
Abstract
The CSF1R gene, located on chromosome 5, encodes a 108 kDa protein and plays a critical role in regulating myeloid cell function. Mutations in CSF1R have been identified as a cause of a rare white matter disease called adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP, also known as CSF1R-related leukoencephalopathy), characterized by progressive neurological dysfunction. This study aimed to broaden the genetic basis of ALSP by identifying novel CSF1R variants in patients with characteristic clinical and imaging features of ALSP. Genetic analysis was performed through whole-exome sequencing or panel analysis for leukodystrophy genes. Variant annotation and classification were conducted using computational tools, and the identified variants were categorized following the recommendations of the American College of Medical Genetics and Genomics (ACMG). To assess the evolutionary conservation of the novel variants within the CSF1R protein, amino acid sequences were compared across different species. The study identified six previously unreported CSF1R variants (c.2384G>T, c.2133_2919del, c.1837G>A, c.2304C>A, c.2517G>T, c.2642C>T) in seven patients with ALSP, contributing to the expanding knowledge of the genetic diversity underlying this rare disease. The analysis revealed considerable genetic and clinical heterogeneity among these patients. The findings emphasize the need for a comprehensive understanding of the genetic basis of rare diseases like ALSP and underscored the importance of genetic testing, even in cases with no family history of the disease. The study’s contribution to the growing spectrum of ALSP genetics and phenotypes enhances our knowledge of this condition, which can be crucial for both diagnosis and potential future treatments.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The CSF1R gene spans approximately 60 kb on chromosome 5 of the human genome, comprises 21 exons, and encodes a 108 kDa protein [1, 2]. CSF1R protein is a class III receptor tyrosine kinase consisting of an extracellular domain, a transmembrane domain and an intracellular ATP binding site as well as a tyrosine kinase domain [2]. It is activated by two ligands, colony-stimulating factor 1 (CSF-1) and interleukin 34 (IL-34) [2, 3]. Upon ligand binding, the receptor homodimerizes, which results in intracellular autophosphorylation and downstream signaling [4]. CSF1R is mainly expressed on myeloid cells, including monocytes, tissue macrophages, dendritic cells, osteoclasts, and microglia. The survival, proliferation, and differentiation of cells belonging to this lineage is maintained by CSF1R signaling [5, 6].
In 2012, mutations in CSF1R were identified as one cause for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) [7]. The inheritance of CSF1R mutations in ALSP is autosomal-dominant; a negative family history, however, does not exclude ALSP, as frequent de novo mutations occur, and penetrance is incomplete [8, 9]. Most of the CSF1R mutations associated with ALSP are missense mutations located in the tyrosine kinase domain of the receptor [7, 10]. To date, 106 CSF1R mutations causing ALSP have been identified [9]. In addition to CSF1R, the gene alanyl-transfer RNA synthetase 2 (AARS2) is known to cause a similar disease phenotype [11, 12].
In the brain, CSF1R is predominantly expressed on microglia [13,14,15]. Since CSF1R mutations in ALSP can cause microglia dysfunction [16,17,18], ALSP is classified as primary microgliopathy [19]. Despite the comprehensive knowledge about CSF1R function in physiological conditions, the exact pathomechanism of ALSP remains unknown.
ALSP patients show a severe neurological disease characterized by substantial cognitive and motor dysfunction due to progressive leukoencephalopathy and brain atrophy [9, 10]. Patients commonly present with progressive cognitive symptoms, including memory impairment, executive dysfunction, and aphasia. Motor deficits such as gait disturbances due to spasticity or parkinsonism are frequently observed, and occasionally tremors or ataxia. Neuropsychiatric symptoms, including depression, apathy and personality changes, are also prevalent. Additionally, patients often exhibit signs of frontal lobe dysfunction, such as disinhibition and impaired judgment. In an advanced state of the disease, epileptic seizures may occur [9, 20]. Neuroimaging typically reveals white matter hyperintensities sparing the temporal lobe and brain atrophy; it may also show persistent diffusion restrictions, corpus callosum abnormalities and white matter calcification [20,21,22]. The mean age at onset is 43 years but ranges from early adulthood to the 8th decade and occurs earlier in female patients compared to male patients [10, 20]. The mean survival after symptom onset is 7 years [9, 10]. At early stages of the disease, hematopoietic stem cell transplantation represents a therapeutic option, after thorough risk–benefit evaluation [23,24,25]. Beyond this experimental treatment, no specific therapies for ALSP exist to date.
While the identification of neurofilament light chain (NfL) and chitotriosidase as biomarkers for ALSP has facilitated the diagnostic work-up of the disease [26, 27], diagnostic certainty requires genetic testing and the detection of a pathogenic variant in CSF1R. By gene panel and whole exome sequencing, respectively, we identified six novel CSF1R variants in seven patients presenting with typical clinical and imaging features of ALSP, which we report here.
Materials and methods
Participants and data collection
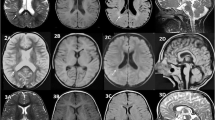
Patients were recruited during routine clinical care at the neurological department of the University Hospital Tübingen, Germany. Information on age, sex, age at symptom onset, and clinical manifestations were collected from all patients, and a detailed neurological examination was performed by a neurologist (Table 1). All individuals underwent cerebral magnetic resonance imaging (MRI) including T2 fluid-attenuated inversion recovery (FLAIR) and diffusion-weighted imaging (DWI) using 3.0 Tesla devices (Fig. 2).
Genetic analysis
Sequencing (whole-exome sequencing or panel analysis) was conducted in routine clinical care either at the Institute of Medical Genetics and Applied Genomics Tübingen, Germany (Patients 3 and 6), the CeGaT Institute Tübingen, Germany (Patients 1, 2 and 4), the Institute of Human Genetics Leipzig, Germany (Patient 5) or the Institut für Klinische Genetik und Tumorgenetik Bonn, Germany (Patients 7 and 8).
