
Abstract
New diagnostic criteria for myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) have recently been proposed, distinguishing this syndrome from other inflammatory diseases of the central nervous system. Seropositivity status for MOG-IgG autoantibodies is important for diagnosing MOGAD, but only in the context of robust clinical characterization and cautious interpretation of neuroimaging. Over the last several years, access to cell-based assay (CBA) techniques has improved diagnostic accuracy, yet the positive predictive value of serum MOG-IgG values varies with the prevalence of MOGAD in any given patient population. For this reason, possible alternative diagnoses need to be considered, and low MOG-IgG titers need to be carefully weighted. In this review, cardinal clinical features of MOGAD are discussed. Key challenges to the current understanding of MOGAD are also highlighted, including uncertainty regarding the specificity and pathogenicity of MOG autoantibodies, the need to identify immunopathologic targets for future therapies, the quest to validate biomarkers that facilitate diagnosis and detect disease activity, and the importance of deciphering which patients with MOGAD require long-term immunotherapy.
Similar content being viewed by others


Introduction
Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) is a relatively new addition to the category of central nervous system (CNS) inflammatory demyelinating diseases [1, 2]. CNS inflammatory demyelinating conditions, including multiple sclerosis (MS) and neuromyelitis optica spectrum disorders (NMOSD), are differentiated based on severity, clinical phenotype, imaging, laboratory, and pathological findings [2] (Table 1). While patients with MS, MOGAD and NMOSD may present with similar clinical manifestations, such as optic neuritis and myelitis, those with MOGAD lack a clear sex predilection, and more commonly experience a monophasic course [2,3,4,5]. MOGAD also has the greatest predilection in children, representing 20–30% of inflammatory CNS syndromes in this population as compared to approximately 5% in adults. The current estimated range of incidence in the pediatric population is 3.1 per 1 million, as compared to 1.6 and 2.39 per 1 million among adults [3]. Notably, these numbers, along with the estimated worldwide prevalence of 20 million [5], are likely to increase with growing recognition of the disease and improved availability of serological testing. Unlike NMOSD, cases of MOGAD are not strongly associated with other systemic autoimmune disorders or circulating autoantibodies [3]. Yet, disease manifestations may occur after prodromal infections, particularly those caused by viral pathogens, such as influenza, Epstein–Barr virus, herpes simplex virus, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), to name a few [3]. Occasionally, patients with MOGAD have an overlap syndrome with anti-NMDA receptor encephalitis, characterized by clinical features of demyelination associated with encephalopathy, seizures, dyskinesias, or psychosis [5]. As the clinical spectrum of MOGAD continues to expand, so too does our appreciation for diagnostic and management challenges associated with this enigmatic condition. Key areas of ongoing research include determining the specificity and pathogenicity of MOG autoantibodies, identifying immunopathologic targets for future therapies, discovering and validating biomarkers that detect disease activity, and deciphering which patients with MOGAD require long-term immunotherapy.
MOGAD: the evolving clinical spectrum
Our growing appreciation of the full clinical spectrum of MOGAD will likely mirror the NMOSD experience. Once considered to be a severe form of MS targeting the optic nerves and spinal cord, NMOSD has since been recognized as a distinct autoimmune astrocytopathy with pathognomonic clinical features [2, 6]. Similarly, MOGAD has now been identified as a separate entity from both MS and NMOSD. Recently, diagnostic criteria proposed by an international panel of experts highlight optic neuritis, myelitis, acute disseminated encephalomyelitis (ADEM), cerebral mono-focal or multifocal deficits, brainstem or cerebellar syndromes, and cerebral cortical encephalitis (often with seizures) as cardinal features of MOGAD (Tables 2 and 3) [5]. Unlike MS, neurological deterioration does not typically progress in the absence of relapses [5]. In real-world settings, there will be challenges in diagnosing MOGAD despite having clearly proposed criteria for the disease, because, as mentioned, many clinical manifestations of MOGAD overlap with other CNS inflammatory syndromes, including but not limited to MS and NMOSD. Diagnostic confusion may also arise from the interpretation of radiological and serological findings since these features may all be caused by other etiologies and are not specific to MOGAD. For this reason, it will be important to not overweigh any single finding in isolation. As a rule of thumb, clinical features of MOGAD, particularly acute optic neuritis, may closely resemble those of NMOSD with severe vision loss (often bilateral) at its nadir. Patients with MOGAD, however, often show greater therapeutic response to high-dose corticosteroid treatment and may also demonstrate significant spontaneous improvement [7,8,9,10]. Importantly, the phenotypic expression of MOGAD may vary with age, such that ADEM-like lesions are more likely to affect children, whereas optic neuritis and isolated myelitis tend to be more common among adults [5]. In addition, relapse risk in MOGAD patients is often higher in adults than children [3].
