
Abstract
Idiopathic basal ganglia calcification (IBGC) or primary familial brain calcification is a rare genetic condition characterized by an autosomal dominant inheritance pattern and the presence of bilateral calcifications in the basal ganglia, thalami, cerebellum and cerebral subcortical white matter. The syndrome is genetically and phenotypically heterogeneous. Causal mutations have been identified in four genes: SLC20A2, PDGFRB, PDGFB and XPR1. A variety of progressive neurological and psychiatric symptoms have been described, including cognitive impairment, movement disorders, bipolar disorder, chronic headaches and migraine, and epilepsy. Here we describe a family with a novel SLC20A2 mutation mainly presenting with neurological symptoms including cortical myoclonus and epilepsy. While epilepsy, although rare, has been reported in patients with IBGC associated with SLC20A2 mutations, cortical myoclonus seems to be a new manifestation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Idiopathic basal ganglia calcification (IBGC) or primary familial brain calcification (PFBC) is a rare autosomal dominant genetic condition characterized by the hallmark of bilateral calcifications in the basal ganglia, thalami, cerebellum and subcortical white matter. The clinical presentation can be widely heterogeneous even among members of the same family. While the imaging phenotype shows 100% penetrance (all patients carrying mutation have calcifications by the age of 40–50 years), the clinical phenotype only shows 61% penetrance [11, 19, 24]. The clinical phenotype typically includes neurological and psychiatric features [11, 18, 19, 24].
The condition is also heterogeneous at the genetic level, mutations in four genes have been reported: SLC20A2, PDGFRB, PDGFB and XPR1 [16, 19]. Mutations in SLC20A2 seem to be the major cause of IBGC, accounting for as many as 41% of the familial cases [11]. This gene encodes the inorganic phosphate (Pi) transporter PiT2 which is expressed ubiquitously in the brain, mainly in the sites where calcifications tend to occur (the globus pallidus, thalamus and cerebellum) [6]. Slc20a2 knockout mice show elevated cerebrospinal fluid (CSF) [Pi], suggesting that the sodium-dependent phosphate transporter-2 (PiT2) is essential for [Pi] export from the CSF, and thus regulation of CSF [Pi] [13].
Mutations or deletion in SLC20A2 are associated with psychiatric conditions (56.9%) including bipolar disorder, mood disturbances and psychosis [2, 19] and neurological symptoms mainly affecting cognitive abilities (58.8%) and motor function (54.9%), including dystonia, chorea, parkinsonism and ataxia [19]. Migraine seems also to be common, but the specific prevalence is difficult to ascertain given the high prevalence in the general population [20]. Epilepsy has been reported more rarely (7.8%) [17, 19]. Given the co-occurrence of movement disorders, neurophysiology (video-polygraphy) is essential to classify epilepsy and differentiate seizures from abnormal movements [14].
Here we report on a family affected by IBGC, showing an autosomal dominant inheritance and a newly reported SLC20A2 mutation. The clinical presentation of this family is complex and includes cortical myoclonus and epilepsy as the main features. While epilepsy has been reported in seven cases with IBGC and SLC20A2 mutations [1, 8, 11, 14, 25, 30], cortical myoclonus has not been reported to our knowledge.
Methods
The study was approved by the appropriate ethics committee. Written informed consent was provided by the family.
The proband was clinically evaluated through a comprehensive neurological and psychiatric examination at the National Hospital for Neurology and Neurosurgery, London. Brain Computed Tomography (CT) scan, electroencephalography (EEG), and peripheral nerve assessment were performed. Genomic DNA of the patient and her mother was extracted from peripheral blood. The father was not available for sampling.
An exonic library was amplified by Nextera Exome Kit (Illumina), and sequenced on a HiSeq 2500 platform (Illumina). Alignment of reads was carried out against the reference human genome (GRCh37). Candidate genes were analyzed by vcf tools [7] and annotated using Annovar [28]. Sanger sequencing was performed to confirm the variant.
