on either attenuation of the KATP channels or V1 vasopressin receptor.
In an attempt to answer the question why the MCA does not dilate in response to extracellular normocapnic acidosis in chronic hyponatremia in our study, performed in the artificial buffer with no CO2 and HCO3−, one has to consider the effect of a decrease of extracellular pH (pHo) on the intracellular pH (pHi) and the participation of pHi in vasodilation during extravascular acidosis. According to Tian et al. [31] and Aalkjaer and Poston [1], decrease of pHo results in a fall of pHi in smooth muscle cells but steady-state relaxation of the MCA of the rat depends on the decrease of pHo. Moreover, according to Tian et al., decrease of pHi results in the increase of vascular wall tension in acute state, i.e., does not cause vasodilation [31]. This observation was later confirmed on smooth muscle cells isolated from the MCA of the rat by Apkon et al. [6].
It should be stressed that most of the studies on the mechanisms of acidotic vasodilation, including these cited above, were performed under physiological conditions in which the cells defend themselves against intracellular acidification by extruding H+ ions. There are at least two mechanisms which are activated by intracellular acidosis and are responsible for the extrusion of the protons. These are voltage-gated proton channels (Hv1) and Na+/H+ exchanger [8, 9]. Both of them were shown to be localized in vascular smooth muscles [9, 19]. The operation of Na+/H+ exchanger requires physiological transmembrane Na+ concentration gradient which is disturbed in hyponatremia and might result in less effective regulation of pHi in smooth muscle cells [2]. Under such conditions, one may expect greater impact of pHi than pHo on the tension of vascular wall during normocapnic acidosis. This aspect of the regulation of acidotic vasodilation in cerebral circulation during chronic hyponatremia is interesting and requires further studies.
The correction of hyponatremia in our study (i.e., return to the solution with Na+ concentration of 145 mM and osmolality of 300 mOsm/kg H2O) did not restore the response of the MCA to acidosis, at least within 2 h following the normalization of osmolality and sodium concentration. It seems that the presence of vasopressin for 3.5 days during induction of chronic hyponatremia resulted in a selective limitation of the functional adaptation to hyponatremic conditions. Another study showed that vasopressin limited the regulation of cell volume during hyponatremia [14, 33]. It should also be stressed, that at 2 h after normalization of osmolality and natremia, the cells of the MCA wall might be still in the phase of regulatory volume increase (RVI) [10]. The reversion to normoosmolality after 3.5 days of hypoosmolality is sensed by the cells as a change from normal to hyperosmolar environment which results in osmotic shrinkage and subsequent RVI.
The restoration of natremia and osmolality to physiological values in the present study caused vasodilation of the MCA. Another study showed that correction of chronic hyponatremia increased cerebral blood flow, without changes in arterial CO2 tension and mean arterial blood pressure [3]. Although, cerebral blood flow measured in the study by Adler’s et al. [3] represents a different level of cerebral circulation than the MCA used in our experiments, an increase in the diameter of the MCA will undoubtedly result in an increase in the flow through microcirculation in the area supplied by this artery.
In conclusion, presented results demonstrate that in chronic vasopressin-induced hyponatremia, the response of the MCA to increased extracellular H+ concentration is impaired despite the preservation of other important mechanisms of adjustability such as autoregulation or endothelium-dependent regulation. As acidosis is one of the main factors regulating cerebral blood flow, impaired response of the cerebral blood vessels to an increased concentration of H+ would have an adverse effect on neurological patients with chronic hyponatremia. This is a particularly important issue as those patients usually have symptoms of hyponatremic encephalopathy with brain acidosis [22]. Further studies need to be performed to elucidate the mechanisms behind the lack of functional adaptation of the cerebral blood vessels to chronic vasopressin-induced hyponatremia.