MOG autoantibodies: specificity, pathogenicity, and interpreting titers
Seropositivity status for MOG-IgG autoantibodies is key to applying the recently proposed criteria for MOGAD (Table 3) [5]. It is also imperative to exclude alternate diagnoses, just as we do for patients with suspected MS or NMOSD. Without rigorous clinical characterization and thoughtful deliberation, individuals with true MOGAD run the risk of being under-treated, whereas those without MOGAD might be inappropriately exposed to long-term immunotherapies or incorrect therapies (Case vignettes here contrasting two scenarios of overlooked MOGAD [Case 1] and false positive [Case 2]). As highlighted by the recently proposed MOGAD criteria [5], there are challenges to relying on a serum MOG-IgG antibody with imperfect specificity and uncertain pathogenicity because causation can be confused with association. Notably, cell-based assays (CBA) are used to establish a MOGAD diagnosis, with live CBA techniques performing slightly better than fixed-based assays. Importantly, enzyme-linked immunosorbent assay (ELISA) testing should not be used to make a diagnosis, as findings are less specific and often discordant with CBA results [32]. While access to CBA techniques has improved diagnostic accuracy, the positive predictive value (the probability that a patient with a positive serum MOG-IgG result harbors MOGAD) still varies with the prevalence of MOGAD in any given patient population. Investigators at the Mayo Clinic have reported a positive predictive value (PPV) of MOG-IgG testing by live CBA of 72% using a cut-off of 1:20 despite having an overall specificity of 98% [33]. Their study found there was a positive correlation between serum MOG-IgG titer cut-off value and PPV, such that the PPV was 100% for 1:1000, 82% for 1:100, 51% for antibody titers 1:20–1:40 [33]. Notably, a ≥ 1:40 titer “cut-off,” (n = 65) yielded a PPV of 93.8%, but lowered sensitivity [34]. Subsequently, Manzano and colleagues also performed an institutional cohort study to determine the PPV of serum MOG-IgG for clinically defined MOGAD, using the same live MOG-IgG CBA [34]. Among 1,877 patients tested, 78 (4.2%) patients tested positive for MOG-IgG with titer ≥ 1:20; of those who were seropositive, 67 had a validated MOGAD diagnosis, yielding a PPV of 85.9%. Undoubtedly, low positive titers need to be interpreted with caution. Yet, it is worth mentioning that excluding patients with MOG IgG titers of 1:20 without proper clinical analysis may lead to a significant number of missed diagnoses of MOGAD [33,34,35], further reiterating the need for thoughtful clinical judgment. Ideally, patients should be tested for MOG-IgG sero-status at their incident event, and when possible, before administration of corticosteroids, immunoglobulins, or apheresis [5]. When there are concerns regarding a false positive MOG-IgG result, repeat serum testing should be performed after 3 months or at the time of relapse [5]. Finally, like aquaporin 4 (AQP4)-IgG, MOG autoantibodies are induced in the peripheral circulation, though the triggering cause for their production remains unclear [2, 4]. Serological testing with CBA techniques is more sensitive for detecting MOG autoantibodies than CSF sampling. Occasionally, patients with MOGAD may have negative serum MOG-IgG results, but show seropositivity with CSF testing [5, 36, 37]. For this reason, it is reasonable to consider CSF testing for MOG-IgG to support the diagnosis of MOGAD when the serum results are negative, but the clinical and MRI findings are otherwise highly suggestive of the diagnosis [36,37,38,39,40].
When considering specificity, MOG autoantibodies are uncommon in patients with MS (0.4%) [4]. However, because of the high prevalence of MS compared to MOGAD, many false positive results may occur if all patients with possible MS are tested for MOG-IgG, therefore discretion is required [33]. Co-existing seropositivity for MOG-IgG is also uncommon in cases of AQP4-IgG-positive NMOSD; when this occurs, AQP4 IgG titers are often higher, and the patient usually follows a NMOSD phenotype [5, 41]. Yet the high prevalence of MOG autoantibodies among patients with AQP4-IgG-seronegative NMOSD has created some diagnostic confusion, such that MOGAD patients have been erroneously labeled as seronegative NMOSD. With the newly proposed MOGAD criteria (Table 3), patients with presenting features of NMOSD who are negative for AQP4-IgG should be tested for MOG-IgG, as part of their diagnostic evaluation for MOGAD [5]. In fact, with more accessible use of reliable CBA for MOG-IgG, rigorous classification of clinical phenotypes and improved understanding of pathognomonic radiological findings, many patients previously diagnosed with autoimmune demyelinating diseases including seronegative NMOSD, atypical MS, chronic relapsing inflammatory optic neuropathy (CRION), and ADEM may be re-classified as MOGAD.
Going forward, it will be important to consider whether clinical manifestations in patients with suspected MOGAD reflect pathogenic effects of MOG autoantibodies, as opposed to a scenario in which MOG-IgG seropositivity more likely represents a “bystander” phenomenon [42]. The pathogenicity of MOG autoantibodies is the subject of ongoing debate. In vivo and in vitro studies suggest that MOG autoantibodies may cause primary demyelination in the CNS with loss of microtubule cytoskeleton in oligodendrocytes and altered expression of proteins [3, 4]. Evidence of the pathogenicity of MOG autoantibodies has also been inferred from models of experimental autoimmune encephalitis (EAE) [3, 4]. Specifically, serum from individuals with MOGAD administered in rat models of EAE has been associated with worse clinical disease and increased evidence of demyelination and axonal loss [3, 4]. In other experimental work, serum derived from pediatric patients with MOGAD has coincided with disruption to F-actin and β-tubulin networks in immortalized oligodendrocyte cells [3]. These data support potential pathologic roles for MOG antibodies in the clinical expression of MOGAD, but further research is needed.
Established and emerging biomarkers for the diagnosis and monitoring of MOGAD [3, 4, 33]
The clinical features of MS, MOGAD, and NMOSD significantly overlap. Therefore, biomarkers will be needed to distinguish these diagnoses accurately and reliably, especially in acute care settings where antibody results are often delayed (Table 4). Future research will also need to explore the impact of patient-related factors (biological sex, racial background, and pregnancy status), co-morbidities (infections), and treatment effects (long-term immunotherapies) on relapse risk and severity. In the path toward patient-centered “precision-based medicine”, it would be beneficial to identify biomarkers that not only facilitate diagnosis and predict relapses, but also prognosticate recovery for those with MOGAD.
