
Abstract
Introduction
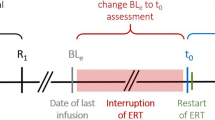
Although not curative, enzyme replacement therapy (ERT) with recombinant human acid alpha-glucosidase enzyme has shown to be effective in the treatment of late-onset Pompe disease (LOPD). For this potentially life-long treatment, little is known on the clinical effect of cessation and resuming ERT. Due to a Swiss supreme court decision on ERT reimbursement, a temporary stop of ERT occurred in our study population. The aim of this study was to report the 36-months follow-up assessments after resuming ERT.
Methods
After resuming ERT, seven patients suffering from genetically and enzymatically confirmed LOPD had periodic, mandatory, prospective assessments of pulmonary function tests, muscle strength summary scores, distances walked in timed walking tests, and patient-reported questionnaires. Data were statistically analyzed for significant differences between time points at ERT cessation, at ERT resuming, and 36 months thereafter.
Results
After resuming ERT forced vital capacity (p = 0.007) and distance walked in the 6 min walk test (6-MWT, p = 0.011) significantly increased at 36 months. Compared to before ERT cessation, distance walked in 6-MWT at 36 months still remained significantly lower (p = 0.005). Self-reported scores in the fatigue severity scale significantly declined at 36 months after resuming ERT (p = 0.019). No other functional or reported parameter significantly changed at 36 months after resuming ERT.
Conclusions
Our data suggests that long-term interruption of ERT in LOPD may lead to deterioration of clinical meaningful parameters and quality of life. In addition, a clinical restoration after ERT cessation is possible for most of the LOPD patients within a 36 months follow-up.




Similar content being viewed by others

References
Raben N, Wong A, Ralston E, Myerowitz R (2012) Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten. Am J Med Genet C Semin Med Genet 160C:13–21
Raben N, Roberts A, Plotz PH (2007) Role of autophagy in the pathogenesis of Pompe disease. Acta Myol Myopathies Cardiomyopath Off J Mediter Soc Myol 26:45–48
Chan J, Desai AK, Kazi ZB et al (2017) The emerging phenotype of late-onset Pompe disease: a systematic literature review. Mol Genet Metab 120:163–172
Kishnani PS, Corzo D, Leslie ND et al (2009) Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res 66:329–335
Kuperus E, Kruijshaar ME, Wens SCA et al (2017) Long-term benefit of enzyme replacement therapy in Pompe disease: a 5-year prospective study. Neurology 89:2365–2373
Kishnani PS, Corzo D, Nicolino M et al (2007) Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 68:99–109
van der Ploeg AT, Clemens PR, Corzo D et al (2010) A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 362:1396–1406
Hundsberger T, Rohrbach M, Kern L, Rösler KM (2013) Swiss national guideline for reimbursement of enzyme replacement therapy in late-onset Pompe disease. J Neurol 260:2279–2285
(2010) BGE 136 V 395 see https://www.bger.ch
Hundsberger T, Rösler KM, Findling O (2014) Cessation and resuming of alglucosidase alfa in Pompe disease: a retrospective analysis. J Neurol 261:1684–1690
Bonita R, Beaglehole R (1988) Recovery of motor function after stroke. Stroke 19:1497–1500
ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories (2002) ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 166:111–117
Duncan PW, Sullivan KJ, Behrman AL et al (2007) Protocol for the Locomotor Experience Applied Post-stroke (LEAPS) trial: a randomized controlled trial. BMC Neurol 7:39
Krupp LB, LaRocca NG, Muir-Nash J, Steinberg AD (1989) The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch Neurol 46:1121–1123
Merkies ISJ, Schmitz PIM, Van Der Meché FGA et al (2002) Psychometric evaluation of a new handicap scale in immune-mediated polyneuropathies. Muscle Nerve 25:370–377
Strothotte S, Strigl-Pill N, Grunert B et al (2010) Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J Neurol 257:91–97
Regnery C, Kornblum C, Hanisch F et al (2012) 36 months observational clinical study of 38 adult Pompe disease patients under alglucosidase alfa enzyme replacement therapy. J Inherit Metab Dis 35:837–845
de Vries JM, van der Beek NAME, Hop WCJ et al (2012) Effect of enzyme therapy and prognostic factors in 69 adults with Pompe disease: an open-label single-center study. Orphanet J Rare Dis 7:73
Bembi B, Pisa FE, Confalonieri M et al (2010) Long-term observational, non-randomized study of enzyme replacement therapy in late-onset glycogenosis type II. J Inherit Metab Dis 33:727–735
Angelini C, Semplicini C, Ravaglia S et al (2012) Observational clinical study in juvenile-adult glycogenosis type 2 patients undergoing enzyme replacement therapy for up to 4 years. J Neurol 259:952–958
Schoser B, Stewart A, Kanters S et al (2017) Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol 264:621–630
Lin D-S, Chiang M-F, Ho C-S et al (2013) Low-frequency enzyme replacement therapy in late-onset Pompe disease. Muscle Nerve 47:612–613
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
TH and KMR served as consultants, received funding for travel expenses and received honoraria from serving on a scientific advisory board from Genzyme, Switzerland. OF received travel expenses and honoraria from serving on a scientific advisory board from Genzyme, Switzerland. OS and DL received funding for travel expenses from Genzyme, Switzerland. RS declared no conflict of interest.
Ethical standards
The local ethics committee approved this retrospective analysis, and written informed consent was obtained from all participants.
Rights and permissions
About this article

Cite this article
Scheidegger, O., Leupold, D., Sauter, R. et al. 36-Months follow-up assessment after cessation and resuming of enzyme replacement therapy in late onset Pompe disease: data from the Swiss Pompe Registry. J Neurol 265, 2783–2788 (2018). https://doi.org/10.1007/s00415-018-9065-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-9065-7