
Abstract
The MRE11 complex (MRE11, RAD50, and NBS1) is a central component of the DNA damage response, governing both double-strand break repair and DNA damage response signaling. To determine the functions of the MRE11 complex in the development and maintenance of oocytes, we analyzed ovarian phenotypes of mice harboring the hypomorphic Mre11 ATLD1 allele. Mre11 ATLD1/ATLD1 females exhibited premature oocyte elimination attributable to defects in homologous chromosome pairing and double-strand break repair during meiotic prophase. Other aspects of meiotic progression, including attachment of telomeres to the nuclear envelope and recruitment of RAD21L, a component of the meiotic cohesin complex to the synaptonemal complex, were normal. Unlike Dmc1 −/− and Trp13 Gt/Gt mice which exhibit comparable defects in double-strand break repair and oocyte depletion by 5 days post-partum, we found that oocyte attrition occurred by 12 weeks in Mre11 ATLD1/ATLD1. Disruption of the oocyte checkpoint pathway governed by Chk2 gene further enhanced the survival of Mre11 ATLD1/ATLD1 follicles. Together our data suggest that the MRE11 complex influences the elimination of oocytes with unrepaired meiotic double-strand breaks post-natally, in addition to its previously described role in double-strand break repair and homologous synapsis during female meiosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sexual reproduction requires the formation of haploid germ cells which are produced by the process of meiosis. Meiosis consists of two rounds of cell division preceded by a single phase of DNA replication. The first meiotic division (MI) is preceded by prophase during which homologous chromosomes align, pair, and recombine. Prophase is divided into five stages based on the level of homologous chromosome pairing (leptonema, zygonema, pachynema, diplonema, and diakinesis). In many species, including yeast and mammals, homologous chromosome pairing requires and is coincident with the formation and repair of meiosis-specific DNA double-strand breaks (DSBs) (reviewed in Keeney (2001)).
Meiotic DSBs are catalyzed by the dimeric topoisomerase II-like enzyme SPO11, which leaves a monomer covalently attached to the 5′ ends of the DSB end upon generation of the break (de Massy et al. 1995; Keeney and Kleckner 1995). Subsequently, MRE11 endonuclease activity releases a SPO11-coupled oligonucleotide from DSB ends to permit the resolution of meiotic DSBs by homology-directed repair (HDR) between homologous chromosomes (Farah et al. 2009; Hartsuiker et al. 2009; Milman et al. 2009; Rothenberg et al. 2009).
A critical outcome of HDR-mediated repair is the formation of crossovers. This exchange of maternal and paternal chromatid segments is required to establish physical connections between maternal and paternal chromosomes, which in turn ensures proper chromosome segregation during the first meiotic division. Only a small minority of DSBs are repaired with the formation of crossovers, but each chromosome pair must have at least one crossover to prevent missegregation and the subsequent creation of dysfunctional gametes (Petronczki et al. 2003).
DSB formation and chromosome pairing are initiated during leptonema, progress throughout zygonema, and are completed at the pachytene stage. Throughout these stages, synapsis is coordinated with the formation of the synaptonemal complex (SC), a proteinaceous structure that runs parallel to the axes of the paired homologous chromosomes (Fawcett 1956; Moses 1956). The SC consists of lateral elements along the chromosomal axes of each homolog and a central connecting element (reviewed in Yang and Wang (2009)). The formation and repair of meiotic DSBs are essential to achieve synapsis, as evidenced by the severe chromosome pairing abnormalities that are observed in mouse lacking the SPO11 enzyme and in mice that carry mutations in (meiotic) DSB-repair genes such as Dmc1 and Rad51C (Kuznetsov et al. 2007; Pittman et al. 1998; Yoshida et al. 1998; Baudat et al. 2000; Romanienko and Camerini-Otero 2000).
The Mre11 hypomorphic mutant, Mre11 ATLD1, is one of the several Mre11 alleles that cause the ataxia–telangiectasia like disorder (A-TLD) (Stracker and Petrini 2011). Mre11 ATLD1 encodes a nonsense mutation at amino acid 633, causing a C-terminal truncation that deletes one of the two putative MRE11 DNA binding domains (Stracker and Petrini 2011; Stewart et al. 1999; Usui et al. 1998). Mitotic Mre11 ATLD1/ATLD1 cells closely phenocopy ATM-deficient cells and exhibit cell cycle checkpoint defects, genome instability, and hypersensitivity to ionizing radiation (Stewart et al. 1999; Theunissen et al. 2003). These phenotypic outcomes are consistent with the role of the MRE11 complex in the activation of ATM.
Unlike males, Mre11 ATLD1/ATLD1 females are infertile and frequently produce blastocysts that appear to die at or before the cavitation stage, indicating that the MRE11 complex is critical for early embryonic cell divisions (Theunissen et al. 2003). This prompted us to examine meiotic recombination in Mre11 ATLD1 mice. We observed a pronounced delay in the progression of meiotic prophase and sexually dimorphic crossover behavior: Males exhibited increased and females decreased recombination (Cherry et al. 2007). In this study, we present a detailed analysis of female meiosis and oogenesis in Mre11 ATLD1/ATLD1 mice. We observe frequent heterologous synapsis and asynapsis, persistent DSBs, reduced crossover formation, and a subsequent elimination of oocytes during folliculogenesis. The elimination of Mre11 ATLD1/ATLD1 oocytes was CHK2 dependent as oocyte numbers were rescued in Mre11 ATLD1/ATLD1 Chk2 −/− females. The data presented here reveal that the functions of the MRE11 complex and CHK2 are interdependent in the response to meiotic DSBs during oogenesis and folliculogenesis.