Management of MOGAD patients
Acute treatment
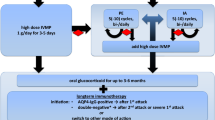
The Optic Neuritis Treatment Trial (ONTT) has informed our understanding regarding the relative risks and benefits of high-dose corticosteroid therapy for patients presenting with acute optic neuritis [15]. This seminal study reported that administering IVMP (1 g/day for 3 days, followed by 1 mg/kg for 11 days) to patients presenting with acute ON expedited visual recovery, but it did not improve long-term visual outcomes [15]. Yet, the lessons of the ONTT may not be extendable to patients with MOGAD, due to study design (bilateral ON was a basis of exclusion). Notably, an examination of serum samples obtained from 177 out of the 448 ONTT participants revealed that only 3 (1.7%) were positive for MOG-IgG antibody [8]. Thus, the conclusions drawn from the ONTT regarding the role of corticosteroids in optic neuritis cannot be extrapolated to predict long-term visual outcomes in MOGAD patients. MOGAD attacks are often very steroid responsive and therefore most experts recommend high-dose corticosteroids as first-line treatment for patients presenting with acute MOGAD attacks [54,55,56,57]. Like the established NMOSD treatment approach, there is observational evidence suggesting that early management of MOGAD-related ON with IVMP is crucial for optimal visual recovery, as it has been shown that early steroid treatment is associated with better visual outcomes and less thinning of the peri-papillary retinal nerve fiber layer [9, 57,58,59]. This signifies a shift in the treatment paradigm of ONTT for ON, as it is felt that in NMOSD and MOGAD, time is of the essence to preserve vision [60]. However, it is important to note that there are cases of MOGAD ON with spontaneous improvement without steroid treatment [9, 10].
Despite the lack of a universally accepted high-dose steroid treatment regimen, the use of IVMP at a dosage of 1 g/day for 5 days is the most prevalent strategy [61, 62]. While no study has compared the efficacy of IVMP to oral corticosteroid in treating acute MOGAD attacks, Morrow et al. have reported that bioequivalent doses of oral corticosteroids are noninferior to IVMP in treating acute ON [63], and therefore 1250 mg of oral prednisone is sometimes offered as an alternative to IVMP. Further research is needed to establish the role of oral corticosteroids in the acute management of MOGAD manifestations. Patients suffering from a recent MOGAD attack are commonly considered to be at risk for relapse upon cessation or reduction of corticosteroid therapy. Therefore, considering multiple observational studies, to mitigate relapse risk, it is recommended that corticosteroids are tapered gradually over a period of 1–2 months [57, 64,65,66,67].
Most MOGAD attacks are responsive to corticosteroids, yet a subset of MOGAD patients do not respond well to IVMP, necessitating further interventions. Both plasmapheresis (PLEX) and immuno-adsorption (IA) have been proposed as therapeutic options for those failing IVMP treatment. PLEX and IA have been successfully utilized to treat immune-mediated neurological diseases, including acute NMOSD attacks [68,69,70,71,72,73,74,75]. An international multicenter retrospective study of 92 MOGAD ON attacks treated with PLEX demonstrated significant improvement in all attacks except for one eye that was treated over 6 months after the initial attack [76]. While preliminary reports are encouraging, prospective studies are required to confirm the efficacy of PLEX for acute MOGAD attacks [35]. Intravenous immunoglobulin (IVIG) is another therapeutic option that has been employed in other demyelinating conditions, such as NMOSD. While no published studies have shown IVIG efficacy in treating acute MOGAD [69, 77], a multicenter retrospective study looking at IVIG for the treatment of acute MOGAD suggests it may be associated with improved outcomes (unpublished data). Further research is required to uncover the utility and role of PLEX, IA, and IVIG in the acute management of MOGAD patients.
Maintenance therapy
The best maintenance therapy for MOGAD patients remains a topic of ongoing debate, particularly considering that approximately 50% of MOGAD patients exhibit monophasic disease, and may not require long-term immunosuppression [9, 14]. Unfortunately, it is difficult to predict if a newly diagnosed patient is destined to develop a relapsing course. Longitudinal MOG-IgG seropositivity was found to be associated with relapsing MOGAD, but many of the patients who remain seropositive do not relapse. Roughly 25% of MOGAD patients will become seronegative and are more likely to have monophasic disease [78,79,80,81]. Furthermore, findings by Cobo-Calvo et al., suggest that adults are at a higher risk of relapse and may experience worse clinical outcomes than children [80]. Long-term immunotherapy is typically initiated after a second MOGAD attack [61]. However, the type of maintenance treatment for recurrent MOGAD remains disputed, and has been extrapolated from NMOSD studies [60]. This is not ideal due to the well-described differences in pathophysiology, demographics, severity, and prognosis between the two disease entities. Common maintenance therapies employed may include oral steroids, azathioprine (AZA), mycophenolate mofetil (MMF), and B cell-targeting biologics, such as rituximab (RTX), and tocilizumab, all of which have been employed in NMOSD, in addition to maintenance IVIG, which is not a common treatment for NMOSD [60, 62].
Multiple studies have shown that maintenance therapy with oral corticosteroids has been associated with a reduction in relapse rate [64, 82, 83]. Bridging immunosuppressive agents with oral steroids is linked with better therapeutic outcomes [56, 64]. However, the potential benefits of chronic corticosteroids use must be weighed against the health risks associated with their long-term use.
Broad spectrum immunosuppressants, such as AZA and MMF, are well tolerated and have been proposed to be effective maintenance therapies for MOGAD patients [84,85,86]. Chen and colleagues reported that MOGAD patients maintained on AZA had a significant reduction in the annualized relapse rate (ARR) from 1.6 (range: 0–9.7) to 0.2 (range: 0–3.2) [84]. A modest reduction in relapse rates was noted for those treated with MMF [84]. Additionally, a prospective observational cohort study provided Class IV evidence that MMF therapy reduces relapse in MOGAD patients [87]. When initiating AZA and MMF, it is recommended to bridge these patients with prednisolone for the first few months until a decrease in lymphocyte counts is observed [56, 62]. While observational studies have shown that AZA and MMF are associated with a reduction in relapses, it is important to note that relapses can still occur and there is no randomized clinical trial data in MOGAD patients.