At the Institute of Medical Genetics and Applied Genomics Tübingen, exome sequencing libraries were generated from genomic DNA using Agilent SureSelect XT Human All Exon V5/V7 enrichment kits (Agilent Technologies) or the TruSeq DNA PCR-Free Kit (Illumina), respectively. Sequencing was performed on a HiSeq2500 or NovaSeq 6000 system (Illumina) as paired-end reads. The sequence data were analyzed using the megSAP pipeline (https://github.com/imgag/megSAP) and aligned to the GRCh37 reference genome.
At the CeGaT Institute Tübingen, genomic DNA was enriched using a custom design Agilent SureSelect in solution kit. Sequencing was performed using barcoded libraries on the SOLiD 5500xl platform (Applied Biosystems by Life Technology). The primary data analysis was performed using Lifescope (versions v2.5-r0 and v2.5-r2.5.1). Only variants (single nucleotide variants (SNVs)/small indels) with a minor allele frequency (MAF) of ≤ 1% in coding and flanking intronic regions (± 8 bp) and the untranslated regions (UTRs) were evaluated.
At the Institute of Human Genetics in Leipzig, exome libraries were generated from genomic DNA using the Illumina TruSeq Rapid Exome Library Preparation kit. Sequencing was performed on the Illumina NextSeq System. Coding sequences ± 8 base pairs of flanking intronic regions were covered by ≥ 20 reads/base per sample. Sequence data were analyzed using the Varvis software (Limbus Medical Technologies GmbH).
At the Institut für Klinische Genetik und Tumorgenetik Bonn, Next-Generation Sequencing (NGS) was performed using enrichment methods. The evaluation of sequence variants (SNVs, coding regions as well as adjacent splice sites (± 5 bp)) was carried out using certified pipeline technology (Varvis, Limbus Medical Technologies GmbH). The analysis of larger deletions/duplications was performed through quantitative NGS data analysis (Varvia, Limbus Medical Technologies GmbH). Classification and interpretation of sequence variants were performed using QIAGEN Clinical Insight-Interpret Software (QCI).
Variant rating and classification
The variants were annotated using wANNOVAR (wannovar.wglab.org) [28] and analyzed using multiple computational software programs to predict the effect of these variants (Functional Analysis Through Hidden Markov Models (FATHMM) [29], MutationTaster [30], Sorting Intolerant from Tolerant (SIFT) [31], Polymorphism Phenotyping v2, Human Variants Database (Polyphen2 HVAR) [32], Likelihood Ratio Test (LRT), Combined Annotation Dependent Depletion (CADD) [33], Mendelian Clinically Applicable Pathogenicity (M-CAP) [34]). Identified variants were classified following the recommendations of the American College of Medical Genetics and Genomics (ACMG) [35]. The current literature (up to June 2024) was searched using the NCBI National Library of Medicine (PubMed) to determine that the variants were not reported previously. Genetic data are summarized in Fig. 1 and Table 2.

Genomic organization and protein domain structure of CSF1R with characteristics of novel CSF1R variants. The CSF1R gene spans more than 60 kbp and contains 21 exons (transcript ID: ENST00000675795.1). It encodes a protein of 972 amino acids (protein ID: ENSP00000501699.1), containing five extracellular immunoglobulin domains, a transmembrane domain as well as an intracellular tyrosine kinase domain. The six novel variants are all located in the tyrosine kinase domain. Their respective amino acids are evolutionarily conserved, as confirmed through alignment with several species. The schematic displays the names and RefSeq IDs of the species used for the alignment with the human reference sequence of the CSF1R protein
Protein alignment
To determine the evolutionary conservation of the CSF1R protein, the amino acid sequence was compared among different species (human, RefSeq NP_001275634.1; chimpanzee, RefSeq XP_016809515.1; rhesus macaque, RefSeq XP_001107711.4; cattle, RefSeq NP_001068871.2; dog, RefSeq XP_546306.2; cat, NP_001009231.1; sheep, RefSeq XP_027826197.2; rat, RefSeq NP_001025072.1; mouse, RefSeq NP_001032948.2; chicken, RefSeq NP_001308446.2; frog, RefSeq NP_001008181.1) using Jalview (jalview.org) [36] (Fig. 1).
Case reports
Patient 1
This patient presented at the age of 47 years with a 2-year history of motor, behavioral and sensory symptoms. After a fall she had developed pain in her right knee and gait difficulties. The gait disturbance worsened, and she was diagnosed with multiple sclerosis 1 year after symptom onset. As the symptoms worsened despite corticosteroid treatment, she became wheelchair-bound within the following year, forcing her to retire and move to a nursery home. Further symptoms included indifferent behavior and impaired impulse control with compulsive shopping and hoarding, requiring the appointment of a legal guardian. In addition, she reported sensory disturbances in her left hand, which were interpreted as carpal tunnel syndrome. The patient’s mother was reported to have had a stroke in her late sixties, her father had no known neurological disease. Her sister was reported to be healthy.
In the neurological examination two years after symptom onset, the patient exhibited spastic hypertonia of the lower limbs and a slight action tremor that was more evident on the right side. She showed severe gait apraxia with difficulties standing up and walking; she could only take single steps when being supported. The Babinski reflex, Gordon’s sign and Oppenheim sign were positive. She also exhibited emotionless behavior.
Cognitive testing using Montreal Cognitive Assessment (MoCA) [37] revealed cognitive deficits (23/30 points). Cerebral magnetic resonance imaging (MRI) showed generalized confluent white matter lesions, brain atrophy, and diffusion restrictions. Laboratory blood and cerebrospinal fluid (CSF) analyses for routine parameters and neurosyphilis serology were unremarkable. Plasma levels for arylsulfatase A, β-galactosidase, β-hexosaminidase A, total β-hexosaminidase A & B, β-galactocerebrosidase and β-glucocerebrosidase were normal.