Results
Premature elimination of oocytes in Mre11 ATLD1/ATLD1 mice
We showed previously that Mre11 ATLD1/ATLD1 females exhibit a high rate of embryonic lethality (72 % if bred with a wild-type male and 90 % with Mre11 ATLD1/ATLD1 male) at embryonic day 3.5 (E3.5) (Theunissen et al. 2003). This observation indicated that oocytes produced in Mre11 ATLD1/ATLD1 females are largely incapable of producing viable zygotes. To gain insight regarding this outcome, we examined folliculogenesis and oocyte development in Mre11 ATLD1/ATLD1 females.
Over the course of the first 5 days after birth, oocytes become surrounded by a single layer of granulosa cells to form primordial follicles, which represent a quiescent pool of oocytes arrested at the dictyate stage, which is maintained until just before ovulation. Folliculogenesis and development of primordial follicles into mature follicles is marked by the proliferation of granulosa cells, which support the dictyate oocytes (Elvin and Matzuk 1998).
We assessed the number and developmental features of follicles in Mre11 ATLD1/ATLD1 females from E17.5 to adult. Ovary sections from E17.5 and post-natal day 1 embryos were prepared and stained with VASA, a marker of oocytes (Fujiwara et al. 1994). In embryonic stage E17.5 (Fig. 1a, f) and post-natal day 1 (Fig. 1b, g), wild-type and Mre11 ATLD1/ATLD1 ovaries contained similar numbers of oocytes (E17.5/wild-type = 419, Mre11 ATLD1/ATLD1 = 342, p = 0.5. Day 1/wild-type = 329, Mre11 ATLD1/ATLD1 = 221, p = 0.5) (Fig. 1k). TUNEL staining of post-natal day 1 mice from both genotypes was unremarkable (data not shown), suggesting that apoptotic attrition of oocytes was not occurring at this stage.

Premature elimination of Mre11 ATLD1/ATLD1 oocytes. a–j Representative images of anti-VASA-stained mid-ovary sections in a–e wild-type and f–j Mre11 ATLD1/ATLD1 ovaries, from E17.5 (a and f), day 1 (b and g), 3-week-old (c and h), 6-week-old (d and i), and 12-week-old (e and j). Bar = 100 μm. k Quantification of the number of follicles. Bars denote the average ± standard deviation (SD). Age and the number of ovaries analyzed were shown below the x-axis. P-value was determined by unpaired t-test. Blue and white bars indicate wild-type and Mre11 ATLD1/ATLD1, respectively
Mre11 ATLD1/ATLD1 ovaries exhibited a precipitous loss of follicles, with complete depletion observed by 12 weeks. At 3 weeks, the number of primordial follicles declined by 12-fold in Mre11 ATLD1/ATLD1 (wild-type = 53.9, Mre11 ATLD1/ATLD1 = 4.4), while the number of mature follicles was unchanged in wild-type and Mre11 ATLD1/ATLD1 (wild-type = 35.4, Mre11 ATLD1/ATLD1 = 35.3) (Fig. 1c, h). By 6 weeks, attrition of mature follicles was also observed in Mre11 ATLD1/ATLD1 (Fig. 1i, k) relative to wild-type (Fig. 1d, k) (wild-type = 66.5, Mre11 ATLD1/ATLD1 = 5.4). At 12 weeks, a single follicle was detected in Mre11 ATLD1/ATLD1 (Fig. 1j, and enlarged image), in contrast to wild-type ovaries in which mature follicles were readily detected (Fig. 1e, k). There were several corpora lutea observed at this point in Mre11 ATLD1/ATLD1, suggesting the presence of antecedent mature oocytes. These data indicated that similar numbers of oocytes are produced in wild-type and Mre11 ATLD1/ATLD1 females, but experience accelerated attrition in the latter context. This depletion likely underlies the infertility of Mre11 ATLD1/ATLD1 females.
Failure in homologous synapsis during meiotic prophase I in Mre11 ATLD1/ATLD1
The failure to repair DSBs induced by SPO11 during meiotic prophase I has previously been shown to cause oocyte attrition. For example, in Atm −/− and Dmc1 −/− ovaries, oocytes are lost within 5 days post-partum (Di Giacomo et al. 2005). Female meiosis is initiated during embryonic development around E13.5. Oocytes progress through the leptotene and zygotene stages, and at E17.5, the vast majority of oocytes reached the pachytene stage, where homologous chromosomes pair and form the SC. Complete synapsis is essential for proper meiotic development (reviewed in Inagaki et al. (2010)).
We examined the state of meiotic progression in Mre11 ATLD1/ATLD1 at time points corresponding to wild-type pachynema (E17.5) and dictyate stage (newborn). Consistent with our previous analyses (Cherry et al. 2007), Mre11 ATLD1/ATLD1 exhibited only 5 % complete synapsis, while 31 % of oocytes were still in zygonema (Fig. 2b, f), indicating that Mre11 ATLD1/ATLD1 shows a delay in meiotic progression. We noted that 65 % of Mre11 ATLD1/ATLD1 nuclei exhibited pachytene-like stage with heterologous synapsis (Fig. 2c, d; enlarged drawings) or partial asynapsis (Fig. 2e) was observed. We noted that partial heterologous synapsis often appeared to initiate from one of the telomeric ends (Fig. 2c, d, enlarged drawings). The degree of asynapsis was variable from two to ten homologous chromosomes (Fig. 2e). Mre11 ATLD1/ATLD1 nuclei often exhibited both classes of aberrancy (Fig. 2c, d). These data indicate that the MRE11 complex promotes homologous synapsis in female meiosis.