The efficacy of RTX in reducing relapses and EDSS scores in NMOSD patients has been well-established through RCTs [88, 89]. However, RTX’s efficacy in MOGAD patients remains ambiguous as there are no randomized clinical trials evaluating its effectiveness in this patient population. Meta-analyses have suggested that RTX reduces relapses and EDSS scores in MOGAD, though to a lesser extent than in NMOSD [90, 91]. Additionally, retrospective studies have also reported varying degrees of ARR reduction for MOGAD patients treated with RTX [61, 84, 92]. In addition, MOGAD relapses can occur despite depletion of memory B cells [93].
More recently, IVIG has emerged as a promising maintenance therapy for MOGAD patients [55]. A multicenter retrospective cohort study conducted by Chen et al. reported that maintenance IVIG therapy was associated with a reduced relapse rate in adult MOGAD patients, especially when administered at a dose of 1–2 g/kg every 4 weeks [84, 94]. Other retrospective studies have suggested maintenance IVIG therapy is effective in pediatric MOGAD patients [55]. Looking forward, IL-6 inhibitors may play a role in MOGAD maintenance therapy with growing evidence suggesting that tocilizumab might be effective for highly relapsing MOGAD patients, and satralizumab is being investigated in a multicenter randomized clinical trials [95, 96]. Additionally, rozanolixizumab, an anti-neonatal Fc receptor inhibitor, is also a promising therapy that is currently in randomized clinical trials for the treatment of relapsing MOGAD patients [14]. As evidences for therapeutic approaches continue to accumulate, randomized clinical trials with sufficient follow-up are necessary to uncover the most efficacious and well-tolerated maintenance therapy for patients with recurrent MOGAD.
Prognosis
The duration of immunosuppressive treatment for MOGAD remains uncertain, as the field currently lacks robust biomarkers to inform therapy. Specifically, it is challenging to identify patients who will remain monophasic after their first attack. Several indicators of disease activity have been proposed, including attack severity and frequency, MOG-IgG sero-status and titers, patient age, relapse-free interval, spinal cord involvement, and resultant neurological disability [61, 97,98,99]. The identification of prognostic factors indicating the likelihood of relapse and disability in a patient would catalyze a personalized treatment approach for MOGAD patients.
Adult MOGAD patients tend to have more recurrent episodes and poorer functional recovery compared to pediatric patients [82, 97, 100, 101]. MOG-IgG sero-status, particularly in adult patients, is proposed to be a predictor of disease course, with ‘seroreversion’ (from positive to negative) being indicative of a low relapse risk [67, 79, 82]. Yet, the utility of MOG-IgG titers for treatment planning remains debatable [102]. Nevertheless, recurrent disease has been associated with elevated initial MOG-IgG titers, while transiently low titers have been shown to be associated with a monophasic course [80, 82, 103].
Long-term outcomes and disability are quite challenging to predict as studies with long follow-up are sparse for patients with MOGAD. While it is believed that, on average, even frequently relapsing MOGAD results in less disability than NMOSD, inter-patient variability has been documented [16, 104, 105]. Indeed, the clinical heterogeneity of MOGAD is an increasingly recognized phenomenon [97]. Sufficiently powered prospective studies with extended follow-up would enable a more comprehensive understanding of the natural history of MOGAD, identify predictors of its course and establish guidelines for its treatment.


Data availability
Data Availaiblity statement is not applicable as this review article is based exclusively on published work.
References
Cañellas AR, Gols AR, Izquierdo JR et al (2007) Idiopathic inflammatory-demyelinating diseases of the central nervous system. Neuroradiology 49:393–409. https://doi.org/10.1007/s00234-007-0216-2
Costello F (2022) Neuromyelitis optica spectrum disorders. Contin Lifelong Learn Neurol 28:1131–1170. https://doi.org/10.1212/CON.0000000000001168
Longbrake E (2022) Myelin oligodendrocyte glycoprotein-associated disorders. Contin Lifelong Learn Neurol 28:1171–1193. https://doi.org/10.1212/CON.0000000000001127
Marignier R, Hacohen Y, Cobo-Calvo A et al (2021) Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol 20:762–772. https://doi.org/10.1016/S1474-4422(21)00218-0
Banwell B, Bennett JL, Marignier R et al (2023) Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet Neurol. https://doi.org/10.1016/S1474-4422(22)00431-8
Wingerchuk DM, Banwell B, Bennett JL et al (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85:177–189. https://doi.org/10.1212/WNL.0000000000001729
Bennett JL, Costello F, Chen JJ et al (2023) Optic neuritis and autoimmune optic neuropathies: advances in diagnosis and treatment. Lancet Neurol 22:89–100. https://doi.org/10.1016/S1474-4422(22)00187-9
Chen JJ, Tobin WO, Majed M et al (2018) Prevalence of myelin oligodendrocyte glycoprotein and aquaporin-4–IgG in patients in the optic neuritis treatment trial. JAMA Ophthalmol 136:419. https://doi.org/10.1001/jamaophthalmol.2017.6757
