
Abstract
Background
Dimethyl fumarate and fingolimod are oral disease modifying treatments (DMTs) that reduce relapse activity and slow disability worsening in relapsing–remitting multiple sclerosis (RRMS).
Objective
To compare the effectiveness of dimethyl fumarate and fingolimod in a real-world setting, where both agents are licensed as a first-line DMT for the treatment of RRMS.
Methods
We identified patients with RRMS commencing dimethyl fumarate or fingolimod in the Swiss Federation for Common Tasks of Health Insurances (SVK) Registry between August 2014 and July 2019. Propensity score-matching was applied to select subpopulations with comparable baseline characteristics. Relapses and disability outcomes were compared in paired, pairwise-censored analyses.
Results
Of the 2113 included patients, 1922 were matched (dimethyl fumarate, n = 961; fingolimod, n = 961). Relapse rates did not differ between the groups (incident rate ratio 1.0, 95%CI 0.8–1.2, p = 0.86). Moreover, no difference in the hazard of 1-year confirmed disability worsening (hazard ratio [HR] 0.9; 95%CI 0.6–1.6; p = 0.80) or disability improvement (HR 0.9; 95%CI 0.6–1.2; p = 0.40) was detected. These findings were consistent both for treatment-naïve patients and patients switching from another DMT.
Conclusion
Dimethyl fumarate and fingolimod have comparable effectiveness regarding reduction of relapses and disability worsening in RRMS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Dimethyl fumarate and fingolimod are oral drugs, which are approved as first-line disease modifying treatment (DMT) for patients with relapsing–remitting multiple sclerosis (RRMS) in Switzerland [1, 2]. Both agents effectively reduce relapse activity and delay short-term disability worsening in patients with RRMS compared to placebo [3,4,5,6,7]. While the results from the pivotal phase III studies suggested a similar efficacy of dimethyl fumarate and fingolimod compared to placebo, a direct comparison with a head-to-head randomised controlled trial (RCT) has not been performed, yet. A matching-adjusted study of pooled data from four phase III trials found comparable effects on relapses and disability outcomes of the two compounds [8]. However, RCTs have specific inclusion and exclusion criteria and are hence of limited generalizability. An alternative strategy is the use of observational data with appropriate statistical methodology [9, 10]. To date, a few observational studies have compared the real-world effectiveness of dimethyl fumarate and fingolimod with conflicting results: While several studies demonstrated a similar effectiveness of both drugs on relapse rates [11,12,13,14,15], a recent a study from the international MSBase registry found that fingolimod was superior to dimethyl fumarate in suppressing relapses [16], and a multicentre study from Italy showed that fingolimod-treatment resulted in a lower risk of relapses and disability worsening compared to dimethyl fumarate in patients who had switched from a platform injectable drug [17].
Hence, our aim was to compare the effectiveness of dimethyl fumarate and fingolimod in different treatment scenarios in a real-world setting, in which regulatory requirements are identical for both compounds.
Methods
Database
Data were derived from the registry of the Swiss Federation for Common Tasks of Health Insurances (SVK) that controls reimbursement for DMTs in Switzerland [18] The SVK registry contains data from 15,552 patients who started DMTs between February 1995 and July 2019. Individual patient data are provided annually by board-certified neurologists and recorded in a prospective manner as part of the routine centralised reimbursement approval process. The data included standardised information on disease type, date of disease onset, relapse rate, and neurological status assessed using the Expanded Disability Status Scale (EDSS). The case record forms were checked for completeness and internal plausibility by a data manager at the SVK and entered into a database. Queries about missing or inconsistent data were issued by the SVK under the supervision of an independent physician, who was responsible for reimbursement decisions. The application for reimbursement was valid for 1 year. Patients were followed-up until they discontinued treatment or changed health insurance to a company that was not associated to the SVK. From 1995 to 2008 about 85% of all health insurance companies in Switzerland used the SVK forms for DMT reimbursement. This number decreased to about 65% in the year 2013 [19]. Written informed consent was obtained from all patients in accordance with the Declaration of Helsinki. The Ethics Committee of North-West and Central Switzerland confirmed that this study does not fall under the Swiss Federal Act on Research involving Human Beings, due to the administrative nature of the data and adequate anonymisation (Req-2019-00470).