Synaptonemal complex assembly defects in Mre11 ATLD1/ATLD1 oocytes. a–e Representative images of double-staining with SYCP3 (red) and SYCP1 (green) in a wild-type and b–e Mre11 ATLD1/ATLD1 oocyte nuclei at E17.5. c and d Enlarged images and drawings were shown in the next to the merged panel. In the drawings, each SYCP3 staining was marked with different colors. f Quantification of the distribution of meiotic prophase substages in wild-type and Mre11 ATLD1/ATLD1 oocytes. The number of oocytes and mice analyzed were indicated. Bars denote the average ± SD. P-value was determined by unpaired t-test. Blue and white bars indicate wild-type and Mre11 ATLD1/ATLD1, respectively
Defective DSB processing in the Mre11 ATLD1/ATLD1 oocytes
Aberrant synapses such as those observed in Mre11 ATLD1/ATLD1 mice are also observed in SPO11-deficient mice (Baudat et al. 2000; Romanienko and Camerini-Otero 2000). However, SPO11-induced DSBs are formed at normal levels in Mre11 ATLD1/ATLD1 in leptonema quantified with the number of DMC1 foci (wild-type = 141 ± 2.4, Mre11 ATLD1/ATLD1 = 174 ± 16.5; p = 0.07 (Online Resource 1). DMC1 foci persisted in Mre11 ATLD1/ATLD1 pachytene-like oocytes and were generally restricted to asynapsed chromosomal segments (Fig. 3d–f, enlarged images), reflecting compromised DSB repair.

Presence of SPO11-induced meiotic DSBs in Mre11 ATLD1/ATLD1 oocytes. a–f Representative images of double-staining with SYCP3 (red) and DMC1 (green) in (a and b) wild-type and (c-f) Mre11 ATLD1/ATLD1 oocyte nuclei at a E15.5 and b–f E17.5. Substages of the meiotic prophase are shown next to the panel. d and e Enlarged images and drawings were shown in the next to the merged panel. In the drawings, each SYCP3 staining was marked with different colors. Over 100 nuclei were analyzed from three independent mice
MLH1 foci in Mre11 ATLD1/ATLD1 pachytene-like oocytes were reduced relative to wild-type pachytene oocytes (wild-type = 23.8 ± 2.3, Mre11 ATLD1/ATLD1 = 13.3 ± 4.6; p = 0.02, Fig. 4e) consistent with our previous data (Fig. 4a–e) (Cherry et al. 2007). In contrast to our previous study, we quantified all oocytes with MLH1 foci. As some oocytes exhibited severely synaptonemal complex defects (Online Resource 2), the number of MLH1 foci compared to our previous data is reduced (Cherry et al. 2007). No MLH1 foci were detected on asynapsed chromosomes, whereas the most of partially paired chromosomes exhibited MLH1 foci in Mre11 ATLD1/ATLD1 (Fig. 4c, d), suggesting marking positions of crossovers between heterologous chromosomes since 65 % of Mre11 ATLD1/ATLD1 nuclei exhibited pachytene-like stage with heterologous synapsis or partial asynapsis (Fig. 2).

Reduced crossovers in Mre11 ATLD1/ATLD1 oocytes. a–d Representative images of double-staining with SYCP3 (red) and MLH1 (green) in a wild-type and b–d Mre11 ATLD1/ATLD1 oocyte nuclei at E17.5. The number of the MLH1 foci is indicated in the images. The white dashed lines in a and b indicate the boundary of the nucleus. e Quantification of the MLH1 foci in wild-type and Mre11 ATLD1/ATLD1 oocytes. Bars denote the average ± SD. P-value was determined by unpaired t-test. The number of oocytes and mice analyzed were indicated. Blue and white bars indicate wild-type and Mre11 ATLD1/ATLD1, respectively
In newborn mice, fewer than 2 % of wild-type nuclei contained DMC1 foci (Fig. 5a, e). In contrast, more than 30 % of nuclei were positive with DMC1 foci in Mre11 ATLD1/ATLD1 (Fig. 5e), indicating the persistence of DSBs, that exhibited hallmarks of zygotene or pachytene-like stages (Fig. 5b–d). Consistent with the presence of DSBs in oocytes of newborn Mre11 ATLD1/ATLD1 mice, 80 % of Mre11 ATLD1/ATLD1 oocytes were γH2AX-positive compared with 10 % of wild-type oocytes (Online Resource 3). These results suggest that unrepaired DSBs and aberrant synapsis underlie the premature depletion of follicles in Mre11 ATLD1/ATLD1 ovaries.