References
Aalkjaer C, Poston L (1996) Effects of pH on vascular tension: which are the important mechanisms? J Vasc Res 33:347–359. https://doi.org/10.1159/000159163
Adler S, Simplaceanu V (1989) Effect of acute hyponatremia on rat brain pH and rat brain buffering. Am J Physiol 256:113–119. https://doi.org/10.1152/ajprenal.1989.256.1.F113
Adler S, Verbalis JG, Meyers S, Simplaceanu E, Williams DS (2000) Changes in cerebral blood flow and distribution associated with acute increases in plasma sodium and osmolality of chronic hyponatremic rats. Exp Neurol 163:63–71. https://doi.org/10.1006/exnr.2000.7376
Aleksandrowicz M, Dworakowska B, Dolowy K, Kozniewska E (2017) Restoration of the response of the middle cerebral artery of the rat to acidosis in hyposmotic hyponatremia by the opener of large-conductance calcium sensitive potassium channels (BKCa). J Cereb Blood Flow Metab 37:3219–3230. https://doi.org/10.1177/0271678X16685575
Aleksandrowicz M, Koźniewska E (2013) Disturbed regulation of the isolated middle cerebral artery in acute hyponatremia. Folia Neuropathol 51:227–234
Apkon M, Weed RA, Boron WF (1997) Motor responses of cultured rat cerebral vascular smooth muscle cells to intra- and extracellular pH changes. Am J Physiol 273:434–445. https://doi.org/10.1152/ajpheart.1997.273.1.H434
Arieff AI, Kozniewska E, Roberts TP, Vexler ZS, Ayus JC, Kucharczyk J (1995) Age, gender, and vasopressin affect survival and brain adaptation in rats with metabolic encephalopathy. Am J Physiol 268:1143–1152. https://doi.org/10.1152/ajpregu.1995.268.5.R1143
Asuaje A, Smaldini P, Martín P, Enrique N, Orlowski A, Aiello EA, Gonzalez León C, Docena G, Milesi V (2017) The inhibition of voltage-gated H+ channel (HVCN1) induces acidification of leukemic Jurkat T cells promoting cell death by apoptosis. Pflugers Arch 469:251–261. https://doi.org/10.1007/s00424-016-1928-0
Bkaily G, Jacques D (2017) Na+-H+ exchanger and proton channel in heart failure associated with Becker and Duchenne muscular dystrophies. Can J Physiol Pharmacol 95:1213–1223. https://doi.org/10.1139/cjpp-2017-0265
Cossins AR, Gibson JS (1997) Volume-sensitive transport systems and volume homeostasis in vertebrate red blood cells. J Exp Biol 200:343–352
Faraco G, Wijasa TS, Park L, Moore J, Anrather J, Iadecola C (2014) Water deprivation induces neurovascular and cognitive dysfunction through vasopressin-induced oxidative stress. J Cereb Blood Flow Metab 34:852–860. https://doi.org/10.1038/jcbfm.2014.24
Fraser CL, Arieff AI (1997) Epidemiology, pathophysiology, and management of hyponatremic encephalopathy. Am J Med 102:67–77
Fraser CL, Kucharczyk J, Arieff A, Rollin C, Sarnacki P, Norman D (1989) Sex differences result in increased morbidity from hyponatremia in female rats. Am J Physiol 256:880–885. https://doi.org/10.1152/ajpregu.1989.256.4.R880
Kanda F, Arieff AI (1994) Vasopressin inhibits calcium-coupled sodium efflux system in rat brain. Am J Physiol 266:1169–1173. https://doi.org/10.1152/ajpregu.1994.266.4.R1169
Koshimizu TA, Nakamura K, Egashira N, Hiroyama M, Nonoguchi H, Tanoue A (2012) Vasopressin V1a and V1b receptors: from molecules to physiological systems. Physiol Rev 92:1813–1864. https://doi.org/10.1152/physrev.00035.2011
Koźniewska E, Roberts TPL, Vexler ZS, Oseka M, Kucharczyk J, Arieff A (1995) Hormonal dependence of the effects of metabolic encephalopathy on cerebral perfusion and oxygen utilization in the rat. Circ Res 76:551–558
Koźniewska E, Szczepańska-Sadowska E (1990) V2-like receptors mediate cerebral blood flow increase following vasopressin administration in rats. J Cardiovasc Pharmacol 15:579–585
Lindauer U, Vogt J, Schuh-Hofer S, Dreier JP, Dirnagl U (2003) Cerebrovascular vasodilation to extraluminal acidosis occurs via combined activation of ATP-sensitive and Ca2+-activated potassium channels. J Cereb Blood Flow Metab 23:1227–1238. https://doi.org/10.1097/01.WCB.0000088764.02615.B7
Little PJ, Cragoe EJ Jr, Bobik A (1986) Na-H exchange is a major pathway for Na influx in rat vascular smooth muscle. Am J Physiol 251:707–712. https://doi.org/10.1152/ajpcell.1986.251.5.C707