Study population
For this analysis, we selected patients with RRMS who started or switched to treatment with dimethyl fumarate or fingolimod. The minimum required dataset consisted of complete data for MS disease type, date of disease onset, date of last relapse, the EDSS and Functional System (FS) scores and at least one subsequent on-treatment visit. For the analysis of 12-months confirmed disability worsening and disability improvement, at least two annual follow-up evaluations were required. To ensure the validity of the matching process, we restricted the analysis to patients who switched DMTs after August 2014 when dimethyl fumarate became available in Switzerland and excluded patients switching from fingolimod to dimethyl fumarate or vice versa [2].
Study endpoints
The primary study endpoints were the time to first relapse and annualised relapse rate (ARR). ARR was calculated as the annualised number of relapses between treatment initiation and censoring. As secondary study endpoints, we used disability worsening, which was defined as an increase of ≥ 1.5 points from an EDSS score of 0.0, ≥ 1.0 point from an EDSS score of 1.0–5.5 or ≥ 0.5 point from an EDSS score ≥ 6.0 confirmed in the following year and disability improvement, which was defined as a decrease of ≥ 1 EDSS step (≥ 1.5 points if baseline EDSS was ≤ 1.5 and ≥ 0.5 points if baseline EDSS was ≥ 6.0) also confirmed after at least 1 year [20]. Disease duration was calculated from the first demyelinating event.
Matching and statistical analysis
Matching and statistical analysis was conducted by P.B. using R [21]. The included patients were matched on their propensity for receiving dimethyl fumarate vs. fingolimod using the MatchIt package [22]. The propensity score was based on a multivariable logistic regression model with treatment allocation as the outcome variable and the demographic and clinical variables available to treating neurologists at the time of the treatment decision as the independent variables. These comprised age, gender, disease duration, baseline EDSS and FS scores, EDSS change in the preceding year, number of previous DMTs, time on first-line DMT, time since last visit, time since last relapse, number of relapses in the year before treatment switch and region in Switzerland.
Patients were matched in a 1:1 ratio using nearest neighbour matching within a calliper of 0.2 standard deviations of the propensity score. Patients who switched or discontinued treatment were censored. The pairwise on-treatment follow-up was determined as the shorter of the two individual follow-up periods for each matched patient pair (pairwise censoring) to control for attrition bias [10].
Time to relapse, EDSS worsening and improvement as well as treatment persistence were explored using Kaplan–Meier plots and analysed using marginal proportional hazards models, the cluster term indicating the matched patient pairs. The proportional hazards assumption was examined visually and by testing Schoenfeld’s residuals. After assessing the data distribution, the annualised relapse rate was estimated using a negative binomial regression model with the log of the follow-up time as offset variable.
The primary analysis included all eligible subjects, whereas the two secondary analyses were restricted to patients (A) on first-line and (B) on second-line treatment. In addition, we performed three sensitivity analyses: (1) matching with a calliper of 0.1, (2) omitting pairwise censoring and (3) also including patients starting fingolimod before August 2014.
Observed differences were considered significant if two-tailed p ≤ 0.05.
Results
We identified 2113 eligible patients treated with dimethyl fumarate or fingolimod. Of these, 1222 (58%) had sufficient follow up to analyse the secondary endpoints (Fig. 1). Baseline characteristics of the cohorts are shown in Table 1 and in Supplementary Table 1 for the dataset used to analyse disability outcomes. Several demographic factors and markers of disease activity differed between the unmatched patient groups. The logistic model used to estimate the propensity scores showed that fewer relapses in the year before treatment start, a higher number of previous DMTs as well as being treated in the Eastern region of Switzerland were associated with a higher probability of treatment with dimethyl fumarate (Supplementary Table 2).

CONSORT flowchart of patient disposition. DMF dimethyl fumarate, FTY fingolimod
The propensity-score matching procedure for the primary analysis retained 961 patients on dimethyl fumarate (93%) and 961 on fingolimod (89%). The matching procedure significantly improved the overall balance as indicated by the baseline characteristics and distribution of the propensity scores (Table 1, Supplementary Figure).