Persistent meiotic DSBs in Mre11 ATLD1/ATLD1 newborn oocytes. a–d Representative images of double-staining with SYCP3 (red) and DMC1 (green) in a wild-type and b–d Mre11 ATLD1/ATLD1 oocyte nuclei from newborn. e Quantification of the distribution of meiotic prophase substages and DMC1 positive nuclei in wild-type and Mre11 ATLD1/ATLD1 oocytes. Bars denote the average ± SD. P-value was determined by unpaired t-test. The number of oocytes and mice analyzed were indicated. Blue and white bars indicate wild-type and Mre11 ATLD1/ATLD1, respectively
Normal telomere attachment and RAD21L recruitment in Mre11 ATLD1/ATLD1 oocytes
Two additional possible sequelae of MRE11 complex hypomorphism that could account for oocyte attrition were assessed: defective telomere attachment and impaired cohesin recruitment. Attachment of telomeres to the nuclear envelope from early leptonema to pachynema is required for homologous chromosome pairing, SC formation, and folliculogenesis (Ding et al. 2007). We examined whether the aberrant synapsis observed in Mre11 ATLD1/ATLD1 was attributed to irregular localization of telomeres. Fluorescence in situ hybridization (FISH) analysis of telomeres (Tel-FISH) in cryo-sections of embryonic E17.5 ovaries was carried out. Three-dimensional image reconstructions revealed that telomeric signals were confined to the nuclear periphery (Fig. 6a, b). Both wild-type and Mre11 ATLD1/ATLD1 exhibited all Tel-FISH signals that were localized at the nuclear peripheral in more than 80 % of the oocytes (Fig. 6c). In the remaining 20 % of the nuclei, a single Tel-FISH signal was mislocalized and appeared within the nucleus. These data excluded the possibility of irregular telomere localization as a cause of aberrant synapsis in Mre11 ATLD1/ATLD1.

Normal attachment of telomeres to the nuclear envelope in Mre11 ATLD1/ATLD1 oocytes. a and b Representative images of telomere FISH (red) in wild-type oocyte nucleus at E17.5 in a two-dimension and b three-dimension. c Quantification of telomere attachment to the nuclear envelop in wild-type and Mre11 ATLD1/ATLD1 oocytes. Shown is the percentage of the oocytes that show all detectable telomere FISH signals attach to the nuclear envelop. The number of oocytes and mice analyzed were shown below the x-axis. Bars denote the average ± SD. Blue and white bars indicate wild-type and Mre11 ATLD1/ATLD1, respectively
MRE11 complex-dependent recruitment of cohesin complexes has been described in yeast and human cell lines (Kim et al. 2002; Tittel-Elmer et al. 2012). In meiotic cells, cohesin promotes homology search and recombination between homologous chromatids by assembly of the SC (reviewed in Inagaki et al. (2010)). Deletion of meiosis-specific cohesin subunits in mice (Rec8 −/−, Smc1b −/− and Rad21L −/−) showed defects in synapsis and infertility (Bannister et al. 2004; Revenkova et al. 2004; Xu et al. 2005) and premature reproductive senescence in Rad21L −/− females associated with a defect in primordial follicles (Herran et al. 2011).
Accordingly, we examined recruitment of RAD21L to axial elements in Mre11 ATLD1/ATLD1. RAD21L was localized along the SYCP3 from early zygonema to late pachynema at synapsed and asynapsed parts, and the localization was essentially identical in wild-type and Mre11 ATLD1/ATLD1 (Fig. 7a–f). Despite aberrant synapses in Mre11 ATLD1/ATLD1 pachytene-like nuclei, RAD21L was observed at paired axial element regions (Fig. 7f), suggesting that aberrant synapsis observed in Mre11 ATLD1/ATLD1 oocytes was not due to the irregular localization of RAD21L.

No detectable difference of RAD21L recruitment to axial elements in Mre11 ATLD1/ATLD1 oocytes. a–f Representative images of double-staining with SYCP3 (red) and RAD21L (green) in a–c wild-type and d–f Mre11 ATLD1/ATLD1 oocyte nuclei at (a, b, d, and e) E15.5 and (c and f) E17.5. Substages of the meiotic prophase are shown next to the panel. More than 300 nuclei were analyzed from three independent mice
CHK2-dependent elimination of Mre11 ATLD1/ATLD1 oocytes
CHK2 governs a p53- and p63-dependent DNA damage checkpoint response which promotes elimination of oocytes harboring unrepaired meiotic DSBs ( Bolcun-Filas et al. 2014). Previous studies of Mre11 ATLD1/ATLD1 Chk2 −/− mice have shown that CHK2 suppresses the oncogenic potential of DNA damage arising in S phase. This CHK2 function was revealed by the observation that CHK2 deficiency in Mre11 ATLD1/ATLD1 mice—which exhibit pronounced chromosome fragility during DNA replication—leads to a broad spectrum of tumorigenesis (Stracker et al. 2008). This observation indicated that CHK2 is required to mitigate the effects of DNA damage accumulating as a result of MRE11 complex hypomorphism.
A similar relationship appears to be operative in meiotic tissue. CHK2 deficiency markedly enhanced the survival of developing follicles in 3-, 6-, and 9-week-old Mre11 ATLD1/ATLD1 ovaries as the total number of follicles in Mre11 ATLD1/ATLD1 Chk2 −/− were similar to the wild-type level (wild-type = 58, Mre11 ATLD1/ATLD1 = 6, Mre11 ATLD1/ATLD1 Chk2 −/− = 44) (Fig. 8a–d). This outcome supports the conclusion that oocyte attrition in Mre11 ATLD1/ATLD1 is caused by CHK2-dependent elimination of oocytes that harbor unresolved meiotic DSBs. As expected, CHK2 deficiency had no effect on the disrupted synaptic phenotype in Mre11 ATLD1/ATLD1 oocytes (Fig. 8e, f), and the fertility of Mre11 ATLD1/ATLD1 was not rescued (data not shown).