Six months later, she required help in most activities of daily living such as cooking and personal hygiene. She also showed symptoms of depression despite antidepressant medication. Her gait disturbance had progressed and she was unable to stand or walk independently. She also showed slightly hypometric saccadic eye movements and an overshoot in the finger-to-finger test as well as moderate dysdiadochokinesia. Her rapidly progressive cognitive impairment over the course of six months became evident in her MoCA score, which was then 12/30 points.
The electrophysiological examination revealed a lesion of the pyramidal tract to the left arm and leg. The magnetic resonance imaging (MRI) of the brain showed supratentorial atrophy and white matter lesions as well as diffusion restriction (Fig. 2A). The neurodegenerative marker NfL was highly increased in serum and CSF (serum: 251 pg/mL, CSF: 19,081 pg/mL; controls [26]: serum: 24.3 ± 3.4 pg/mL, CSF 1052 ± 208.5 pg/mL).

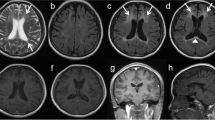
Cerebral magnetic resonance imaging. Magnetic resonance (MR) T2-weighted fluid-attenuated inversion recovery (FLAIR) as well as diffusion-weighted images (DWI) of every patient are depicted. The hallmark features of ALSP in MR imaging are patchy to confluent T2-hyperintensities mainly in the frontoparietal white matter while sparing the temporal lobe, supratentorial global brain atrophy accentuated in the frontal region as well as dispersed foci of restricted diffusion in the periventricular white matter on DWI sequences. All patients had white matter lesions and marked brain atrophy. In five of the patients, diffusion restriction was detected (Patients 1, 4 (circle), 5, 6, 7). All sites of diffusion restriction are hypointense in the apparent diffusion coefficient (ADC) sequence (data not shown)
Over the next 5 years, her condition deteriorated further; she was bedridden, unable to move, and mutistic at the age of 52 years.
Genetic testing revealed a missense variant in the CSF1R gene leading to an amino acid exchange in the CSF1R protein, c.2384G>T, p.Gly795Val (Fig. 1; Table 2).
DNA samples of the parents were not available; family history might suggest an affected mother, who was described to have had a stroke in her sixties. The variant of the patient was absent from the Genome Aggregation Database (gnomAD) [38] (PM2) and is located in the tyrosine kinase domain of the protein, which is known as a mutational hot spot (PM1). The amino acid glycine at this position (795) is conserved across different species [39] and is classified as deleterious by several in silico prediction programs (PP3). An amino acid exchange at one position before (794) has been described as likely pathogenic/pathogenic in the context of ALSP [40]. The clinical and MRI phenotype (diffusion restriction) is highly specific for ALSP (PP4). Taken together, the variant of the patient was classified as likely pathogenic according to ACMG criteria (PM1, PM2, PP3, and PP4).
Patient 2
This patient presented at the age of 40 years with a 3-year history of progressive memory deficits, social withdrawal, indifference, aggressive behavior, poverty of speech, and incontinence for urine and stool. There was no family history of neurological disease. The patient had one sister and the parents were of Turkish origin.
In the clinical evaluation she showed orientation deficits, reserved behavior, executive dysfunction, apraxia and a slight dysdiadochokinesia on the left side. An MRI from the same year showed leukoencephalopathy, especially of the frontal lobe, atrophy of the corpus callosum as well as global atrophy. Infection serology, microbiologic, and virologic tests as well as CSF routine analysis and electrophysiology revealed no abnormalities. Plasma levels for arylsulfatase A, β-galactosidase, β-hexosaminidase A, total β-hexosaminidase A & B, β-galactocerebrosidase and β-glucocerebrosidase were normal.
One year later, at the age of 41 years, the patient needed help with most activities of daily living. She occasionally stumbled when climbing stairs. The patient sometimes left the house without notice and returned hours later. The neurological examination revealed a positive applause sign, marked poverty of speech, slight apraxia, dysdiadochokinesia and a slightly dysmetric heel–knee test. She had marked cognitive impairment (MoCA 8/30 points). NfL was highly elevated in serum and CSF (serum: 252 pg/mL, CSF: 14,786 pg/mL).
Over the course of the next 2 years, the gait worsened with parkinsonian features, apraxia became prominent, and cognitive impairment progressed (MoCA 3/30 points). The MRI of the brain showed white matter lesions and global atrophy, but no diffusion restriction (Fig. 2B).
Genetic testing revealed a heterozygous deletion of exon 15–21 in the CSF1R gene (Fig. 1; Table 2). The deletion affects a large part of the tyrosine kinase domain of the protein (PM1, PM4) and is absent from gnomAD (PM2). Taken together, the variant was classified as likely pathogenic according to ACMG criteria (PM1, PM2, and PM4).
Patient 3
This patient experienced first symptoms at the age of 46 years, including poverty of speech, slowness in daily activities, and indifference. He also had difficulties executing his work as a forklift operator due to concentration deficits. The family history revealed neurologically healthy parents, but the patient’s maternal grandmother had suffered from early-onset dementia, categorized as arteriosclerotic, and had died at the age of 59 years.
When presenting at our institution 2 years later, the patient had no spontaneous speech production, and answered only to direct questions with “yes” or “no”. He needed help in many activities of daily living, such as shaving and showering, and suffered from a slight urinary incontinence. He was able to walk independently, although he was careful and slow when climbing stairs. In the neurological examination, he presented with pronounced apathy, apraxia, and slight action tremor of the right hand. The gait pattern was smooth, but the patient had a slightly stooped posture.
Cerebral MRI showed white matter lesions especially in the frontal lobe as well as global brain atrophy, most pronounced in the frontal lobe. Diffusion restriction was not detected (Fig. 2C). NfL was increased in serum and CSF (serum: 81 pg/mL, CSF: 6840 pg/mL). There were no other abnormalities in the routine CSF analysis and blood tests.