Family description
This is a two-generation family of British origin (Fig. 1a). The proband (II:3) is a 22-year old woman, the third child of unrelated parents. She was born at term through a natural delivery after an uneventful pregnancy. The psychomotor milestones were normal. At the age of eight she was noticed to have myoclonic jerks affecting her hands and legs, particularly in the morning. The interictal EEG at that time showed bursts of bilateral spike and slow wave activity. The jerks were resistant to topiramate and levetiracetam (LEV) treatment. A concomitant slowing in school performance was reported. From the age of 11 she experienced anxiety and phobias. At the age of 14, she underwent a comprehensive psychiatric evaluation for autism and intellectual disabilities: she was assessed using the Autism Diagnostic Observation Schedule-Generic (ADOS-G) and the Wechsler Intelligence Scale for Children, 4th edition (WISC-IV). The diagnostic criteria for Autism Spectrum Disorders were not met, although social communication and social interaction difficulties were present. Her IQ fell within the low average range. At the age of 14 she had her first GTCS and these recurred every few months. Around the same age, she also developed dystonic attacks involving her upper limbs and spreading to the neck and torso. They could last from 1 h up to 24 h. These involuntary movements worsened her mood disturbances and had a significant impact on her mobility, requiring her to use a wheelchair when not at home.

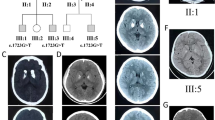
a pedigree of the family; filled symbols indicate subjects with symptoms; ± indicates screened individuals with heterozygous SLC20A2 mutation; b CT scans of the proband showing calcification of the basal ganglia and caudate nuclei. Further diffuse calcification is noted within the region of the centrum semi-ovale on the right; c electropherogram from the proband (lower box) and her mother (upper box)
At the age of 18, she was first admitted to our hospital and underwent a comprehensive evaluation. Generalized tonic–clonic seizures (GTCSs) had ceased, but she had very frequent dystonic attacks. These affected all four limbs, her head, back, and abdomen. Sometimes these were triggered by environmental light and were always preceded by generalized jerks and a feeling of fear. On admission, she was on treatment with valproate (VPA) 1100 mg/day, LEV 1000 mg/day and clonazepam 3 mg/day.
On examination, she was able to walk unassisted but preferred to use a wheelchair outdoors. Her gait was slightly ataxic, without upper limb ataxia, but rare myoclonic upper limbs jerks were noted. Cogwheel rigidity was present in both arms.
Psychometry revealed a low average score on Verbal Comprehension (85), Perceptual Reasoning (84) and working memory (83) subscales and a borderline score in the processing speed (76) subscale of the WAIS-IV (Wechsler Adult Intelligence Scale). The full scale IQ score was 79.
CSF, including testing for autoantibodies associated with encephalitic presentations, was unremarkable. Blood and urine organic acids went normal. Serum calcium and parathyroid hormone levels were normal. A brain MRI showed bilateral symmetrical signal abnormality of the globus pallidus, head of caudate and nucleus accumbens, consistent with abnormal mineralization.
At the age of 21, a CT scan was performed, showing calcification of the basal ganglia and caudate nuclei. Further diffuse calcification was noted within the region of the centrum semi-ovale on the right (Fig. 1b).
The awake EEG showed brief high voltage spikes and slow waves. Several episodes characterized by four limb shaking with retained consciousness were captured, without any EEG correlate, suggesting these episodes were not epileptic.
A multichannel EEG-EMG recording showed a pattern of frequent myoclonic jerks of short duration with craniocaudal progression and intermittent silence of muscle activity, suggestive of positive and negative cortical myoclonus (cortical tremor; Fig. 2a, b). A giant SEP was also found supporting the presence of cortical hyperexcitability (Fig. 2c).