Restored follicles by depletion of Chk2 in adult Mre11 ATLD1/ATLD1 ovaries. (a-c) Representative images of anti-VASA-stained mid-ovary sections in (a) wild-type, (b) Mre11 ATLD1/ATLD1, and (c) Mre11 ATLD1/ATLD1 Chk2 −/− at 9-week-old. Enlarged images show the restored number of primordial follicles by Chk2 deletion. Bar = 100 μm. (d) Quantification of the number of follicles. The number of ovaries analyzed was indicated below the x-axis. (e) Quantification of the distribution of meiotic prophase substages at E17.5 in wild-type, Mre11 ATLD1/ATLD1 and Mre11 ATLD1/ATLD1 Chk2 −/−. Pachytene stages are classified to complete and aberrant synapsis. (f) Quantification of the distribution of meiotic prophase substages and DMC1 positive nuclei from newborn oocytes in wild-type, Mre11 ATLD1/ATLD1 and Mre11 ATLD1/ATLD1 Chk2 −/−. (d-f) Bars denote the average ± SD. P-value was determined by unpaired t-test. The number of nuclei and mice analyzed were indicated. Blue, white and green bars indicate wild-type, Mre11 ATLD1/ATLD1 and Mre11 ATLD1/ATLD1 Chk2 −/−, respectively
Discussion
The Mre11 ATLD1 allele is one of several Mre11 alleles underlying the human A-TLD in which attenuated ATM activation essentially phenocopies ATM deficiency (Stracker and Petrini 2011). In this study, we used the Mre11 ATLD1 allele to examine the MRE11 complex and ATM-dependent DNA damage response in female meiosis. Meiotic progression was delayed in Mre11 ATLD1/ATLD1 mice and associated with failure in homologous synapsis, persistence of DSBs, and reduced crossover frequency, consistent with defective homologous recombination during meiotic prophase (Cherry et al. 2007).
Presumably due to the presence of multiple persistent DSBs in meiotic prophase, Mre11 ATLD1/ATLD1 oocytes were arrested in a pachytene-like stage, with normal number of oocytes present in newborn ovaries. However, we observed attrition of oocytes from adult ovaries, as observed in Dmc1 −/− mutants (Di Giacomo et al. 2005). In Dmc1 −/− mutants, CHK2 deficiency sharply reduced oocyte attrition, without rescuing fertility ( Bolcun-Filas et al. 2014). Unlike Dmc1 −/− mice in which precipitous attrition is observed (Di Giacomo et al. 2005), oocyte attrition in Mre11 ATLD1/ATLD1 occurred gradually, suggesting that the MRE11 complex influences the rate of post-natal oocyte attrition. Although that mechanism is largely dependent on ATR and CHK2 (Pacheco et al. 2015; Bolcun-Filas et al. 2014), the rate of attrition in Mre11 ATLD1/ATLD1 raises the possibility that ATM may also influence CHK2 in the oocyte surveillance pathway. Collectively, these data underscore the importance of the MRE11 complex for the development and maintenance of oocytes.
The MRE11 complex in meiotic DSB repair and homologous synapsis
Meiotic prophase in Mre11 ATLD1/ATLD1 oocytes was characterized by asynapsis and heterologous synapsis (Fig. 2c–f). As homologous synapsis depends on meiotic recombination (Bolcun-Filas and Schimenti 2012), such impaired homologous synapsis may be attributed to compromised DSB repair in Mre11 ATLD1/ATLD1 oocytes. The repair defects associated with the Mre11 ATLD1 allele are likely to synergize with the reduction of ATM activity. For example, Mre11 ATLD1/ATLD1 spermatocytes exhibited a twofold increase in SPO11–oligonucleotide complexes compared to wild-type spermatocytes (Pacheco et al. 2015). If a similar increase were to occur in oocytes, the manifestations of defective DSB repair, including impaired synapsis, would likely be more pronounced. However, we did not observe increased DMC1 foci in Mre11 ATLD1/ATLD1 leptotene oocytes that argues against this interpretation (Online resource 1), although we cannot exclude the possibility that the transient nature of leptonema limits our ability to reliably determine smaller differences in cytological outcomes. It is clear, in any case from the persistence of RAD51 and DMC1 in pachynema, that DSB repair is defective in Mre11 ATLD1/ATLD1 oocytes (Fig. 3c–f and Cherry et al. (2007)).
Meiotic DNA damage response leads to follicle elimination in post-natal ovaries
Genetic analyses suggest the existence of distinct checkpoints for DNA damage, synapsis, and crossover formation in oocytes. The data presented herein suggest that the process of oocyte elimination is influenced by the MRE11 complex. Oocytes showing persistent DSBs during meiotic prophase such as Trip13 Gt/Gt (normal synapsis) (Li and Schimenti 2007), Rec8 −/− (Xu et al. 2005) and Sun1 −/− (Ding et al. 2007) (asynapsis) or Dmc1 −/− and Atm −/− (asynapsis and heterologous synapsis) (Di Giacomo et al. 2005) exhibit follicular elimination by 5 days post-partum. Spo11 −/− oocytes in which DSBs are not formed exhibit failure in synapsis (heterologous synapsis and asynapsis) but persist in follicles until 6 weeks post-partum (Di Giacomo et al. 2005). SPO11 deficiency is epistatic to Dmc1 −/− and Atm −/− with respect to oocyte attrition (Di Giacomo et al. 2005), suggesting that DSB formation as well as the ensuing synapsis and repair are the events that determine oocyte elimination in post-natal ovaries. This is in contrast to Mlh1 −/− oocytes which do not exhibit follicular attrition. In that setting, DSB formation, DSB repair, and synapsis are normal, but crossover formation does not occur, resulting in infertility (Edelmann et al. 1996). Unrepaired DSBs are abundant in Mre11 ATLD1/ATLD1 oocytes, and gross defects in synapsis are evident, yet the elimination of follicles occurred at 12 weeks post-partum, markedly later than the examples above. We propose two distinct hypotheses based on this observation.