Genetic testing revealed a heterozygous missense variant in the CSF1R gene, c.1837G>A, p.Val613Met (Fig. 1; Table 2). The patient’s mother carried the same CSF1R variant but had not developed any symptoms at the time of examination (aged 68 years); however, the maternal grandmother had early-onset dementia (see above), suggesting an autosomal-dominant inheritance with incomplete penetrance in this family. The variant is located in the ATP binding site of the tyrosine kinase domain (PM1). It is not present in gnomAD (PM2). The affected amino acid is evolutionary highly conserved and classified as deleterious by several in silico prediction programs (PP3). Chu et al. described a variant in CSF1R at the same position, but with a different amino acid exchange (c.1837G>T, p.V613L) in the context of ALSP (PM5) [41]. Also, Berdowski et al. generated a zebrafish model carrying this variant, which exhibits pathological features found in ALSP brain tissue [42]. In addition, we found a second patient presenting with classical symptoms of ALSP and carrying the same variant (see Patient 6). The variant in our patient was classified as likely pathogenic according to ACMG criteria (PM1, PM2, PM5, and PP3).
Patient 4
This patient had experienced first symptoms at the age of 40 years, including gait disturbances and impaired fine motor skills. Over the next 3 years, the symptoms had intensified. He was now wheelchair-bound and needed help with daily activities such as eating and body care. He had also developed swallowing difficulties and blurred speech. Further symptoms included depression, restlessness, anxiety, loss of appetite and weight, obstipation, and urinary urge. The family history war negative for neurological disorders.
In the clinical examination the patient showed marked signs of Parkinsonism with rigidity and strong freezing. He was not able to sit or stand up independently and was unable to walk due to freezing. In addition, reduced facial expression, dysarthria, apraxia, and dysdiadochokinesis were observed. He had marked anxiety. MoCA testing revealed cognitive impairment (17/30 points).
The cerebral MRI showed global atrophy, moderate white matter lesions, especially in the parietal lobe, and one site of diffusion restriction (Fig. 2D). Routine CSF analysis and blood tests were normal. NfL in serum and CSF was strongly elevated (serum: 42 pg/mL, CSF: 7363 pg/mL).
Genetic testing revealed a heterozygous missense variant in the CSF1R gene, c.2304C>A, p.Phe768Leu (Fig. 1; Table 2). Segregation analysis uncovered the absence of this variant in the parent’s genome, i.e., we assume the de novo occurrence of the variant in our patient (PM6). The variant is located in the tyrosine kinase domain of the protein (PM1) and is not listed in gnomAD (PM2). The amino acid at the corresponding location is highly conserved and the variant was rated as deleterious by several in silico prediction programs (PP3). The variant of the patient was classified as likely pathogenic according to ACMG criteria (PM1, PM2, PM6, and PP3).
Patient 5
The patient started to have orientation difficulties when driving at the age of 46 years. His cognitive abilities rapidly deteriorated and he had to resign from his work as truck driver. His condition further worsened and he was transferred to a nursing home at the age of 48 years, where we first met him 1 year later. By then, aged 49 years, he had no spontaneous speech production and only communicated with nodding or “no”. He also suffered from epileptic seizures. Family history revealed that the patient’s mother suffered from slight memory deficits from the age of 75 years.
Clinical examination was limited due to the severity of his status. He had slight left-sided facial palsy. He was able to move his arms and legs spontaneously, but was unable to walk, even with assistance, most likely due to apraxia. He had no prominent swallowing difficulties. In the following 2 years, he lost his residual ability to communicate as well as his ability to sit and was confined to bed. He developed severe dysphagia. He died shortly after his 51st birthday.
Cerebral MRI at the age of 46 and 47 years showed bilateral white matter hyperintensities on FLAIR sequence, multiple areas of diffusion restriction in subcortical and central white matter on DWI as well as global brain and corpus callosum atrophy (Fig. 2E). NfL in serum and CSF was strongly elevated (serum: 352 pg/mL, CSF: 14,563 pg/mL).
Genetic testing uncovered a heterozygous missense variant in the CSF1R gene, c.2517G>T, p.Trp839Cys (Fig. 1; Table 2). Segregation analysis revealed the same variant in the mother, who was clinically unaffected at the time of genetic testing. She developed slight memory deficits at the age of 75 years. The variant is located in the tyrosine kinase domain of the protein (PM1) and is not listed in gnomAD (PM2). The amino acid at the corresponding location is highly conserved and the variant was rated as deleterious by several in silico prediction programs (PP3). MRI showed diffusion restriction which together with the clinical course is very specific for ALSP (PP4). The variant of the patient was classified as likely pathogenic according to ACMG criteria (PM1, PM2, PP3, and PP4).
Patient 6
The patient first noticed a limited walking distance at the age of 51 years and pain in her legs after exertion. The gait difficulties worsened and she additionally developed fine motor impairment. At her first visit in our outpatient clinic, aged 54 years, the patient was only able to walk with a walker. She reported being clumsy and having an unclear handwriting; she also suffered from progressive dysarthria for six months. She had urge symptoms and infrequent urge incontinence for urine. She was still able to perform all activities of daily living. Family history revealed that the patient’s mother had developed similar symptoms at around 50 years of age including dysarthria and gait disturbance. She died at the age of 56 years.
Clinical examination of our patient revealed saccadic eye movements and hypermetric saccades. She had a mixed dysarthria with pseudobulbar and cerebellar components. Motor examination showed mild weakness in arm and hip abduction, and pyramidal signs (mild spasticity, increased reflexes, bilaterally positive Babinski sign). She had mild dysmetria with discrete intention tremor, bradydysdiadochokinesia as well as bradykinesia and decrement in finger tapping. Her walking was broad-based with small steps and reduced arm swinging.
When the patient presented again 1 year later, she was no longer able to walk; she had been permanently dependent on a wheelchair since the age of 55 years. The fine motor impairment, particularly of the left hand, had worsened further, and she needed assistance with eating. Due to her progressive dysarthria, the patient no longer spoke on the phone. Her husband reported pathological laughter and mood swings with increased verbal aggression. The patient and her husband had not noticed any memory deficits. Previously, she had undergone treatment with up to 600 mg of L-Dopa for 1 month, but it proved ineffective.