Neurophysiology study of the proband and her brother II:2. a, b EEG-EMG recording of the proband showing positive and negative myoclonic jerks when extending arms against gravity. a The trace (1.0 s/division) shows the ongoing 20 Hz myoclonic jerks; b the trace (0.1 s/division) demonstrates craniocaudal progression of atonic periods on surface EMG (bipolar EEG channels; EMG channels: ROrbOr right orbicularis oris, LBicep left bicep, REI right extensor indicis, RECR right extensor carpi radialis, RRectF right rectus femoris). c Somatosensory evoked potential of the proband, recorded from the median nerve showing a giant evoked potential (P25–N35 = 52 µV) at the scalp electrodes (C4-Fz). d, e EEG-EMG recording of the brother II:2 showing positive and negative myoclonic jerks when extending arms against gravity. d The trace (1.0 s/division) shows the ongoing 20 Hz myoclonic jerks; e the trace (0.1 s/division) demonstrates craniocaudal progression and atonic periods on surface EMG channels. [EEG channels (F4, FC4, Cz, FC3, F3, FPZ) referenced to linked earlobes]; EMG channels top–bottom: LDeltoi left deltoid, LBicep left bicep, LECR left extensor carpi radialis, LFDIO left first dorsal interosseous, LRectF left rectus femoris, LTibAn left tibialis anterior). f Evoked potential recording showing ‘giant’ (P25–N35 = 25 µV) response at the scalp electrodes (C3-Fz) and cortical-reflex (C-reflex, 50.6 ms) from the surface abductor pollicis brevis electrode
The eldest brother (II:1) was born by natural delivery after an uneventful pregnancy; he is now 27 years old. He had normal developmental milestones. He was diagnosed with high-functioning social communication disorder (Asperger’s Syndrome) at the age of 12 years. He developed myoclonic jerks at the age of 11 years. At that time, EEG showed bilateral spike and polyspike and wave activity; these abnormalities increased on photic stimulation at flash frequencies between 15 and 20 Hz. He also reported blackouts, but absences were never clinically observed nor caught on EEG recording. During treatment with carbamazepine (CBZ) he had GTCSs. He was then switched to VPA (1600 mg/day) with a significant reduction of the jerks. However, because of weight gain, VPA had to be discontinued, and was substituted with LEV (up to 1500 mg/day). He underwent a brain MRI twice (13 years old and 22 years old): both exams were reportedly normal.
The second brother (II:2) is now 24 years old. He had normal developmental milestones. At the age of 10 years, he had his first nocturnal GTCS and from the age of 12 he noted small myoclonic jerks of his hands. He was initially treated with CBZ, which failed to control seizures and worsened the jerks. He was then switched to VPA; LEV was added as seizure control was not complete. EEG performed at the age of 11 showed bursts of high voltage poly-spike and slow wave discharges, more frequent during drowsiness and during intermittent photic stimulation. From the age of 17 he had daytime GTCS and myoclonic jerks worsened. Clonazepam (up to 2 mg/day) was added to VPA (1200 mg/day) and LEV (500 mg/day), but myoclonus has never been controlled. Propranolol (40 mg/day) was added at the age of 20. A brain MRI performed at the age of 18 was normal. A neurophysiology study was performed at the age of 23 including an EEG-EMG (Fig. 2d, e). No myoclonic jerks were seen at rest. When holding his arms against gravity and during action, 20 Hz myoclonic jerks lasting about 20 ms, and episodes of sudden loss of tone of about 100 ms, were recorded in all the EMG channels. These abnormal findings together with a positive C-reflex and giant SSEP, were consistent with cortical myoclonus (Fig. 2f).
The mother I:2 has never reported psychiatric nor neurological symptoms.
Genetic results
A heterozygous mutation in SLC20A2 was identified in two members (see Fig. 1a: proband II:3 and mother I:2). DNA was not available from father and brothers (Fig. 1a: II:1 and II:1, II:2, respectively).
The deletion (c.1583delA) located in exon 9 causes a frameshift (p.Q528fsD) (see Fig. 1c) predicting a truncated protein. This mutation is absent from public databases (ExAc, Exome Variant Server, 1000Genomes, gnomAD) and was predicted as damaging by SIFT. Polyphen-2 is only available for amino acid substitutions; it is not available for this mutation but the predicted CADD score is 35. Sanger sequencing confirmed the mutation in both individuals. The screening of a curated list of 198 epilepsy-related genes did not reveal any other pathogenic variants in this family. Total coverage was calculated for both whole exomes. The percentage of exonic bases with more than 10 × coverage was 69.4% in the proband and 84.7% in the mother.
Discussion
Mutations in SLC20A2 account for the majority of cases with IBGC [11, 19]. This condition is characterized by phenotypic variability spanning from lack of symptoms to a variety of progressive psychiatric and neurological symptoms [19].
Epilepsy has been reported only in seven such cases [1, 8, 11, 14, 25, 30], but only two reports focused on the epilepsy phenotype [8, 14]. Fjaer et al. reported two siblings with GTCSs easily controlled on antiepileptic medications. Both the patients also had a second pathogenic variant, in CHRNB2, a known epilepsy gene that might have contributed to the phenotype [8]. Knowles et al. reported refractory focal epilepsy in a pediatric patient who also had another variant, of unknown significance, in SCN2A. Thus the SLC20A2 mutation contribution to the phenotype in this case remains uncertain. This individual also presented with dystonic events affecting the same hand that was also affected by seizures; the authors suggested that an accurate neurophysiology assessment was essential to classify epilepsy and differentiate it from the motor disorder [14].