Chen JJ, Flanagan EP, Bhatti MT, et al (2022) Details and outcomes of a large cohort of MOG-IgG associated optic neuritis. Mult Scler Relat Disord 68:104237. https://doi.org/10.1016/j.msard.2022.104237
Vosoughi AR, Muccilli A, Schneider R, et al (2022) Recovery of Vision in Myelin Oligodendrocyte Glycoprotein–IgG Optic Neuritis Without Treatment: A Case Series. J Neuro-Ophthalmology Publish Ah:3–5. https://doi.org/10.1097/WNO.0000000000001583
Levy M, Fujihara K, Palace J (2021) New therapies for neuromyelitis optica spectrum disorder. Lancet Neurol 20:60–67. https://doi.org/10.1016/S1474-4422(20)30392-6
Sechi E, Krecke KN, Messina SA et al (2021) Comparison of MRI lesion evolution in different central nervous system demyelinating disorders. Neurology 97:e1097–e1109. https://doi.org/10.1212/WNL.0000000000012467
McDonald WI, Compston A, Edan G et al (2001) Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol 50:121–127. https://doi.org/10.1002/ana.1032
Sechi E, Cacciaguerra L, Chen JJ, et al (2022) Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD): A Review of Clinical and MRI Features, Diagnosis, and Management. Front Neurol 13:885218. https://doi.org/10.3389/fneur.2022.885218
Beck RW, Cleary PA, Anderson MM et al (1992) A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med 326:581–588. https://doi.org/10.1056/nejm199202273260901
Chen JJ, Flanagan EP, Jitprapaikulsan J et al (2018) Myelin oligodendrocyte glycoprotein antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol 195:8–15. https://doi.org/10.1016/j.ajo.2018.07.020
Gaitán MI, Paday Formenti ME, Calandri I, et al (2022) The central vein sign is present in most infratentorial multiple sclerosis plaques. Mult Scler Relat Disord 58:103484. https://doi.org/10.1016/j.msard.2021.103484
Costello F, Scott JN (2019) Imaging in Neuro-ophthalmology. Contin Lifelong Learn Neurol 25:1438–1490. https://doi.org/10.1212/CON.0000000000000783
Myers KA, Nikolic A, Romanchuk K et al (2017) Optic neuropathy in the context of leukemia or lymphoma: diagnostic approach to a neuro-oncologic emergency. Neuro-Oncology Pract 4:60–66. https://doi.org/10.1093/nop/npw006
Abrams RMC, Safavi F, Tuhrim S et al (2021) MRI negative myelopathy post mild SARS-CoV-2 infection: vasculopathy or inflammatory myelitis? J Neurovirol 27:650–655. https://doi.org/10.1007/s13365-021-00986-w
Tobin WO, Guo Y, Krecke KN et al (2017) Diagnostic criteria for chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS). Brain 140:2415–2425. https://doi.org/10.1093/brain/awx200
Guo J, Bu Y, Liu W (2022) Case Report: A Case With MOGAD and Anti-NMDAR Encephalitis Overlapping Syndrome Mimicing Radiological Characteristics of CLIPPERS. Front Immunol 13:832084. https://doi.org/10.3389/fimmu.2022.832084
Banks SA, Morris PP, Chen JJ et al (2021) Brainstem and cerebellar involvement in MOG-IgG-associated disorder versus aquaporin-4-IgG and MS. J Neurol Neurosurg Psychiatry 92:384–390. https://doi.org/10.1136/jnnp-2020-325121
Pike M (2013) Opsoclonus–myoclonus syndrome. In: Handbook of Clinical Neurology. Elsevier, pp 1209–1211
Stunkel L, Kung NH, Wilson B et al (2018) Incidence and causes of overdiagnosis of optic neuritis. JAMA Ophthalmol 136:76. https://doi.org/10.1001/jamaophthalmol.2017.5470
Kunchok A, Krecke KN, Flanagan EP et al (2020) Does area postrema syndrome occur in myelin oligodendrocyte glycoprotein-IgG–associated disorders (MOGAD)? Neurology 94:85–88. https://doi.org/10.1212/WNL.0000000000008786
Jonathan Maran J, Sharpe C, Carroll S (2023) Journal Pre-proof Paediatric MOG-antibody disease presenting with intracranial hypertension and unilateral vision loss without radiological evidence of optic neuritis. J Neuroimmunol. https://doi.org/10.1016/j.jneuroim.2023.578083
Marignier R (2019) Unusual presentations of MOG antibody-associated central nervous system demyelination: expanding the spectrum. Mult Scler J 25:128–129. https://doi.org/10.1177/1352458518804127
Valdrighi A, Russ J, Waubant E, et al (2021) Atypical myelin oligodendrocyte glycoprotein antibody disease presenting with isolated elevated intracranial pressure. Neuroimmunol Reports 1:100028. https://doi.org/10.1016/j.nerep.2021.100028
Vosoughi AR, Ling J, Tam KT et al (2021) Ophthalmic manifestations of myelin oligodendrocyte glycoprotein-IgG-associated disorder other than optic neuritis: a systematic review. Br J Ophthalmol 105:1591–1598. https://doi.org/10.1136/bjophthalmol-2020-317267
Costello F (2014) Inflammatory optic neuropathies. Continuum (Minneap Minn) 20:816–37. https://doi.org/10.1212/01.CON.0000453316.60013.52
Reindl M, Schanda K, Woodhall M, et al (2020) International multicenter examination of MOG antibody assays. Neurol—Neuroimmunol Neuroinflammation 7:e674. https://doi.org/10.1212/NXI.0000000000000674
Sechi E, Buciuc M, Pittock SJ et al (2021) Positive predictive value of myelin oligodendrocyte glycoprotein autoantibody testing. JAMA Neurol 78:741. https://doi.org/10.1001/jamaneurol.2021.0912
Manzano GS, Salky R, Mateen FJ et al (2022) Positive predictive value of MOG-IgG for clinically defined MOG-AD within a real-world cohort. Front Neurol 13:1–4. https://doi.org/10.3389/fneur.2022.947630