During clinical evaluation, the patient mostly communicated in 1–2-word sentences. The clinical examination was limited due to apraxia, motor initiation difficulties, and moderate aphasia. She had frontal release signs (applause sign, palmomental reflex, pathological laughter). Dysarthria was mixed, with occasional words difficult to understand. She showed slight involuntary movements and an increased muscle tone in her left side. She had pyramidal signs (brisk reflexes with clonic responses, Babinski’s sign on the left). The finger-nose test revealed a slight intention tremor on the right side and was apractic on the left side. Fingertapping was irregular on the right side, while it was not possible on the left side due to apraxia/motor initiation impairment. The patient was unable to independently transfer from wheelchair to bed. She was only able to stand and walk with significant support from two people due to a combination of gait-induced spasticity, leg apraxia, extrapyramidal-motor disorder, and significant retropulsion. Cognitive performance assed by MoCA was impaired (23/30 points).
Cerebral MRI showed an advanced neurodegenerative process with significant atrophy of gray and white matter, including the corpus callosum, as well as diffusion restriction (Fig. 2F). Except for protein (72 mg/dl), routine CSF parameters were unremarkable, but NfL was highly increased (9756 pg/ml).
Genetic testing revealed a heterozygous missense variant in the CSF1R gene, c.1837G>A, p.Val613Met (Fig. 1; Table 2). This is the same variant we found in Patient 3 (PM1, PM2, PM5, and PP3). MRI showed diffusion restriction which together with the clinical course and the family history is very specific for ALSP (PP4). The variant was classified as likely pathogenic according to ACMG criteria (PM1, PM2, PM5, PP3, and PP4).
Patient 7
The patient had first symptoms at the age of 44 years with a change in personality including physical aggression. In addition, he was hospitalized twice due to depression. Shortly thereafter, he lost his job as electrician since he could no longer meet the demands of the work. He further developed a significant lack of drive, requiring him to move back in with his parents. He also showed signs of an impulse control disorder with uncontrolled purchases on the internet. Activities of daily living could still be performed independently. Family history revealed that his mother had no apparent motor deficits, but slight memory difficulties at the time of presentation, aged 68 years, that later progressed to severe dementia, requiring her translocation to a nursing home. The maternal grandfather of the patient died at the age of 53 years; the death was attributed to long-term consequences of his service at war. The patient’s maternal grandmother, originally from Slovenia, died at the age of 61 years due to heart disease.
When presenting the first time at our hospital at the age of 46 years, neurological examination revealed no physical deficits but moderate apraxia as well as cognitive deficits (MoCA 15/30 points).
Cerebral MRI showed white matter lesions in the frontal and parietal lobe, global atrophy as well as prominent diffusion restriction (Fig. 2G). NfL in CSF was highly increased (7043 pg/ml).
Genetic testing uncovered a heterozygous variant c.2642C>T, p.Ala881Val in the CSF1R gene (Fig. 1; Table 2). The patient’s mother carried the same variant. Her cerebral MRI revealed extensive confluent hyperintensities in the superior frontal gyri and global brain atrophy accentuated in the frontal region; no diffusion restriction was detected (Fig. 2H). The variant was absent from gnomAD (PM2) and is located in the tyrosine kinase domain of the protein, which is known as a mutational hot spot (PM1). The amino acid alanine at this position (881) is conserved across different species [39] and is classified as deleterious by several in silico prediction programs (PP3). The clinical and MRI phenotype (diffusion restriction) is highly specific for ALSP (PP4). Taken together, the variant was classified as likely pathogenic according to ACMG criteria (PM1, PM2, PP3, and PP4).
Discussion
Since the identification of CSF1R as the causal gene for ALSP, over 100 pathogenic/likely pathogenic variants have been reported in association with the disorder [7, 9]. Comprehensive understanding of the range of pathogenic variants and the diversity of symptoms they elicit is essential in rare genetic diseases such as ALSP. To this end, we report seven patients carrying six hitherto undescribed CSF1R variants associated with ALSP, and thereby contribute to the expanding spectrum of ALSP genetics and phenomenology.
Five of the six novel variants reported here are missense variants located either in the ATP-binding site or the tyrosine kinase domain of the receptor (Fig. 1). Given that these variants occur in evolutionarily conserved positions, they are considered critical for maintaining proper receptor function, i.e., the autophosphorylation of the intracellular domain. This is further substantiated by the predictions made by the applied rating tools (FATHMM, MutationTaster, SIFT, Polyphen2, LRT, CADD, M-CAP), most of which indicated that all of the missense variants were deleterious (Table 2). Although missense mutations in the kinase domain of CSF1R are presumed to cause loss-of-function [43,44,45], their heterozygous presence is sufficient to manifest the disease. This indicates a dominant-negative mode of action, possibly due to the receptor's obligate dimeric configuration. However, the pathomechanism of the missense mutations remains a topic of ongoing debate [42, 44, 46,47,48]. In one patient (Patient 2), a heterozygous deletion spanning exon 15–21 was identified. This large deletion has the potential to cause mRNA decay or generate a truncated protein. Theoretically, mRNA decay would lead to haploinsufficiency, whereas the production of a truncated protein would likely cause a dominant-negative effect since the deletion includes the kinase domain. Notably, Patient 2 exhibits a comparatively slow progression of the disease; this could be indicative of mRNA decay with resultant haploinsufficiency as a possible scenario in this patient. However, a comprehensive analysis of molecular genetics in conjunction with clinical phenotype and disease progression in larger cohorts is required to establish a correlation between genotype and phenotype in CSF1R-related ALSP.