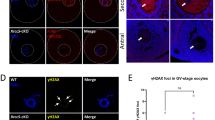
We interpret this observation to reflect the contribution of the MRE11 complex to promoting the elimination of oocytes with synapsis and DSB repair defects. The oocyte elimination pathway depends on ATR and CHK2 ( Bolcun-Filas et al. 2014). Accordingly, inactivation of CHK2 in Trip13 Gt/Gt, Dmc1 −/−, Atm −/− (Bolcun-Filas et al. 2014), Mre11 ATLD1/ATLD1, and Rad50 +/46, which harbors a mutation in the Rad50 hook domain and exhibits ovarian atrophy (Roset et al. 2014) (Online Resource 4b and c), suppresses the attrition of defective oocytes. The slower rate of attrition in Mre11 ATLD1/ATLD1 ovaries could reflect that the MRE11 complex influences ATR activation, as has been suggested previously (Duursma et al. 2013; Shiotani et al. 2013). Alternatively, as ATM activation is compromised but not abolished in Mre11 ATLD1/ATLD1, the data may reveal that ATM contributes to the pathway as well. In this scenario, we speculate that complete loss of ATM is pleiotropic, whereas the ATM functional deficit in Mre11 ATLD1/ATLD1 would selectively affect the presumptive role in oocyte elimination. Precedence for circumscribed loss of certain ATM functions in MRE11 complex mutants comes from the Nbs1 ∆C/∆C mouse in which ATM-dependent apoptosis is selectively impaired (Stracker et al. 2007). It is notable that the Nbs1 ∆C/∆C also revealed that ATM and CHK2 act in parallel to regulate thymocyte apoptosis, making it conceivable that if ATM has a role in oocyte depletion, it could be CHK2 independent. It must also be noted that we cannot exclude the possibility that the female meiotic DNA damage checkpoint and synapsis checkpoint function in a dose-dependent manner and that the apparently slower oocyte attrition reflects a lower level of persistent DNA damage and aberrant synapsis in Mre11 ATLD1/ATLD1 oocyte than previously characterized mutants.
Moreover, we obtained evidence in Rad50 +/46 Chk2 −/− mice that CHK2 may promote depletion of oocytes harboring persistent DSBs in meiotic prophase (Online Resource 4d). Despite exhibiting follicular attrition, Rad50 +/46 mice did not exhibit overt defects in DNA repair or homologous synapsis (Online Resource 4a and d) (Roset et al. 2014). However, in Rad50 46/+ Chk2 −/− double mutants, aberrant heterologous synapses were readily observed at E17.5, suggesting that as in spermatocytes (Pacheco et al. 2015), CHK2 may play a role in the surveillance of meiotic recombination in female meiotic prophase.
Materials and methods
Mice
Mre11 ATLD1 and Rad50 +/46 mice were previously described (Theunissen et al. 2003; Roset et al. 2014). Chk2 −/− mice were obtained from T. Mak. All mice were maintained on mixed 129/SvEv and C57BL6 background.
Mice were housed in a ventilated rack caging in a pathogen-free facility. The Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center approved animal use protocols.
Antibodies
For primary antibodies, SYCP3 (goat anti-mouse monoclonal provided by S. West), SYCP1 (goat anti-rabbit, Novus), MLH1 (goat anti-mouse monoclonal, BD Pharmingen), DMC1 (goat anti-rabbit polyclonal, Santa Cruz), and RAD21L (goat anti-mouse monoclonal provided by T. Hirano) were used. For secondary antibodies, goat anti-rabbit IgG Alexa 488/564 and goat anti-mouse Alexa IgG 488/564 (Molecular Probes) were used.
Histological sample preparation, staining, and analysis
Tissue samples for histological analyses were fixed overnight at 4 °C in 4 % paraformaldehyde, rinsed, stored at 4 °C in 70 % ethanol, and then processed for paraffin embedding. Eight-micrometer sections were prepared, and slides were processed and stained at the Memorial Sloan Kettering molecular cytogenetics core facility for hematoxylin and eosin (H&E), TUNEL, anti-VASA, and anti-γH2AX.
H&E-stained and immunohistochemically stained slides were digitally scanned using a Mirax scanner. Anti-VASA-positive cells and anti-γH2AX-positive cells per slide were manually scored.
Meiotic spread nuclei preparations, immunocytochemistry, and analysis
Ovaries were processed to obtain spread nuclei for immunocytochemistry. Briefly, embryonic ovaries were dissected and placed in phosphate-buffered saline (PBS), pH7.4, at room temperature. Adherent extra tissue was removed in PBS. The ovaries were placed in a hypotonic extraction buffer containing 30 mM Tris, 50 mM sucrose, 17 mM trisodium citrate dehydrate, 5 mM EDTA, 0.5 mM DTT, and 0.5 mM phnylmethylsuphonyl fluoride, pH 8.2, for 20–40 min. Subsequently, an oocyte suspension was made in 10 μl of 50 mM sucrose and 10 μl of PBS by puncturing a single ovary with needles on a clean glass slide. The cell suspension was fixed with 1 % paraformaldehyde, pH 9.2 (set by using 10 mM sodium borate pH 9.2), containing 0.15 % Triton X-100 for 1.5 h. The slides were washed twice for 2 min in 0.4 % Photoflo (Kodak) and dried at room temperature.