Benard-seguin E, Costello F (2023) Optic neuritis : current challenges in diagnosis and management. 36:10–18. https://doi.org/10.1097/WCO.0000000000001128
Carta S, Cobo Calvo Á, Armangué T et al (2023) Significance of myelin oligodendrocyte glycoprotein antibodies in CSF. Neurology 100:e1095–e1108. https://doi.org/10.1212/WNL.0000000000201662
Dinoto A, Sechi E, Flanagan EP et al (2022) Serum and cerebrospinal fluid biomarkers in neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein associated disease. Front Neurol 13:1–13. https://doi.org/10.3389/fneur.2022.866824
Pace S, Orrell M, Woodhall M et al (2022) Frequency of MOG-IgG in cerebrospinal fluid versus serum. J Neurol Neurosurg Psychiatry 93:334–335. https://doi.org/10.1136/jnnp-2021-326779
Kwon YN, Kim B, Kim J-S, et al (2022) Myelin Oligodendrocyte glycoprotein-immunoglobulin G in the CSF. Neurol—Neuroimmunol Neuroinflammation 9:e1095. https://doi.org/10.1212/NXI.0000000000001095
Mariotto S, Gajofatto A, Batzu L et al (2019) Relevance of antibodies to myelin oligodendrocyte glycoprotein in CSF of seronegative cases. Neurology 93:e1867–e1872. https://doi.org/10.1212/WNL.0000000000008479
Kunchok A, Chen JJ, McKeon A et al (2020) Coexistence of myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies in adult and pediatric patients. JAMA Neurol 77:257. https://doi.org/10.1001/jamaneurol.2019.3656
Lerch M, Bauer A, Reindl M (2023) The Potential pathogenicity of myelin oligodendrocyte glycoprotein antibodies in the optic pathway. J Neuro-Ophthalmology 43:5–16. https://doi.org/10.1097/WNO.0000000000001772
Traboulsee A, Simon JH, Stone L et al (2016) Revised recommendations of the consortium of MS centers task force for a standardized MRI protocol and clinical guidelines for the diagnosis and follow-up of multiple sclerosis. Am J Neuroradiol 37:394–401. https://doi.org/10.3174/ajnr.A4539
Bacioglu M, Maia LF, Preische O et al (2016) Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron 91:56–66. https://doi.org/10.1016/j.neuron.2016.05.018
Khalil M, Teunissen CE, Otto M et al (2018) Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 14:577–589. https://doi.org/10.1038/s41582-018-0058-z
Luo W, Chen Y, Mao S et al (2022) Serum neurofilament light chain in adult and pediatric patients with myelin oligodendrocyte glycoprotein antibody-associated disease: Correlation with relapses and seizures. J Neurochem 160:568–577. https://doi.org/10.1111/jnc.15549
Chang X, Huang W, Wang L, et al (2021) Serum Neurofilament Light and GFAP Are Associated With Disease Severity in Inflammatory Disorders With Aquaporin-4 or Myelin Oligodendrocyte Glycoprotein Antibodies. Front Immunol 12:647618. https://doi.org/10.3389/fimmu.2021.647618
Kim H, Lee E-J, Kim S, et al (2020) Serum biomarkers in myelin oligodendrocyte glycoprotein antibody–associated disease. Neurol—Neuroimmunol Neuroinflammation 7:e708. https://doi.org/10.1212/NXI.0000000000000708
Barro C, Chitnis T, Weiner HL (2020) Blood neurofilament light: a critical review of its application to neurologic disease. Ann Clin Transl Neurol 7:2508–2523. https://doi.org/10.1002/acn3.51234
De Lott LB, Bennett JL, Costello F (2022) The changing landscape of optic neuritis: a narrative review. J Neurol 269:111–124. https://doi.org/10.1007/s00415-020-10352-1
Oertel FC, Sotirchos ES, Zimmermann HG et al (2022) Longitudinal retinal changes in <scp>MOGAD</scp>. Ann Neurol 92:476–485. https://doi.org/10.1002/ana.26440
Yu J, Huang Y, Quan C et al (2021) Alterations in the retinal vascular network and structure in MOG antibody-associated disease: an optical coherence tomography angiography study. J Neuro-Ophthalmology 41:e424–e432. https://doi.org/10.1097/WNO.0000000000001116
Yao Y, Li X, Xu Y et al (2022) The difference of the retinal structural and microvascular characteristics in patients with MOGAD-ON and AQP4-ON. BMC Neurol 22:323. https://doi.org/10.1186/s12883-022-02848-2
Wynford-Thomas R, Jacob A, Tomassini V (2019) Neurological update: MOG antibody disease. J Neurol 266:1280–1286. https://doi.org/10.1007/s00415-018-9122-2
Hacohen Y, Banwell B (2019) Treatment approaches for MOG-Ab-associated demyelination in children. Curr Treat Options Neurol 21:2. https://doi.org/10.1007/s11940-019-0541-x
Jarius S, Ruprecht K, Kleiter I, et al (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation 13:280. https://doi.org/10.1186/s12974-016-0718-0
Stiebel-Kalish H, Hellmann MA, Mimouni M, et al (2019) Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurol—Neuroimmunol Neuroinflammation 6:e572. https://doi.org/10.1212/NXI.0000000000000572
Soelberg K, Specovius S, Zimmermann HG et al (2018) Optical coherence tomography in acute optic neuritis: a population-based study. Acta Neurol Scand 138:566–573. https://doi.org/10.1111/ane.13004
Rode J, Pique J, Maarouf A, et al (2022) Time to steroids impacts visual outcome of optic neuritis in MOGAD. J Neurol Neurosurg Psychiatry jnnp-2022–330360. https://doi.org/10.1136/jnnp-2022-330360
Chwalisz BK, Levy M (2022) The treatment of myelin oligodendrocyte glycoprotein antibody disease: a state-of-the-art review. J Neuro-Ophthalmology 42:292–296. https://doi.org/10.1097/WNO.0000000000001684