The family histories of the patients described here reflect the heterogeneity of inheritance and penetrance of CSF1R mutations in ALSP. In two families (Patients 2 and 4), family history was informative and negative. This raises the possibility of a de novo CSF1R mutation in these patients. De novo mutations in ALSP are a known phenomenon and account for up to 30% of ALSP cases [8, 10]. In one of these families (Patient 4), segregation analysis confirmed the de novo occurrence of the CSF1R variant by excluding the variant in both parents. Genetic testing was performed in three other families (Patient 3, Patient 5, and Patient 7). The results showed that the three mothers were carriers of the CSF1R variant. Two of them, the mothers of Patient 3 and Patient 5, were both in their late 60 s and asymptomatic at the time of testing. The third, mother of Patient 7, had slight memory difficulties aged 68 years, and later developed severe dementia. Her MRI exhibited classical features of ALSP with confluent white matter lesions and frontally accentuated brain atrophy (Fig. 2H). Clinical presentation with progressive cognitive impairment and MRI features is compatible with findings in ALSP, even though onset would be comparatively late and progression slow. The mother of Patient 5 developed mild memory and walking deficits at the age of 75 years. While in these two persons, especially in the mother of Patient 7, cognitive decline could indicate the manifestation of ALSP, it is also possible that the impairment resulted from another form of dementia. As the average age at onset for ALSP in women is 40 years [9, 10], and there is a known age-dependent penetrance with 95% of the carriers diseased by the age of 60 years [10], a manifestation beyond the age of 65 years seems unlikely, but is not impossible. In one of the families with the mother as asymptomatic carrier (Patient 3), the maternal grandmother was likely affected by ALSP, as she suffered from early-onset dementia and died at the age of 59 years, suggesting an incomplete penetrance in the patient’s asymptomatic mother. Only in one family (Patient 6), even though not confirmed genetically, there was a clear pattern of dominant inheritance with the mother showing typical ALSP symptoms at an early age. In conclusion, the study’s findings on the differences in family history, inheritance, and penetrance, highlight the intricate nature of ALSP genetics, emphasizing the importance of performing CSF1R genetic testing, even in cases where the family history is negative or suggests a recessive inheritance. Moreover, the high percentage of asymptomatic, oligosymptomatic, or late-onset carriers in this cohort is intriguing and demands further investigation into the protective factors that take effect in these individuals.
From a clinical perspective, the hallmark features of ALSP consist of cognitive impairment and motor dysfunction [9, 10]. Among the seven patients examined in this study, four displayed cognitive impairment as the first sign of the disease, while the remaining three exhibited gait difficulties as their initial symptom (Table 1). With the exception of one patient (Patient 6), who was only examined in the early stage of her disease course, all other patients are known to have progressed to severe dementia. All patients developed motor impairment. The combination of cognitive and motor impairment lead to a significant nursing effort and to wheelchair dependence, or even confinement to bed. Additionally, this study frequently observed symptoms congruent with cases reported in the literature, including apraxia as well as pyramidal and extrapyramidal signs such as spasticity and parkinsonism [9, 10, 49,50,51,52,53]. As the disease progressed, the patients frequently suffered from dysarthria, dysphagia, incontinence, and, in one case, seizures, which is also in line with previously described cases of ALSP [9, 10, 50, 51, 54]. Interestingly, one patient exhibited pronounced anxiety, and another showed pathological laughing, both of which have not yet been described as typical symptoms in ALSP.
Cerebral MRI scans of all seven patients showed frontoparietal white matter lesions and brain atrophy, which are typical features of ALSP [9, 10, 45, 53, 55]. Additionally, diffusion restriction was detected in five of the patients, which is another characteristic finding of the disease [9, 10, 21]. Besides the typical clinical signs and characteristic features in MR imaging, the determination of NfL in serum and/or CSF constitutes the third pillar in the diagnostic workup of ALSP. While NfL serves as a general neuronal biomarker found elevated in various neurological conditions [56], it is exceptionally increased in ALSP [26]. The CSF NfL levels of the patients included in this study ranged from 6840 to 19,081 pg/mL, which significantly exceeds the upper standard value of approximately 1000 pg/mL [26], reflecting the rapid neuronal loss in these patients.
In summary, our study expands the genetic spectrum of ALSP by identifying six previously undescribed variants in seven patients displaying characteristic symptoms of the disease. The observed variability in genetic background and penetrance across the studied families underscores the crucial role of genetic testing, even in cases with no prior family history. Since rare diseases are frequently caused by genetic alterations, genetic testing is often the only way to establish a definitive diagnosis, highlighting its importance in the diagnostic process. Therefore, we emphasize the significance of early inclusion of genetic testing in the diagnostic workup of rare diseases and especially leukoencephalopathies of undefined origin.
Data availability
Data will be shared upon qualified request.