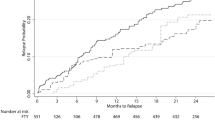
The median follow-up time after pairwise censoring was 0.9 years (interquartile range 0.8–1.8). We did not observe any difference in the time to first relapse between the dimethyl fumarate- and fingolimod-treated groups (HR 1.1, 95%CI 0.9–1.4, p = 0.41, Fig. 2a). Annualised relapse rates dropped in both the dimethyl fumarate and the fingolimod group without a significant between-group difference (incident rate ratio 1.0, 95%CI 0.9–1.5, p = 0.18, Fig. 2b). Moreover, we did not observe any differences regarding the hazard of one-year confirmed disability worsening (HR 0.9, 95%CI 0.6–1.6, p = 0.80, Fig. 3a) or of 1-year confirmed disability improvement (HR 0.9, 95%CI 0.6–1.2, p = 0.40; Fig. 3b) in matched patients with sufficient follow-up (dimethyl fumarate, n = 516; fingolimod, n = 516).

Relapse outcomes. a Proportion of patients free from relapses. Shaded areas represent 95% confidence intervals. b Annualised relapse rates at baseline, 12, 24 and 36 months. Error bars represent 95% confidence intervals. HR hazard ratio, EDSS Expanded Disability Status Scale, DMT disease modifying treatment

Disability outcomes and treatment persistence. a Proportion of patients free from disability worsening. b Proportion of patients with disability improvement. c Proportion of patients still on treatment. Shaded areas represent 95% confidence intervals
The secondary analyses in treatment-naïve patients and treatment switchers did also not show any differences between the groups, except for a higher chance of fingolimod-treated patients to experience confirmed disability improvement in the treatment-switchers (Table 2).
The sensitivity analyses confirmed the results of the primary analysis to full extent (Supplementary Table 3).
After 1 year, the proportion of patients continuing on treatment with fingolimod was higher than on dimethyl fumarate (87%, 95%CI 85–90% vs. 81%, 95%CI 76–83%, p = 0.002), but the difference was no longer statistically significant when considering the entire available follow-up time (HR 1.2, 95%CI 1.0–1.3, p = 0.06, Fig. 3c).
When analysing follow-up treatment after discontinuation of one of the drugs, we found that most patients switched from dimethyl fumarate to fingolimod and vice versa, followed by natalizumab (Table 3). Among the patients switched to natalizumab about two-thirds had experienced a relapse in the year prior to the switch and the average relapse rate was the highest in this group.
Discussion
In this observational, propensity-score matched analysis of a large MS cohort from Switzerland, we found that dimethyl fumarate and fingolimod had comparable effects on relapse activity and disability outcomes. This finding was consistent for treatment-naïve patients and those switching from another DMT. Treatment persistence was lower with dimethyl fumarate after 1 year but not significantly different over the whole follow-up period.
The pivotal phase III trials of dimethyl fumarate, DEFINE and CONFIRM, found a significant reduction in ARR by about 50% compared with placebo and by 29% compared with glatiramer acetate, respectively [6, 7]. In addition, the placebo-controlled DEFINE trial showed a 38% lower risk of 12-week confirmed disability progression for dimethyl fumarate, which could not be seen in comparison with glatiramer acetate. The efficacy of fingolimod was evaluated in three large phase III trials [3,4,5]. In the placebo controlled studies (FREEDOMS/FREEDOMS II), fingolimod was shown to be superior compared with placebo, reducing the ARR by 54% and 48%, respectively. The FREEDOMS trial also demonstrated a reduced risk of 6-months confirmed disability progression by 37% under fingolimod and the active-controlled TRANSFORMS trial could show a superior efficacy of fingolimod over interferon beta-1a with respect to relapse rates [3].
As no data from a randomised head-to-head trial between dimethyl fumarate and fingolimod was available, an attempt to assess the comparative efficacy of the two drugs was made by analysing pooled data from the CONFIRM, DEFINE and FREEDOMS/FREEDOMS II studies with a matching-adjusted, indirect approach [8]. This analysis found a comparable efficacy for both compounds on relapse activity and disability progression. However, the study was subject to unknown or unmeasured confounders similar to observational studies and also of limited generalisability.
An observational study from a single, large academic centre followed 358 patients treated with dimethyl fumarate and 317 treated with fingolimod over the period of 12 months [12]. Interestingly, the study also included a proportion of patients with progressive disease courses (26% and 12%, respectively) and in contrast to our study, rather few patients were on first-line dimethyl-fumarate (8%) and fingolimod treatment (5%), respectively. Both 1:1 propensity score-matching and Average Treatment effect on the Treated (ATT)-weighting based on the propensity score were used to mitigate indication bias. The study found no significant differences in ARR between the treatment groups and similar ARRs compared to phase III therapeutic trials. Moreover, measures of neurologic impairment, namely the Timed 25-Foot Walk and the 9-Hole Peg Test showed no difference between the groups. Also, no difference in a composite measure of MRI activity or new T2 lesions was found, although patients treated with dimethyl fumarate had a higher risk of developing gadolinium enhancing lesions by 12 months. Of note, patients treated with dimethyl fumarate experienced first relapses earlier than fingolimod patients and were more likely to discontinue treatment due to intolerability. A 24-months follow-up study from the same centre largely confirmed these results [13].