Whittam DH, Karthikeayan V, Gibbons E et al (2020) Treatment of MOG antibody associated disorders: results of an international survey. J Neurol 267:3565–3577. https://doi.org/10.1007/s00415-020-10026-y
Healy S, Elhadd KT, Gibbons E et al (2021) Treatment of myelin oligodendrocyte glycoprotein immunoglobulin G–associated disease. Clin Exp Neuroimmunol 12:22–41. https://doi.org/10.1111/cen3.12630
Morrow SA, Fraser JA, Day C et al (2018) Effect of treating acute optic neuritis with bioequivalent oral vs intravenous corticosteroids. JAMA Neurol 75:690. https://doi.org/10.1001/jamaneurol.2018.0024
Ramanathan sudarshini, Mohammad S, Tantsis E, et al (2018) Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry 89:127–137. https://doi.org/10.1136/jnnp-2017-316880
Song H, Zhou H, Yang M et al (2019) Different characteristics of aquaporin-4 and myelin oligodendrocyte glycoprotein antibody-seropositive male optic neuritis in China. J Ophthalmol 2019:1–7. https://doi.org/10.1155/2019/4015075
Tajfirouz DA, Bhatti MT, Chen JJ (2019) Clinical characteristics and treatment of MOG-IgG–associated optic neuritis. Curr Neurol Neurosci Rep 19:100. https://doi.org/10.1007/s11910-019-1014-z
Huda S, Whittam D, Jackson R, et al (2021) Predictors of relapse in MOG antibody associated disease: a cohort study. BMJ Open 11:e055392. https://doi.org/10.1136/bmjopen-2021-055392
Osman C, Jennings R, El-Ghariani K, Pinto A (2020) Plasma exchange in neurological disease. Pract Neurol 20:92–99. https://doi.org/10.1136/practneurol-2019-002336
Kleiter I, Gahlen A, Borisow N et al (2016) Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol 79:206–216. https://doi.org/10.1002/ana.24554
Sokolov AA, Solovyev AG (2014) Russian pioneers of therapeutic hemapheresis and extracorporeal hemocorrection: 100-year anniversary of the world’s first successful plasmapheresis. Ther Apher Dial 18:117–121. https://doi.org/10.1111/1744-9987.12067
Levy M (2018) Plasmapheresis for acute attacks in neuromyelitis optica spectrum disorders. Neurol - Neuroimmunol Neuroinflammation 5:e510. https://doi.org/10.1212/NXI.0000000000000510
Pinching AJ, Peters DK (1976) Remission of myasthenia gravis following plasma-exchange. Lancet (London, England) 2:1373–1376. https://doi.org/10.1016/s0140-6736(76)91917-6
Oji S, Nomura K (2017) Immunoadsorption in neurological disorders. Transfus Apher Sci 56:671–676. https://doi.org/10.1016/j.transci.2017.08.013
Kobayashi M, Nanri K, Taguchi T et al (2015) Immunoadsorption therapy for neuromyelitis optica spectrum disorders long after the acute phase. J Clin Apher 30:43–45. https://doi.org/10.1002/jca.21324
Faissner S, Nikolayczik J, Chan A et al (2016) Immunoadsorption in patients with neuromyelitis optica spectrum disorder. Ther Adv Neurol Disord 9:281–286. https://doi.org/10.1177/1756285616646332
Chen JJ, Flanagan EP, Pittock SJ et al (2023) Visual outcomes following plasma exchange for optic neuritis: an international multicenter retrospective analysis of 395 optic neuritis attacks. Am J Ophthalmol. https://doi.org/10.1016/j.ajo.2023.02.013
Elsone L, Panicker J, Mutch K et al (2014) Role of intravenous immunoglobulin in the treatment of acute relapses of neuromyelitis optica: experience in 10 patients. Mult Scler J 20:501–504. https://doi.org/10.1177/1352458513495938
Oliveira LM, Apóstolos-Pereira SL, Pitombeira MS et al (2019) Persistent MOG-IgG positivity is a predictor of recurrence in MOG-IgG-associated optic neuritis, encephalitis and myelitis. Mult Scler J 25:1907–1914. https://doi.org/10.1177/1352458518811597
Hyun JW, Woodhall MR, Kim SH et al (2017) Longitudinal analysis of myelin oligodendrocyte glycoprotein antibodies in CNS inflammatory diseases. J Neurol Neurosurg Psychiatry 88:811–817. https://doi.org/10.1136/JNNP-2017-315998
Cobo-Calvo A, Ruiz A, Rollot F et al (2021) Clinical Features and Risk of Relapse in Children and Adults with Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease. Ann Neurol 89:30–41. https://doi.org/10.1002/ana.25909
López-Chiriboga AS, Majed M, Fryer J et al (2018) Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG–associated disorders. JAMA Neurol 75:1355. https://doi.org/10.1001/jamaneurol.2018.1814
Jurynczyk M, Messina S, Woodhall MR et al (2017) Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain 140:3128–3138. https://doi.org/10.1093/brain/awx276
Hacohen Y, Wong YY, Lechner C et al (2018) Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol 75:478–487. https://doi.org/10.1001/JAMANEUROL.2017.4601
Chen JJ, Flanagan EP, Bhatti MT et al (2020) Steroid-sparing maintenance immunotherapy for MOG-IgG associated disorder. Neurology 95:e111–e120. https://doi.org/10.1212/WNL.0000000000009758
Whittam DH, Cobo-Calvo A, Lopez-Chiriboga AS, et al (2020) Treatment of MOG-IgG-associated disorder with rituximab: An international study of 121 patients. Mult Scler Relat Disord 44:102251. https://doi.org/10.1016/j.msard.2020.102251
Lu Q, Luo J, Hao H et al (2021) Efficacy and safety of long-term immunotherapy in adult patients with MOG antibody disease: a systematic analysis. J Neurol 268:4537–4548. https://doi.org/10.1007/s00415-020-10236-4