References
Cunningham F, Allen JE, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Austine-Orimoloye O, Azov AG, Barnes I, Bennett R et al (2022) Ensembl 2022. Nucleic Acids Res 50(D1):D988–D995
UniProt C (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49(D1):D480–D489
Lin H, Lee E, Hestir K, Leo C, Huang M, Bosch E, Halenbeck R, Wu G, Zhou A, Behrens D et al (2008) Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 320(5877):807–811
Yu W, Chen J, Xiong Y, Pixley FJ, Dai XM, Yeung YG, Stanley ER (2008) CSF-1 receptor structure/function in MacCsf1r-/- macrophages: regulation of proliferation, differentiation, and morphology. J Leukoc Biol 84(3):852–863
Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL et al (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82(2):380–397
Pollard JW (2009) Trophic macrophages in development and disease. Nat Rev Immunol 9(4):259–270
Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, Lash J, Wider C, Wojtas A, DeJesus-Hernandez M et al (2011) Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 44(2):200–205
Karle KN, Biskup S, Schule R, Schweitzer KJ, Kruger R, Bauer P, Bender B, Nagele T, Schols L (2013) De novo mutations in hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). Neurology 81(23):2039–2044
Papapetropoulos S, Pontius A, Finger E, Karrenbauer V, Lynch DS, Brennan M, Zappia S, Koehler W, Schoels L, Hayer SN et al (2021) Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: review of clinical manifestations as foundations for therapeutic development. Front Neurol 12:788168
Konno T, Yoshida K, Mizuno T, Kawarai T, Tada M, Nozaki H, Ikeda SI, Nishizawa M, Onodera O, Wszolek ZK et al (2017) Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur J Neurol 24(1):37–45
Dallabona C, Diodato D, Kevelam SH, Haack TB, Wong LJ, Salomons GS, Baruffini E, Melchionda L, Mariotti C, Strom TM et al (2014) Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 82(23):2063–2071
Lakshmanan R, Adams ME, Lynch DS, Kinsella JA, Phadke R, Schott JM, Murphy E, Rohrer JD, Chataway J, Houlden H et al (2017) Redefining the phenotype of ALSP and AARS2 mutation-related leukodystrophy. Neurol Genet 3(2):e135
Raivich G, Haas S, Werner A, Klein MA, Kloss C, Kreutzberg GW (1998) Regulation of MCSF receptors on microglia in the normal and injured mouse central nervous system: a quantitative immunofluorescence study using confocal laser microscopy. J Comp Neurol 395(3):342–358
Akiyama H, Nishimura T, Kondo H, Ikeda K, Hayashi Y, McGeer PL (1994) Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer’s disease and amyotrophic lateral sclerosis. Brain Res 639(1):171–174
Chitu V, Gokhan S, Nandi S, Mehler MF, Stanley ER (2016) Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci 39(6):378–393
Oosterhof N, Kuil LE, van der Linde HC, Burm SM, Berdowski W, van Ijcken WFJ, van Swieten JC, Hol EM, Verheijen MHG, van Ham TJ (2018) Colony-stimulating factor 1 receptor (CSF1R) regulates microglia density and distribution, but not microglia differentiation in vivo. Cell Rep 24(5):1203-1217.e6
Tada M, Konno T, Tada M, Tezuka T, Miura T, Mezaki N, Okazaki K, Arakawa M, Itoh K, Yamamoto T et al (2016) Characteristic microglial features in patients with hereditary diffuse leukoencephalopathy with spheroids. Ann Neurol 80(4):554–565
Tian WT, Zhan FX, Liu Q, Luan XH, Zhang C, Shang L, Zhang BY, Pan SJ, Miao F, Hu J et al (2019) Clinicopathologic characterization and abnormal autophagy of CSF1R-related leukoencephalopathy. Transl Neurodegener 8:32
Konno T, Kasanuki K, Ikeuchi T, Dickson DW, Wszolek ZK (2018) CSF1R-related leukoencephalopathy: a major player in primary microgliopathies. Neurology 91(24):1092–1104
Papapetropoulos S, Gelfand JM, Konno T, Ikeuchi T, Pontius A, Meier A, Foroutan F, Wszolek ZK (2024) Clinical presentation and diagnosis of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: a literature analysis of case studies. Front Neurol 15:1320663
Bender B, Klose U, Lindig T, Biskup S, Nagele T, Schols L, Karle KN (2014) Imaging features in conventional MRI, spectroscopy and diffusion weighted images of hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). J Neurol 261(12):2351–2359
Sundal C, Van Gerpen JA, Nicholson AM, Wider C, Shuster EA, Aasly J, Spina S, Ghetti B, Roeber S, Garbern J et al (2012) MRI characteristics and scoring in HDLS due to CSF1R gene mutations. Neurology 79(6):566–574
Mochel F, Delorme C, Czernecki V, Froger J, Cormier F, Ellie E, Fegueux N, Lehericy S, Lumbroso S, Schiffmann R et al (2019) Haematopoietic stem cell transplantation in CSF1R-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J Neurol Neurosurg Psychiatry 90(12):1375–1376
Tipton PW, Kenney-Jung D, Rush BK, Middlebrooks EH, Nascene D, Singh B, Holtan S, Ayala E, Broderick DF, Lund T et al (2021) Treatment of CSF1R-related leukoencephalopathy: breaking new ground. Mov Disord 36(12):2901–2909
Dulski J, Heckman MG, White LJ, Zur-Wyrozumska K, Lund TC, Wszolek ZK (2022) Hematopoietic stem cell transplantation in CSF1R-related leukoencephalopathy: retrospective study on predictors of outcomes. Pharmaceutics 14(12):2778
Hayer SN, Krey I, Barro C, Rossler F, Kortvelyessy P, Lemke JR, Kuhle J, Schols L (2018) NfL is a biomarker for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Neurology 91(16):755–757
Hayer SN, Santhanakumaran V, Bohringer J, Schols L (2022) Chitotriosidase is a biomarker for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Ann Clin Transl Neurol 9(11):1807–1812
Chang X, Wang K (2012) wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet 49(7):433–436
Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, Gaunt TR (2013) Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 34(1):57–65
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11(4):361–362
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC (2016) SIFT missense predictions for genomes. Nat Protoc 11(1):1–9
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7:Unit7 20
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46(3):310–315
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, Bernstein JA, Bejerano G (2016) M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 48(12):1581–1586
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25(9):1189–1191
Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, Cummings JL, Chertkow H (2005) The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53(4):695–699