Another single centre study included 613 patients (dimethyl fumarate, n = 342, fingolimod n = 271) followed for up to 2 years, of which approximately 25% were treatment naïve and 17% had a progressive disease course [14]. The analysis was performed using logistic regression, propensity-score matching and ATT-weighting and found no difference between dimethyl fumarate and fingolimod on a composite outcome including clinical relapses and MRI activity, but increased odds of treatment discontinuation for dimethyl fumarate.
Recently, a pooled analysis of data from these two centres confirmed the results of the previously published studies showing comparable clinical and radiographic effectiveness profiles of both drugs, but demonstrating higher odds of dimethyl fumarate discontinuation compared with fingolimod [15].
A multicentre study from Italy analysed the effect of both compounds on No Evident Disease Activity (NEDA)-3 status, defined as being free from relapses, disability worsening and radiologic activity [17]. The study analysed 550 propensity-score matched patients with relapsing–remitting MS, of which 179 were treatment naïve and 389 switched from platform injectable drugs with a median follow-up of 18 months. While the analysis revealed a similar effectiveness of dimethyl fumarate and fingolimod on NEDA-3 and its subcomponents in the whole cohort, the subgroup analysis of treatment switchers found a 43% increased likelihood of achieving NEDA-3 status in the short-term follow-up for fingolimod compared with dimethyl fumarate driven by fewer relapses and less 6 months-confirmed disability progression.
The largest observational study to date originated from the MSBase registry and analysed 504 propensity-score matched patients on DMF and 1825 on fingolimod, with a mean ARR prior to inclusion of 0.8 and 0.9, respectively and a median follow-up of 1.3 years [16]. The authors demonstrated a lower hazard for relapses under fingolimod compared to DMF, but no difference between the two drugs in terms of confirmed disability worsening or improvement.
In our study, we found a comparable effectiveness of dimethyl fumarate and fingolimod on relapse activity in our analysis of 1922 propensity-score matched patients. In contrast to the study by Prosperini et al., our results were consistent for all subgroups irrespective of previous disease modifying treatment [17]. As the mean pre-switch ARR was substantially higher in this study compared to our subgroup of treatment switchers (1.3 vs. 0.6) one could speculate that fingolimod might have an advantage over DMF in patients with a higher disease activity than in our cohort. We also cannot exclude that unavailability of MRI data for the matching procedure has confounded our results to a certain degree. However, the absolute risk reduction for relapses reported in the study by Prosperini et al. [17] was only of a moderate magnitude (72% vs. 68%). Similarly, the study from MSBase resembling our cohort more closely in terms of baseline characteristics, showed only a very modest absolute risk reduction of approximately 0.06 relapses per year [16].
When analysing 1 year-confirmed disability worsening, we were not able to detect a difference between the two groups, in line with the analysis from MSBase [16]. While disability worsening measured by the EDSS was not analysed in the studies performed by Hersh et al., and Vollmer et al., [13,14,15] the Italian study found a lower risk of 6-months confirmed disability worsening for fingolimod vs. dimethyl fumarate in the whole cohort as well as in treatment switchers [17]. This discrepancy could be explained by our definition of disability worsening, requiring confirmation after at least 1 year, which has been shown to be less sensitive but more specific than shorter confirmation intervals [20].
We found that treatment discontinuation during the first year was slightly more frequent in the dimethyl fumarate than in the fingolimod group (19% vs. 13%) confirming findings from the pivotal RCTs as well as the previously mentioned observational studies. Unfortunately, information about reasons for treatment discontinuation were not documented in our registry. Of note, the difference between the groups diminished after the first year of treatment. Hence, we can assume that lack of tolerability of dimethyl fumarate early after treatment initiation was the strongest driver for discontinuation, as has been shown in previous studies [16, 17].