A progressive myoclonus phenotype has been reported by Lamquet et al. in a patient bearing a likely pathogenic variant in SLC20A2 [15]. However the authors did not specify whether the myoclonus was cortical, subcortical or spinal in origin. Their investigations (EEG and EMG) were uninformative and polygraphy was not performed.
Here we describe a two generations family bearing a newly-reported SLC20A2 mutation. The screening of other candidate epilepsy genes did not reveal other pathogenic variants in this family.
The genetic analysis could only be performed for the proband and her mother. Her mother declined to undergo brain CT scan and did not volunteer any clinical symptoms, thus falling into the category of putatively incomplete penetrance [11, 19]. The proband’s two brothers declined genetic analysis; however, they did present with very similar clinical symptoms. They both underwent brain MRI that failed to show calcium deposition. This does not exclude the IBGC diagnosis as the exam was performed at the age of 18 and 22, respectively, while patients with this condition can first show basal ganglia calcification as late as at 40–50 years old [11, 18]. The brother (II:2) who had neurophysiological evaluation had findings consistent with the presence of cortical myoclonus.
The clinical presentation in this family is complex and includes both psychiatric and neurological conditions. We have focused on the neurological presentation as the presence of dystonia, cortical myoclonus and epilepsy in the proband proved challenging for both diagnosis and treatment strategies. To the best of our knowledge, cortical myoclonus has not been reported in IBGC before, possibly due to association with a new mutation in this pedigree, but possibly also because other cases may have been misdiagnosed with tremor considered part of a parkinsonian presentation. In our proband and in one of her brothers who agreed to testing, the EEG-EMG and the SEP analysis disclosed a myoclonus of cortical origin. The condition familial adult myoclonic epilepsy (FAME) is an autosomal dominant syndrome described in more than 70 families to date. Beyond myoclonus and seizures, psychiatric comorbidities seem to occur in this condition and a slow progression has also been described [3, 4]. Five different loci have been disclosed including one on chromosome 8 (8q24) [23] in Japanese families. The exact genetic defect reported for the Italian, Indian, French and Japanese families is similar and affects the non-coding DNA [5, 9, 12, 29]. For the Japanese families an expansion of an intronic ATTTT pentamer coupled with an insertion of a repeated ATTTC pentamer within the genes SAMD12, TNRC6A, RAPGEF2 has been identified [12]. SLC20A2 is located on 8p11.21, and basal ganglia calcification has never been reported in families with FAME [22]. Thus we do not consider our family has FAME.
Whilst the progressive nature may be reminiscent of some progressive myoclonic epilepsies (PMEs), these are usually associated with autosomal recessive inheritance. Franceschetti et al. reported the phenotypes of patients with PME of undetermined causes, showing they varied widely [10], but none included dystonia or brain calcification as seen in our kindred.
In our cases, seizures were an early symptom in all siblings, with an onset in childhood; seizures were not reported in their mother. Seizures were generalized, myoclonic or tonic–clonic, usually responded to VPA or LEV and worsened under CBZ. The proband presented with other events in adulthood, shown to be not epileptic. Some of them were clinically dystonic attacks, while some others were psychogenic. Myoclonus, on the contrary, was resistant to AED medication or only partially controlled at least in the proband.
The mutation found in our proband affects SLC20A2, a gene that account for the majority of cases with IBGC. This gene encodes for the inorganic phosphate (Pi) transporter PiT2, which is expressed in the whole brain, and mainly in the sites where calcifications tends to occur mostly, namely the globus pallidus, thalamus and cerebellum [6]. In the mouse brain PiT-2 has been detected in astrocytes, endothelial cells and neurons. SLc20a2 Knockout mice show elevated CSF [Pi] suggesting that PiT2 is essential to export Pi from the CSF and thus to regulate the [Pi] in cerebrospinal fluid (CSF) [13]. Although we have not demonstrated directly the involvement of this transporter into the pathophysiology of myoclonus and epilepsy, we can hypothesize that the local cortical environmental ion balance might be altered and responsible for the seizures and myoclonus, or that the effect is indirect, following calcification. Indeed, excess phosphate is detrimental to glycogen structure, and in Lafora disease, there is over-accumulation of covalent phosphate in glycogen leading to a severe progressive myoclonus [26]. Moreover, myoclonus has been reported in basal ganglia calcifications of various other causes (for instance associated with mitochondrial encephalomyopathies, FORL1 deficiency) [21, 27].