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP et al (2021) Author Correction: The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 590(7846):E53
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D (2002) The human genome browser at UCSC. Genome Res 12(6):996–1006
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W et al (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46(D1):D1062–D1067
Chu M, Wang DX, Cui Y, Kong Y, Liu L, Xie KX, Xia TX, Zhang J, Gao R, Zhou AH et al (2021) Three novel mutations in Chinese patients with CSF1R-related leukoencephalopathy. Ann Transl Med 9(13):1072
Berdowski WM, van der Linde HC, Breur M, Oosterhof N, Beerepoot S, Sanderson L, Wijnands LI, de Jong P, Tsai-Meu-Chong E, de Valk W et al (2022) Dominant-acting CSF1R variants cause microglial depletion and altered astrocytic phenotype in zebrafish and adult-onset leukodystrophy. Acta Neuropathol 144(2):211–239
Rosenstein I, Andersen O, Victor D, Englund E, Granberg T, Hedberg-Oldfors C, Jood K, Fitrah YA, Ikeuchi T, Danylaite Karrenbauer V (2022) Four Swedish cases of CSF1R-related leukoencephalopathy: Visualization of clinical phenotypes. Acta Neurol Scand 145(5):599–609
Miura T, Mezaki N, Konno T, Iwasaki A, Hara N, Miura M, Funayama M, Unai Y, Tashiro Y, Okita K et al (2018) Identification and functional characterization of novel mutations including frameshift mutation in exon 4 of CSF1R in patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J Neurol 265(10):2415–2424
Tsai PC, Fuh JL, Yang CC, Chang A, Lien LM, Wang PN, Lai KL, Tsai YS, Lee YC, Liao YC (2021) Clinical and genetic characterization of adult-onset leukoencephalopathy caused by CSF1R mutations. Ann Clin Transl Neurol 8(11):2121–2131
Leng C, Lu L, Wang G, Zhang Y, Xu Y, Lin X, Shen N, Xu X, Qun S, Sun M et al (2019) A novel dominant-negative mutation of the CSF1R gene causes adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Am J Transl Res 11(9):6093–6101
Konno T, Tada M, Tada M, Koyama A, Nozaki H, Harigaya Y, Nishimiya J, Matsunaga A, Yoshikura N, Ishihara K et al (2014) Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology 82(2):139–148
Arreola MA, Soni N, Crapser JD, Hohsfield LA, Elmore MRP, Matheos DP, Wood MA, Swarup V, Mortazavi A, Green KN (2021) Microglial dyshomeostasis drives perineuronal net and synaptic loss in a CSF1R(+/-) mouse model of ALSP, which can be rescued via CSF1R inhibitors. Sci Adv 7(35):eabg1601
Lynch DS, Jaunmuktane Z, Sheerin UM, Phadke R, Brandner S, Milonas I, Dean A, Bajaj N, McNicholas N, Costello D et al (2016) Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry 87(5):512–519
Kawakami I, Iseki E, Kasanuki K, Minegishi M, Sato K, Hino H, Shibuya K, Fujisawa K, Higashi S, Akiyama H et al (2016) A family with hereditary diffuse leukoencephalopathy with spheroids caused by a novel c.2442+2T>C mutation in the CSF1R gene. J Neurol Sci 367:349–355
Zhuang LP, Liu CY, Li YX, Huang HP, Zou ZY (2020) Clinical features and genetic characteristics of hereditary diffuse leukoencephalopathy with spheroids due to CSF1R mutation: a case report and literature review. Ann Transl Med 8(1):11
Wider C, Van Gerpen JA, DeArmond S, Shuster EA, Dickson DW, Wszolek ZK (2009) Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): a single entity? Neurology 72(22):1953–1959
Sundal C, Lash J, Aasly J, Oygarden S, Roeber S, Kretzschman H, Garbern JY, Tselis A, Rademakers R, Dickson DW et al (2012) Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): a misdiagnosed disease entity. J Neurol Sci 314(1–2):130–137
Kinoshita M, Yoshida K, Oyanagi K, Hashimoto T, Ikeda S (2012) Hereditary diffuse leukoencephalopathy with axonal spheroids caused by R782H mutation in CSF1R: case report. J Neurol Sci 318(1–2):115–118
Kleinfeld K, Mobley B, Hedera P, Wegner A, Sriram S, Pawate S (2013) Adult-onset leukoencephalopathy with neuroaxonal spheroids and pigmented glia: report of five cases and a new mutation. J Neurol 260(2):558–571
Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, Barro C, Kappos L, Comabella M, Fazekas F et al (2018) Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 14(10):577–589
Acknowledgements
We thank the patients and their families for participation in this study.
Funding
Open Access funding enabled and organized by Projekt DEAL. Stefanie Hayer was supported by a grant from the Hertie Foundation (P1200021) and the Clinician–Scientist program of the Medical Faculty of the University of Tübingen, Germany (434-0-0). Benjamin Roeben was supported by the Clinician Scientist program of the Medical Faculty of the University of Tübingen, Germany (478-0-0). Wolfgang Köhler is funded by a German Ministry of Health grant “LeukoExpert” (2520DAT94B) and EU grant “Tackle Genetic White Matter Diseases”, supported by tax funds on the basis of the budget adopted by the Saxon state parliament. Ludger Schöls is supported by a grant of the German Ministry of Health to the “LeukoExpert” project (ZMVI1-2520DAT94E).
Author information
Authors and Affiliations
Contributions
ASS: drafting of case reports; JR: literature research, drafting of manuscript; WK: medical care and imaging of Patient 5, revision of manuscript; SK: medical care of Patient 3 and 4; KC: medical care of Patient 7; FR: genetic analysis of Patient 5, revision of manuscript; SB: genetic analysis of Patients 1, 2, and 4; TBH: genetic analysis of Patients 3 and 6; BR: medical care of Patient 2, revision of manuscript; MK: medical care of Patient 6; NR: genetic analysis of Patient 7; TB: imaging of Patient 7 and the mother of Patient 7; JL: genetic analysis of Patient 5; BB: imaging of patients 1, 2, 3, 4, 6, and 7, LS: Medical care of patients 1, 2, 3, 4, 6, 7, revision of the manuscript; HH: data analysis, revision of the manuscript; SNH: medical care of all patients, literature research, data analysis, drafting, revision and finalization of manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical standards
The study was approved by the Institutional Review Board of the University of Tübingen (Vote 690/2011BO1). All individuals involved in this study provided written informed consent for genetic analysis and study participation before enrollment.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schmitz, A.S., Raju, J., Köhler, W. et al. Novel variants in CSF1R associated with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP). J Neurol (2024). https://doi.org/10.1007/s00415-024-12557-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00415-024-12557-0