The main limitations of our study apart from the observational, non-randomised study design are the lack of MRI data and the relatively short median follow-up time of 1 year after pairwise censoring. Although MRI scans are routinely performed as required by clinical practice before treatment initiation, on-treatment and before planned treatment switch, MRI parameters are not recorded in the SVK registry in detail, so that we could not use MRI metrics in the matching process or as an outcome variable. As inclusion of MRI data is beyond the scope of the SVK registry, we plan to utilise the Swiss Multiple Sclerosis Cohort study (SMSC), which is currently performed at tertiary Swiss MS centres for future analysis of systematically acquired clinical data, standardised MRI and body fluid biomarkers (such as serum Neurofilament light chain), once patient numbers and follow-up time are sufficient [23]. In addition, the exclusion of patients without at least one follow-up visit may have introduced a bias towards equivalence between the two treatment groups. However, the number of patients excluded because of insufficient follow-up was relatively low (804; 15%), which argues against a pronounced effect. To mitigate the known treatment indication bias, we employed propensity score-matching. Unlike randomisation, this technique cannot eliminate bias due to unknown or unmeasurable confounders. However, this is unlikely to have a substantial effect on our overall conclusions, as sensitivity analyses with varying inclusion criteria confirmed the results of the primary analysis. We ensured comparison within the same treatment era by excluding patients who commenced fingolimod treatment before dimethyl fumarate was available. Pairwise censoring was applied to control for attrition bias. As yearly visits were mandatory, adjustment for reporting bias by taking-into account the frequency of clinical follow-up was not necessary. Generalisability of our study results was maximised by inclusion of a broad spectrum of patients.
In conclusion, our observational study adds further evidence to the comparable effectiveness of dimethyl fumarate and fingolimod in a real-world setting. Further analyses, ideally including MRI data are needed to assess the comparative long-term effectiveness of the two compounds.
Availability of data
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Swissmedic (2017) Gilenya, Kapseln (Fingolimod), https://www.swissmedic.ch/zulassungen/00153/00189/00200/00927/index.html?lang=de. Accessed 16 Nov 2017
Swissmedic (2017) Tecfidera, magensaftresistente Kapseln (Dimethylis fumaras)https://www.swissmedic.ch/zulassungen/00153/00189/00200/02374/index.html?lang=de. Accessed 16 Nov 2017
Cohen JA, Barkhof F, Comi G et al (2010) Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 362:402–415
Kappos L, Radue E-W, O’Connor P et al (2010) A Placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 362:387–401
Calabresi PA, Radue EW, Goodin D et al (2014) Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 13:545–556
Gold R, Kappos L, Arnold DL et al (2012) Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med 367:1098–1107
Fox RJ, Miller DH, Phillips JT et al (2012) Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med 367:1087–1097
Fox RJ, Chan A, Zhang A et al (2017) Comparative effectiveness using a matching-adjusted indirect comparison between delayed-release dimethyl fumarate and fingolimod for the treatment of multiple sclerosis. Curr Med Res Opin 33:175–183
Trojano M, Tintore M, Montalban X et al (2017) Treatment decisions in multiple sclerosis—insights from real-world observational studies. Nat Rev Neurol 13:105
Kalincik T, Butzkueven H (2016) Observational data: understanding the real MS world. Mult Scler. 22:1642–1648
Boster A, Nicholas J, Wu N et al (2017) Comparative effectiveness research of disease-modifying therapies for the management of multiple sclerosis: analysis of a large health insurance claims database. Neurol Ther 6:91–102
Hersh CM, Love TE, Cohn S et al (2016) Comparative efficacy and discontinuation of dimethyl fumarate and fingolimod in clinical practice at 12-month follow-up. Mult Scler Relat Disord 10:44–52
Hersh CM, Love TE, Bandyopadhyay A et al (2017) Comparative efficacy and discontinuation of dimethyl fumarate and fingolimod in clinical practice at 24-month follow-up. Mult Scler J Exp Transl Clin 3:2055217317715485
Vollmer B, Nair KV, Sillau SH, Corboy J, Vollmer T, Alvarez E (2017) Comparison of fingolimod and dimethyl fumarate in the treatment of multiple sclerosis: two-year experience. Mult Scler J Exp Transl Clin 3:2055217317725102