Spread nuclei of oocytes were stained with the antibodies mentioned above. Before incubation with antibodies, slides were washed in PBS (3×10 min), and non-specific sites were blocked with 0.5 % w/v BSA and 0.5 % w/v milk powder in PBS. Primary antibodies were diluted in 10 % w/v BSA in PBS, and incubations were held overnight at room temperature in a humid chamber. Subsequently, slides were washed (3×10 min) in PBS , blocked in 10 % v/v normal goat serum (Sigma) in blocking buffer (supernatant of 5 % w/v milk powder in PBS centrifuged at 14,000 rpm for 10 min), and incubated with secondary antibodies in 10 % normal goat serum in blocking buffer at RT for 2 h. Finally, slides were washed (3×10 min) in PBS and embedded in Prolong Gold with DAPI (invitrogen).
The number of DMC1 foci was scored using the ImageJ software (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA, [http://rsb.info.nih.gov/ij/]).
References
Bannister LA, Reinholdt LG, Munroe RJ, Schimenti JC (2004) Positional cloning and characterization of mouse mei8, a disrupted allele of the meiotic cohesin Rec8. Genesis. 184–194. doi:10.1002/gene.20085
Baudat F, Manova K, Yuen JP, Jasin M, Keeney S (2000) Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell:989–998
Bolcun-Filas E, Schimenti JC (2012) Genetics of meiosis and recombination in mice. Int Rev Cell Mol Biol. 179–227. doi:10.1016/B978-0-12-394309-5.00005-5
Bolcun-Filas E, Rinaldi VD, White ME, Schimenti JC (2014) Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science. 533–536. doi:10.1126/science.1247671
Cherry SM, Adelman CA, Theunissen JW, Hassold TJ, Hunt PA, Petrini JH (2007) The Mre11 complex influences DNA repair, synapsis, and crossing over in murine meiosis. Curr Biol. 373–378. doi:10.1016/j.cub.2006.12.048
de Massy B, Rocco V, Nicolas A (1995) The nucleotide mapping of DNA double-strand breaks at the CYS3 initiation site of meiotic recombination in Saccharomyces cerevisiae. EMBO J:4589–4598
Di Giacomo M, Barchi M, Baudat F, Edelmann W, Keeney S, Jasin M (2005) Distinct DNA-damage-dependent and -independent responses drive the loss of oocytes in recombination-defective mouse mutants. Proc Natl Acad Sci U S A. 737–742. doi:10.1073/pnas.0406212102
Ding X, Xu R, Yu J, Xu T, Zhuang Y, Han M (2007) SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev Cell. 863–872. doi:10.1016/j.devcel.2007.03.018
Duursma AM, Driscoll R, Elias JE, Cimprich KA (2013) A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol Cell. 116–122. doi:10.1016/j.molcel.2013.03.006
Edelmann W, Cohen PE, Kane M, Lau K, Morrow B, Bennett S, Umar A, Kunkel T, Cattoretti G, Chaganti R, Pollard JW, Kolodner RD, Kucherlapati R (1996) Meiotic pachytene arrest in MLH1-deficient mice. Cell:1125–1134
Elvin JA, Matzuk MM (1998) Mouse models of ovarian failure. Rev Reprod:183–195
Farah JA, Cromie GA, Smith GR (2009) Ctp1 and exonuclease 1, alternative nucleases regulated by the MRN complex, are required for efficient meiotic recombination. Proc Natl Acad Sci U S A. 9356–9361. doi:10.1073/pnas.0902793106
Fawcett DW (1956) The fine structure of chromosomes in the meiotic prophase of vertebrate spermatocytes. J Biophys Biochem Cytol:403–406
Fujiwara Y, Komiya T, Kawabata H, Sato M, Fujimoto H, Furusawa M, Noce T (1994) Isolation of a DEAD-family protein gene that encodes a murine homolog of Drosophila vasa and its specific expression in germ cell lineage. Proc Natl Acad Sci U S A:12258–12262
Hartsuiker E, Neale MJ, Carr AM (2009) Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA. Mol Cell. 117–123. doi:10.1016/j.molcel.2008.11.021
Herran Y, Gutierrez-Caballero C, Sanchez-Martin M, Hernandez T, Viera A, Barbero JL, de Alava E, de Rooij DG, Suja JA, Llano E, Pendas AM (2011) The cohesin subunit RAD21L functions in meiotic synapsis and exhibits sexual dimorphism in fertility. EMBO J. 3091–3105. doi:10.1038/emboj.2011.222
Inagaki A, Schoenmakers S, Baarends WM (2010) DNA double strand break repair, chromosome synapsis and transcriptional silencing in meiosis. Epigenetics:255–266
Keeney S (2001) Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol:1–53
Keeney S, Kleckner N (1995) Covalent protein–DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci U S A:11274–11278
Kim JS, Krasieva TB, LaMorte V, Taylor AM, Yokomori K (2002) Specific recruitment of human cohesin to laser-induced DNA damage. J Biol Chem. 45149–45153. doi:10.1074/jbc.M209123200
Kuznetsov S, Pellegrini M, Shuda K, Fernandez-Capetillo O, Liu Y, Martin BK, Burkett S, Southon E, Pati D, Tessarollo L, West SC, Donovan PJ, Nussenzweig A, Sharan SK (2007) RAD51C deficiency in mice results in early prophase I arrest in males and sister chromatid separation at metaphase II in females. 581–592. J Cell Biol. doi:10.1083/jcb.200608130