Future studies will help explain why calcification is limited to specific parts of the brain, and the functional impact of the disordered phosphate homeostasis in the brain”.
In conclusion, we report one individual with SLC20A2 mutation and a possibly associated new clinical presentation including cortical myoclonus. The variety of neurological symptoms and the overlapping psychiatric manifestation can prove challenging for diagnoses and management. The potential co-occurrence of different paroxysmal events, including epilepsy, dystonia, tremor and cortical myoclonus requires meticulous neurophysiology assessment to elucidate the origin of the events and to offer the best management.
References
Brighina L, Saracchi E, Ferri F, Gagliardi M, Tarantino P, Morzenti S, Musarra M, Patassini M, Annesi G, Ferrarese C (2014) Fahr’s disease linked to a novel SLC20A2 gene mutation manifesting with dynamic aphasia. Neurodegener Dis 14:133–138
Chiriaco C, Novellino F, Salsone M, Gagliardi M, Morelli M, Quattrone A (2018) Neuropsychological heterogeneity in patients with primary familial brain calcification due to a novel mutation in SLC20A2. Neurol Sci 39:379–380
Coppola A, Caccavale C, Santulli L, Balestrini S, Cagnetti C, Licchetta L, Esposito M, Bisulli F, Tinuper P, Provinciali L, Minetti C, Zara F, Striano P, Striano S (2016) Psychiatric comorbidities in patients from seven families with autosomal dominant cortical tremor, myoclonus, and epilepsy. Epilepsy Behav 56:38–43
Coppola A, Santulli L, Del Gaudio L, Minetti C, Striano S, Zara F, Striano P (2011) Natural history and long-term evolution in families with autosomal dominant cortical tremor, myoclonus, and epilepsy. Epilepsia 52:1245–1250
Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, Coppola A, Licchetta L, Franceschetti S, Suppa A, Wenger A, Mei D, Pendziwiat M, Kaya S, Delledonne M, Straussberg R, Xumerle L, Regan B, Crompton D, van Rootselaar AF, Correll A, Catford R, Bisulli F, Chakraborty S, Baldassari S, Tinuper P, Barton K, Carswell S, Smith M, Berardelli A, Carroll R, Gardner A, Friend KL, Blatt I, Iacomino M, Di Bonaventura C, Striano S, Buratti J, Keren B, Nava C, Forlani S, Rudolf G, Hirsch E, Leguern E, Labauge P, Balestrini S, Sander JW, Afawi Z, Helbig I, Ishiura H, Tsuji S, Sisodiya SM, Casari G, Sadleir LG, van Coller R, Tijssen MAJ, Klein KM, van den Maagdenberg A, Zara F, Guerrini R, Berkovic SF, Pippucci T, Canafoglia L, Bahlo M, Striano P, Scheffer IE, Brancati F, Depienne C, Gecz J (2019) Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat Commun 10:4920
da Silva RJ, Pereira IC, Oliveira JR (2013) Analysis of gene expression pattern and neuroanatomical correlates for SLC20A2 (PiT-2) shows a molecular network with potential impact in idiopathic basal ganglia calcification (“Fahr’s disease”). J Mol Neurosci 50:280–283
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, Genomes Project Analysis G (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Fjaer R, Brodtkorb E, Oye AM, Sheng Y, Vigeland MD, Kvistad KA, Backe PH, Selmer KK (2015) Generalized epilepsy in a family with basal ganglia calcifications and mutations in SLC20A2 and CHRNB2. Eur J Med Genet 58:624–628
Florian RT, Kraft F, Leitao E, Kaya S, Klebe S, Magnin E, van Rootselaar AF, Buratti J, Kuhnel T, Schroder C, Giesselmann S, Tschernoster N, Altmueller J, Lamiral A, Keren B, Nava C, Bouteiller D, Forlani S, Jornea L, Kubica R, Ye T, Plassard D, Jost B, Meyer V, Deleuze JF, Delpu Y, Avarello MDM, Vijfhuizen LS, Rudolf G, Hirsch E, Kroes T, Reif PS, Rosenow F, Ganos C, Vidailhet M, Thivard L, Mathieu A, Bourgeron T, Kurth I, Rafehi H, Steenpass L, Horsthemke B, Consortium F, LeGuern E, Klein KM, Labauge P, Bennett MF, Bahlo M, Gecz J, Corbett MA, Tijssen MAJ, van den Maagdenberg A, Depienne C (2019) Unstable TTTTA/TTTCA expansions in MARCH6 are associated with familial adult myoclonic epilepsy type 3. Nat Commun 10:4919