Vollmer B, Ontaneda D, Bandyopadhyay A et al (2018) Discontinuation and comparative effectiveness of dimethyl fumarate and fingolimod in 2 centers. Neurol Clin Pract 8:292–301
Kalincik T, Kubala Havrdova E, Horakova D et al (2019) Comparison of fingolimod, dimethyl fumarate and teriflunomide for multiple sclerosis. J Neurol Neurosurg Psychiatry. 90:458–468
Prosperini L, Lucchini M, Haggiag S et al (2018) Fingolimod vs dimethyl fumarate in multiple sclerosis: a real-world propensity score-matched study. Neurology 91:e153–e161
Lorscheider J, Benkert P, Lienert C et al (2018) Comparative analysis of natalizumab versus fingolimod as second-line treatment in relapsing-remitting multiple sclerosis. Mult Scler 24:777–785
Swiss Federation for Common Tasks of Health Insurances (2014) Annual report 2014. Solothurn, Switzerland. https://svk.org/
Kalincik T, Cutter G, Spelman T et al (2015) Defining reliable disability outcomes in multiple sclerosis. Brain 138:3287–3298
R Core Development Team (2011) R: A language and environment for statistical computing. In: Vienna, Austria: R Foundation for Statistical Computing
Ho D, Imai K, King G, Stuart EA (2011) MatchIt: nonparametric preprocessing for parametric causal inference. J Stat Softw 42:1–28
Disanto G, Benkert P, Lorscheider J et al (2016) The Swiss multiple sclerosis cohort-study (SMSC): a prospective swiss wide investigation of key phases in disease evolution and new treatment options. PLoS One 11:e0152347
Funding
Open access funding provided by University of Basel.
Author information
Authors and Affiliations
Contributions
JL designed the study, interpreted the data, drafted and revised the manuscript. PB contributed to design of the study, analysed and interpreted the data, and revised the manuscript for important intellectual content. CL contributed to data acquisition and revised the manuscript for important intellectual content. PH contributed to data acquisition and revised the manuscript for important intellectual content. JK revised the manuscript for important intellectual content. TD revised the manuscript for important intellectual content. LK contributed to interpretation of data and revised the manuscript for important intellectual content. ÖY contributed to design of the study, interpretation of the data, and reviewed the manuscript for important intellectual content.
Corresponding author
Ethics declarations
Conflicts of interest
JL has received research support from Innosuisse-Swiss Innovation Agency, Biogen and Novartis and served on advisory boards for Roche and Teva. PB reports no conflicts of interest. CL reports no conflicts of interest. PH is an employee of the Swiss Federation for Common Tasks of Health Insurances. TD received speaker fees, research support, travel support, and/or served on Advisory Boards or Steering Committees of Novartis Pharma, Merck Serono, Biogen, Teva, Bayer-Schering, GeNeuro, Mitsubishi Pharma, MedDay, Roche, and Genzyme. JK’s institution received research support from Swiss MS Society, Biogen, Novartis, Roche, Genzyme, and Merck Serono; he received research support from Bayer AG, Genzyme, Novartis, Roche Pharma (Schweiz) AG, Swiss National Research Foundation, ECTRIMS, University of Basel, and Swiss MS Society. The University Hospital Basel, as the employer of L.K., has received and dedicated to research support fees for board membership, consultancy or speaking, or grants in the past 3 years from Actelion, Advancell, Allozyne, Bayer, Bayhill, Biogen Idec, BioMarin, CSL Behring, Eli Lilly EU, Genmab, GeNeuro SA, Gianni Rubatto Foundation, Glenmark, Merck Serono, MediciNova, Mitsubishi Pharma, Novartis, Novartis Research Foundation, Novo Nordisk, Peptimmune, Roche, Roche Research Foundation, Santhera, Sanofi-Aventis, Swiss MS Society, Swiss National Research Foundation, Teva Pharmaceutical Industries Ltd, UCB, and Wyeth. Ö.Y.’s institution (University Hospital Basel) received honoraria for lectures from Teva, Novartis and Bayer Schering exclusively used for funding of research or educational courses.
Ethics approval
The Ethics Committee of North-West and Central Switzerland confirmed that this study does not fall under the Swiss Federal Act on Research involving Human Beings, due to the administrative nature of the data and adequate anonymisation (Req-2019-00470).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lorscheider, J., Benkert, P., Lienert, C. et al. Comparative analysis of dimethyl fumarate and fingolimod in relapsing–remitting multiple sclerosis. J Neurol 268, 941–949 (2021). https://doi.org/10.1007/s00415-020-10226-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-10226-6