Manning M, Stoev S, Bankowski K, Misicka A, Lammek B (1992) Synthesis and some pharmacological properties of potent and selective antagonists of the vasopressor (V1-receptor) response to arginine-vasopressin. J Med Chem 35:382–388
Melton JE, Patlak CS, Pettigrew KD, Cserr HF (1987) Volume regulatory loss of Na+, Cl-, and K+ from rat brain during acute hyponatremia. Am J Physiol 252:661–669. https://doi.org/10.1152/ajprenal.1987.252.4.F661
Milionis HJ, Liamis GL, Elisaf MS (2002) The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ 166:1056–1062
Nelson RJ, Perry S, Burns AC, Roberts J, Pickard JD (1991) The effects of hyponatraemia and subarachnoid haemorrhage on the cerebral vasomotor responses of the rabbit. J Cereb Blood Flow Metab 11:661–666. https://doi.org/10.1038/jcbfm.1991.118
Pasantes-Morales H, Lezama RA, Ramos-Mandujano G, Tuz KL (2006) Mechanisms of cell volume regulation in hypo-osmolality. Am J Med 119:4–11. https://doi.org/10.1016/j.amjmed.2006.05.002
Peng HL, Jensen PE, Nilsson H, Aaljkaer C (1998) Effect of acidosis on tension and [Ca2+]i in rat cerebral arteries: is there a role for membrane potential? Am J Physiol 274:655–662
Rabinstein AA (2006) Vasopressin antagonism: potential impact on neurologic disease. Clin Neuropharmacol 29:87–93
Rabinstein AA, Wijdicks EF (2003) Hyponatremia in critically ill neurological patients. Neurologist 9:290–300. https://doi.org/10.1097/01.nrl.0000095258.07720.89
Shi W, Cui N, Shi Y, Zhang X, Yang Y, Jiang C (2007) Arginine vasopressin inhibits Kir6.1/SUR2B channel and constricts the mesenteric artery via V1a receptor and protein kinase C. Am J Physiol Regul Integr Comp Physiol 293:191–199. https://doi.org/10.1152/ajpregu.00047.2007
Sterns RH, Riggs JE, Schochet SS Jr (1986) Osmotic demyelination syndrome following correction of hyponatremia. N Engl J Med 314:1535–1542. https://doi.org/10.1056/NEJM198606123142402
Takayasu M, Kajita Y, Suzuki Y, Shibuya M, Sugita K, Ishikawa T, Hidaka H (1993) Triphasic response of rat intracerebral arterioles to increasing concentration of vasopressin in vitro. J Cereb Blood Flow Metab 13:304–309. https://doi.org/10.1038/jcbfm.1993.38
Tian R, Vogel P, Lassen NA, Mulvany MJ, Andreasen F, Aaljkaer C (1995) Role of extracellular and intracellular acidosis for hypercapnia-induced inhibition of tension of isolated rat cerebral arteries. Circ Res 76:269–275
Wakatsuki T, Nakaya Y, Inoue I (1992) Vasopressin modulates K+-channel activities of cultured smooth muscle cells from porcine coronary artery. Am J Physiol 263:491–496. https://doi.org/10.1152/ajpheart.1992.263.2.H491
Vajda Z, Pedersen M, Dóczi T, Sulyok E, Stødkilde-Jørgensen H, Frøkiaer J, Nielsen S (2001) Effects of centrally administered arginine vasopressin and atrial natriuretic peptide on the development of brain edema in hyponatremic rats. Neurosurgery 49:697–704
Verbalis JG (1993) Hyponatremia induced by vasopressin or desmopressin in female and male rats. Am J Soc Nephrol 3:1600–1606
Verbalis JG, Drutarosky MD (1988) Adaptation to chronic hypoosmolality in rats. Kidney Int 34:351–360
Verbalis JG, Gullans SR (1991) Hyponatremia causes large sustained reductions in brain content of multiple organic osmolytes in rats. Brain Res 567:274–282
Zerbe R, Stropes L, Robertson G (1980) Vasopressin function in the syndrome of inappropriate antidiuresis. Annu Rev Med 31:315–327. https://doi.org/10.1146/annurev.me.31.020180.001531
Funding
This study was supported by a grant of the Polish Ministry of Science and Higher Education [N401 19032/3924] and by the Mossakowski Medical Research Centre, PAS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Aleksandrowicz, M., Kozniewska, E. Effect of vasopressin-induced chronic hyponatremia on the regulation of the middle cerebral artery of the rat. Pflugers Arch - Eur J Physiol 470, 1047–1054 (2018). https://doi.org/10.1007/s00424-018-2141-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-018-2141-0