Franceschetti S, Michelucci R, Canafoglia L, Striano P, Gambardella A, Magaudda A, Tinuper P, La Neve A, Ferlazzo E, Gobbi G, Giallonardo AT, Capovilla G, Visani E, Panzica F, Avanzini G, Tassinari CA, Bianchi A, Zara F, LsgoP Collaborative (2014) Progressive myoclonic epilepsies: definitive and still undetermined causes. Neurology 82:405–411
Hsu SC, Sears RL, Lemos RR, Quintans B, Huang A, Spiteri E, Nevarez L, Mamah C, Zatz M, Pierce KD, Fullerton JM, Adair JC, Berner JE, Bower M, Brodaty H, Carmona O, Dobricic V, Fogel BL, Garcia-Estevez D, Goldman J, Goudreau JL, Hopfer S, Jankovic M, Jauma S, Jen JC, Kirdlarp S, Klepper J, Kostic V, Lang AE, Linglart A, Maisenbacher MK, Manyam BV, Mazzoni P, Miedzybrodzka Z, Mitarnun W, Mitchell PB, Mueller J, Novakovic I, Paucar M, Paulson H, Simpson SA, Svenningsson P, Tuite P, Vitek J, Wetchaphanphesat S, Williams C, Yang M, Schofield PR, de Oliveira JR, Sobrido MJ, Geschwind DH, Coppola G (2013) Mutations in SLC20A2 are a major cause of familial idiopathic basal ganglia calcification. Neurogenetics 14:11–22
Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, Toyoshima Y, Kakita A, Takahashi H, Suzuki Y, Sugano S, Qu W, Ichikawa K, Yurino H, Higasa K, Shibata S, Mitsue A, Tanaka M, Ichikawa Y, Takahashi Y, Date H, Matsukawa T, Kanda J, Nakamoto FK, Higashihara M, Abe K, Koike R, Sasagawa M, Kuroha Y, Hasegawa N, Kanesawa N, Kondo T, Hitomi T, Tada M, Takano H, Saito Y, Sanpei K, Onodera O, Nishizawa M, Nakamura M, Yasuda T, Sakiyama Y, Otsuka M, Ueki A, Kaida KI, Shimizu J, Hanajima R, Hayashi T, Terao Y, Inomata-Terada S, Hamada M, Shirota Y, Kubota A, Ugawa Y, Koh K, Takiyama Y, Ohsawa-Yoshida N, Ishiura S, Yamasaki R, Tamaoka A, Akiyama H, Otsuki T, Sano A, Ikeda A, Goto J, Morishita S, Tsuji S (2018) Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet 50:581–590
Jensen N, Autzen JK, Pedersen L (2016) Slc20a2 is critical for maintaining a physiologic inorganic phosphate level in cerebrospinal fluid. Neurogenetics 17:125–130
Knowles JK, Santoro JD, Porter BE, Baumer FM (2018) Refractory focal epilepsy in a paediatric patient with primary familial brain calcification. Seizure 56:50–52
Lamquet S, Ramos EM, Legati A, Coppola G, Hemelsoet D, Vanakker OM (2019) A likely pathogenic variant in the SLC20A2 gene presenting with progressive myoclonus. Ann Clin Transl Neurol 6:605–609
Legati A, Giovannini D, Nicolas G, Lopez-Sanchez U, Quintans B, Oliveira JR, Sears RL, Ramos EM, Spiteri E, Sobrido MJ, Carracedo A, Castro-Fernandez C, Cubizolle S, Fogel BL, Goizet C, Jen JC, Kirdlarp S, Lang AE, Miedzybrodzka Z, Mitarnun W, Paucar M, Paulson H, Pariente J, Richard AC, Salins NS, Simpson SA, Striano P, Svenningsson P, Tison F, Unni VK, Vanakker O, Wessels MW, Wetchaphanphesat S, Yang M, Boller F, Campion D, Hannequin D, Sitbon M, Geschwind DH, Battini JL, Coppola G (2015) Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat Genet 47:579–581
Lemos RR, Oliveira MF, Oliveira JR (2013) Reporting a new mutation at the SLC20A2 gene in familial idiopathic basal ganglia calcification. Eur J Neurol 20:e43–e44