Li XC, Schimenti JC (2007) Mouse pachytene checkpoint 2 (trip13) is required for completing meiotic recombination but not synapsis. PLoS Genet. e130. doi:10.1371/journal.pgen.0030130
Milman N, Higuchi E, Smith GR (2009) Meiotic DNA double-strand break repair requires two nucleases, MRN and Ctp1, to produce a single size class of Rec12 (Spo11)-oligonucleotide complexes. Mol Cell Biol. 5998–6005. doi:10.1128/MCB.01127-09
Moses MJ (1956) Chromosomal structures in crayfish spermatocytes. J Biophys Biochem Cytol:215–218
Pacheco S, Marcet-Ortega M, Lange J, Jasin M, Keeney S, Roig I (2015) The ATM signaling cascade promotes recombination-dependent pachytene arrest in mouse spermatocytes. PLoS Genet. e1005017. doi:10.1371/journal.pgen.1005017
Petronczki M, Siomos MF, Nasmyth K (2003) Un menage a quatre: the molecular biology of chromosome segregation in meiosis. Cell:423–440
Pittman DL, Cobb J, Schimenti KJ, Wilson LA, Cooper DM, Brignull E, Handel MA, Schimenti JC (1998) Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol Cell:697–705
Revenkova E, Eijpe M, Heyting C, Hodges CA, Hunt PA, Liebe B, Scherthan H, Jessberger R (2004) Cohesin SMC1 beta is required for meiotic chromosome dynamics, sister chromatid cohesion and DNA recombination. Nat Cell Biol. 555–562. doi:10.1038/ncb1135
Romanienko PJ, Camerini-Otero RD (2000) The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell:975–987
Roset R, Inagaki A, Hohl M, Brenet F, Lafrance-Vanasse J, Lange J, Scandura JM, Tainer JA, Keeney S, Petrini JH (2014) The Rad50 hook domain regulates DNA damage signaling and tumorigenesis. Genes Dev. 451–462. doi:10.1101/gad.236745.113
Rothenberg M, Kohli J, Ludin K (2009) Ctp1 and the MRN-complex are required for endonucleolytic Rec12 removal with release of a single class of oligonucleotides in fission yeast. PLoS Genet:e1000722. doi:10.1371/journal.pgen.1000722
Shiotani B, Nguyen HD, Hakansson P, Marechal A, Tse A, Tahara H, Zou L (2013) Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep. 1651–1662. doi:10.1016/j.celrep.2013.04.018
Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia–telangiectasia-like disorder. Cell:577–587
Stracker TH, Petrini JH (2011) The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 90–103. doi:10.1038/nrm3047
Stracker TH, Morales M, Couto SS, Hussein H, Petrini JH (2007) The carboxy terminus of NBS1 is required for induction of apoptosis by the MRE11 complex. Nature. 218–221. doi:10.1038/nature05740
Stracker TH, Couto SS, Cordon-Cardo C, Matos T, Petrini JH (2008) Chk2 suppresses the oncogenic potential of DNA replication-associated DNA damage. Mol Cell. 21–32. doi:10.1016/j.molcel.2008.04.028
Theunissen JW, Kaplan MI, Hunt PA, Williams BR, Ferguson DO, Alt FW, Petrini JH (2003) Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol Cell:1511–1523
Tittel-Elmer M, Lengronne A, Davidson MB, Bacal J, Francois P, Hohl M, Petrini JH, Pasero P, Cobb JA (2012) Cohesin association to replication sites depends on rad50 and promotes fork restart. Mol Cell. 98–108. doi:10.1016/j.molcel.2012.07.004
Usui T, Ohta T, Oshiumi H, Tomizawa J, Ogawa H, Ogawa T (1998) Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell:705–716
Xu H, Beasley MD, Warren WD, van der Horst GT, McKay MJ (2005) Absence of mouse REC8 cohesin promotes synapsis of sister chromatids in meiosis. Dev Cell. 949–961. doi:10.1016/j.devcel.2005.03.018
Yang F, Wang PJ (2009) The mammalian synaptonemal complex: a scaffold and beyond. Genome Dyn. 69–80. doi:10.1159/000166620
Yoshida K, Kondoh G, Matsuda Y, Habu T, Nishimune Y, Morita T (1998) The mouse RecA-like gene Dmc1 is required for homologous chromosome synapsis during meiosis. Mol Cell:707–718
Acknowledgments
We thank Dr. Hirano and Dr. West for providing antibodies; current members and previous members (especially Dr. Attwooll) of the J.H.J.P. laboratory for critical reading of the manuscript, discussions, and helpful insight; and the molecular cytology core facility in MSKCC for histological staining.
Funding
This work was supported by NIH grant RO1-GM59413 and RO1-GM056888 (both to J.H.J.P.).
Conflict of interest
Akiko Inagaki declares that she has no conflict of interest. Ramon Roset declares that he has no conflict of interest. John H.J. Petrini declares that he has no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 8449 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Inagaki, A., Roset, R. & Petrini, J.H.J. Functions of the MRE11 complex in the development and maintenance of oocytes. Chromosoma 125, 151–162 (2016). https://doi.org/10.1007/s00412-015-0535-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-015-0535-8