Li S, Ren H, Xu Y, et al (2020) Long-term efficacy of mycophenolate mofetil in myelin oligodendrocyte glycoprotein antibody-associated disorders. Neurol - Neuroimmunol Neuroinflammation 7:e705. https://doi.org/10.1212/NXI.0000000000000705
Tahara M, Oeda T, Okada K et al (2020) Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 19:298–306. https://doi.org/10.1016/S1474-4422(20)30066-1
Nikoo Z, Badihian S, Shaygannejad V et al (2017) Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol 264:2003–2009. https://doi.org/10.1007/s00415-017-8590-0
Spagni G, Sun B, Monte G et al (2023) Efficacy and safety of rituximab in myelin oligodendrocyte glycoprotein antibody-associated disorders compared with neuromyelitis optica spectrum disorder: a systematic review and meta-analysis Neuro-inflammation. J Neurol Neurosurg Psychiatry 94:62–69. https://doi.org/10.1136/jnnp-2022-330086
Nepal G, Kharel S, Coghlan MA, et al (2022) Safety and efficacy of rituximab for relapse prevention in myelin oligodendrocyte glycoprotein immunoglobulin G (MOG-IgG)-associated disorders (MOGAD): A systematic review and meta-analysis. J Neuroimmunol 364:577812. https://doi.org/10.1016/j.jneuroim.2022.577812
Barreras P, Vasileiou ES, Filippatou AG, et al (2022) Long-term Effectiveness and Safety of Rituximab in Neuromyelitis Optica Spectrum Disorder and MOG Antibody Disease. Neurology 99:https://doi.org/10.1212/WNL.0000000000201260. https://doi.org/10.1212/WNL.0000000000201260
Durozard P, Rico A, Boutiere C et al (2020) Comparison of the response to rituximab between myelin oligodendrocyte glycoprotein and aquaporin-4 antibody diseases. Ann Neurol 87:256–266. https://doi.org/10.1002/ana.25648
Chen JJ, Huda S, Hacohen Y et al (2022) Association of maintenance intravenous immunoglobulin with prevention of relapse in adult myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol 79:518. https://doi.org/10.1001/jamaneurol.2022.0489
Ringelstein M, Ayzenberg I, Lindenblatt G, et al (2022) Interleukin-6 Receptor Blockade in Treatment-Refractory MOG-IgG–Associated Disease and Neuromyelitis Optica Spectrum Disorders. Neurol - Neuroimmunol Neuroinflammation 9:e1100. https://doi.org/10.1212/NXI.0000000000001100
Elsbernd PM, Hoffman WR, Carter JL, Wingerchuk DM (2021) Interleukin-6 inhibition with tocilizumab for relapsing MOG-IgG associated disorder (MOGAD): A case-series and review. Mult Scler Relat Disord 48:102696. https://doi.org/10.1016/j.msard.2020.102696
Havla J, Pakeerathan T, Schwake C et al (2021) Age-dependent favorable visual recovery despite significant retinal atrophy in pediatric MOGAD: how much retina do you really need to see well? J Neuroinflammation 18:121. https://doi.org/10.1186/s12974-021-02160-9
Satukijchai C, Mariano R, Messina S, et al (2022) Factors Associated With Relapse and Treatment of Myelin Oligodendrocyte Glycoprotein Antibody–Associated Disease in the United Kingdom. JAMA Netw Open 5:e2142780. https://doi.org/10.1001/jamanetworkopen.2021.42780
Bruijstens AL, Breu M, Wendel E-M et al (2020) E.U. paediatric MOG consortium consensus: Part 4—outcome of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol 29:32–40. https://doi.org/10.1016/j.ejpn.2020.10.007
Cobo-Calvo A, Sepúlveda M, D’Indy H et al (2019) Usefulness of MOG-antibody titres at first episode to predict the future clinical course in adults. J Neurol 266:806–815. https://doi.org/10.1007/s00415-018-9160-9
Pineles SL, Repka MX, Liu GT et al (2020) Assessment of pediatric optic neuritis visual acuity outcomes at 6 months. JAMA Ophthalmol 138:1253–1261. https://doi.org/10.1001/JAMAOPHTHALMOL.2020.4231
Carnero Contentti E, Marrodan M, Correale J (2021) Emerging drugs for the treatment of adult MOG-IgG-associated diseases. Expert Opin Emerg Drugs 26:75–78. https://doi.org/10.1080/14728214.2021.1919082
Gastaldi M, Foiadelli T, Greco G, et al (2022) Prognostic relevance of quantitative and longitudinal MOG antibody testing in patients with MOGAD: a multicentre retrospective study. J Neurol Neurosurg Psychiatry 0:jnnp-2022–330237. https://doi.org/10.1136/jnnp-2022-330237
Lopez-Chiriboga AS, Sechi E, Buciuc M et al (2020) Long-term outcomes in patients with myelin oligodendrocyte glycoprotein immunoglobulin g-associated disorder. JAMA Neurol 77:1575. https://doi.org/10.1001/jamaneurol.2020.3115
Deschamps R, Pique J, Ayrignac X et al (2021) The long-term outcome of MOGAD: An observational national cohort study of 61 patients. Eur J Neurol 28:1659–1664. https://doi.org/10.1111/ene.14746
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Abdullah Al-Ani has no conflict of interest. Dr. John J Chen served as a consultant to UCB and Horizon. Dr. Fiona Costello has received advisory board or speaker fees from Alexion, Novartis, Accure Therapeutics, Frequency Therapeutics, and Sanofi.
Ethical standard
An ethics statement is not applicable because this article is based on published literature.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Al-Ani, A., Chen, J.J. & Costello, F. Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): current understanding and challenges. J Neurol 270, 4132–4150 (2023). https://doi.org/10.1007/s00415-023-11737-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-023-11737-8