Nicolas G, Charbonnier C, de Lemos RR, Richard AC, Guillin O, Wallon D, Legati A, Geschwind D, Coppola G, Frebourg T, Campion D, de Oliveira JR, Hannequin D, Collaborators from the French IsG (2015) Brain calcification process and phenotypes according to age and sex: lessons from SLC20A2, PDGFB, and PDGFRB mutation carriers. Am J Med Genet B Neuropsychiatr Genet 168:586–594
Nicolas G, Pottier C, Charbonnier C, Guyant-Marechal L, Le Ber I, Pariente J, Labauge P, Ayrignac X, Defebvre L, Maltete D, Martinaud O, Lefaucheur R, Guillin O, Wallon D, Chaumette B, Rondepierre P, Derache N, Fromager G, Schaeffer S, Krystkowiak P, Verny C, Jurici S, Sauvee M, Verin M, Lebouvier T, Rouaud O, Thauvin-Robinet C, Rousseau S, Rovelet-Lecrux A, Frebourg T, Campion D, Hannequin D, French ISG (2013) Phenotypic spectrum of probable and genetically-confirmed idiopathic basal ganglia calcification. Brain 136:3395–3407
Rubino E, Giorgio E, Gallone S, Pinessi L, Orsi L, Gentile S, Duca S, Brusco A (2014) Novel mutation of SLC20A2 in an Italian patient presenting with migraine. J Neurol 261:2019–2021
Seki A, Nishino I, Goto Y, Maegaki Y, Koeda T (1997) Mitochondrial encephalomyopathy with 15915 mutation: clinical report. Pediatr Neurol 17:161–164
Striano P, Zara F (2016) Autosomal dominant cortical tremor, myoclonus and epilepsy. Epilept Disord 18:139–144
Striano P, Zara F, Striano S (2005) Autosomal dominant cortical tremor, myoclonus and epilepsy: many syndromes, one phenotype. Acta Neurol Scand 111:211–217
Tadic V, Westenberger A, Domingo A, Alvarez-Fischer D, Klein C, Kasten M (2015) Primary familial brain calcification with known gene mutations: a systematic review and challenges of phenotypic characterization. JAMA Neurol 72:460–467
Taglia I, Bonifati V, Mignarri A, Dotti MT, Federico A (2015) Primary familial brain calcification: update on molecular genetics. Neurol Sci 36:787–794
Tagliabracci VS, Heiss C, Karthik C, Contreras CJ, Glushka J, Ishihara M, Azadi P, Hurley TD, DePaoli-Roach AA, Roach PJ (2011) Phosphate incorporation during glycogen synthesis and Lafora disease. Cell Metab 13:274–282
Toelle SP, Wille D, Schmitt B, Scheer I, Thony B, Plecko B (2014) Sensory stimulus-sensitive drop attacks and basal ganglia calcification: new findings in a patient with FOLR1 deficiency. Epilept Disord 16:88–92
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164
Yeetong P, Pongpanich M, Srichomthong C, Assawapitaksakul A, Shotelersuk V, Tantirukdham N, Chunharas C, Suphapeetiporn K, Shotelersuk V (2019) TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain 142:3360–3366
Zhang Y, Guo X, Wu A (2013) Association between a novel mutation in SLC20A2 and familial idiopathic basal ganglia calcification. PLoS One 8:e57060
Acknowledgements
We thank the family for giving us permission to publish their data. This work was partly undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme. We are grateful for funding support from the Epilepsy Society and the Muir Maxwell Trust.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
On behalf of all authors, Sanjay M. Sisodiyastates that there is no conflict of interest.
Ethical standards
The study was conducted in accordance with the ethical standards of the University College London according to the declaration of Helsinki.
Informed consent
Patients provided written informed consent for participation in the study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Coppola, A., Hernandez-Hernandez, L., Balestrini, S. et al. Cortical myoclonus and epilepsy in a family with a new SLC20A2 mutation. J Neurol 267, 2221–2227 (2020). https://doi.org/10.1007/s00415-020-09821-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-09821-4