
Abstract
Recently, the gamma-aminobutyric acid (GABA) system has come into focus for the treatment of anxiety, postpartum depression, and major depressive disorder. Endogenous 3α-reduced steroids such as allopregnanolone are potent positive allosteric modulators of GABAA receptors and have been known for decades. Current industry developments and first approvals by the U.S. food and drug administration (FDA) for the treatment of postpartum depression with exogenous analogues of these steroids represent a major step forward in the field. 3α-reduced steroids target both synaptic and extrasynaptic GABAA receptors, unlike benzodiazepines, which bind to synaptic receptors. The first FDA-approved 3α-reduced steroid for postpartum depression is brexanolone, an intravenous formulation of allopregnanolone. It has been shown to provide rapid relief of depressive symptoms. An orally available 3α-reduced steroid is zuranolone, which also received FDA approval in 2023 for the treatment of postpartum depression. Although a number of studies have been conducted, the efficacy data were not sufficient to achieve approval of zuranolone in major depressive disorder by the FDA in 2023. The most prominent side effects of these 3α-reduced steroids are somnolence, dizziness and headache. In addition to the issue of efficacy, it should be noted that current data limit the use of these compounds to two weeks. An alternative to exogenous 3α-reduced steroids may be the use of substances that induce endogenous neurosteroidogenesis, such as the translocator protein 18 kDa (TSPO) ligand etifoxine. TSPO has been extensively studied for its role in steroidogenesis, in addition to other functions such as anti-inflammatory and neuroregenerative properties. Currently, etifoxine is the only clinically available TSPO ligand in France for the treatment of anxiety disorders. Studies are underway to evaluate its antidepressant potential. Hopefully, neurosteroid research will lead to the development of fast-acting antidepressants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Major depressive disorder (MDD) is a serious condition with a high 12-month prevalence of about 6% and a lifetime incidence of about 16% [1]. Unmet needs include the limited efficacy of antidepressants, particularly their slow onset of action. Most antidepressants commonly described in depression are believed to compensate for monoaminergic and anticholinergic neurotransmitter shifts [2], but it takes several weeks for a clinically meaningful onset of action to occur. Thus, there is a need for rapid-acting antidepressants using novel pharmacologic targets. For example, ketamine has been shown to induce relatively rapid but short-lasting improvement in depressive symptoms via the glutamate system [3]. Benzodiazepines are comparably fast-acting and widely used anxiolytic compounds [3,4,5,6]. However, side effects such as tolerance development and abuse liability limit their use for medium- and long-term treatment [3,4,5,6]. Benzodiazepines do not have sustained antidepressant properties [7,8,9,10]. Therefore, other treatment options, that produce rapid antidepressant and/or anxiolytic effects without the side effects of benzodiazepines are needed for the treatment of affective disorders. Most of the available antidepressants are based on the concept of monoamine reuptake inhibition, e.g., selective serotonin reuptake inhibitor (SSRIs). Recently, however, the GABAergic system has come into focus with the industrial development of GABAergic steroids as novel treatment options for postpartum depression and major depressive disorder. Moreover, given the discrete signs of neuroinflammation in depression as revealed by positron emission tomography (PET) studies for the translocator protein 18 kDa (TSPO) and the ability of TSPO ligands to promote endogenous steroidogenesis [5, 11], TSPO ligands may represent an interesting alternative to the administration of exogenous steroids.
Therefore, we discuss below recent developments in the treatment of depression and postpartum depression using GABAergic steroids in relation to their mechanism of action and in relation to TSPO ligands.
Steroids: synthesis and mechanisms of action
Steroids present in the brain originate from both steroidogenic tissues, e.g., the adrenal cortex and also from a local synthesis [12]. Cholesterol is a substrate for the translocator protein 18 kDa (TSPO), which serves as a channel to transport cholesterol into the mitochondrial matrix [13, 14]. There, steroids are formed from cholesterol (Fig. 1).

Neurosteroidogenic pathway and therapeutic compounds. Left panel: 3α-reduced steroids are metabolites of progesterone. Their synthesis is catalyzed by the 5α-reductase and the 3α-hydroxysteroid oxidoreductase. The activity of the 3α-hydroxysteroid oxidoreductase may work both towards reduction and oxidation [92]. Right panel: Molecule structure of brexanolone and zuranolone. Brexanolone is an intravenous formulation of 3α, 5α-tetrahydroprogesterone (3α, 5α-THP, allopregnanolone). Zuranolone is a synthetic modification of allopregnanolone and can be administered orally [11, 38, 43]. Brexanolone and zuranolone are approved by FDA for the treatment of postpartum depression, while zuranolone is still under investigation for major depressive disorder (MDD) and eventually for anxiety disorders
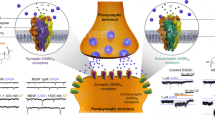
An initial steroid is pregnenolone, which is converted to progesterone and deoxycorticosterone. These steroids are further reduced via the 5α-reductase and then by the 3α-hydroxysteroid oxidoreductase that works in both directions [12] to produce 3α-reduced steroids, such as allopregnanolone and 3α,5α-tetrahydrodeoxycorticosterone (3α5α-THDOC), that are positive allosteric modulators of GABAA receptors. Steroid binding occurs mainly at ß-subunit interfaces [15], whereas the benzodiazepine binding site is determined by the differential composition of α-subunits [16], thereby enhancing GABA-gated chloride currents. GABAA receptors can be differentiated into synaptic and extrasynaptic GABAA receptors. Synaptic GABAA receptors, which are targeted by benzodiazepines and 3α-reduced steroids, show a widespread expression profile and mediate a phasic inhibition. Extrasynaptic GABAA receptors, however, are targeted by 3α-reduced steroids and confer tonic inhibition with a more region-specific expression profile [17] (Fig. 2). As 3α-reduced steroids target both synaptic and extrasynaptic GABAA receptors [18,19,20], this may contribute to their unique pharmacological profile. It is also noteworthy that while benzodiazepines prolong the fast phasic postsynaptic response of γ2 subunit-containing GABAA receptors, 3α-reduced steroids additionally evoke a tonic persistent inhibitory response involving extrasynaptic GABAA receptors containing the δ subunit [21]. These differential effects on the time course and peak amplitude of GABA-evoked chloride currents may explain the differences in efficacy and side effect profile between 3α-reduced steroids and benzodiazepines.

Mechanism of action of etifoxine. Upper box: Molecule structure of etifoxine. Etifoxine has a dual mode of action. It is a TSPO ligand and promotes neurosteroidogenesis, which in turn modulate GABAA receptors, but also directly modulates α2 and α3 containing GABAA receptors (gray dotted arrows). It is approved in France for adjustment anxiety disorders and under investigation for the treatment of depression by our group. Lower boxes: GABAA receptors are expressed both at synaptic and extrasynaptic sites with different expression patterns and physiological and pharmacological properties. While benzodiazepines target only synaptic receptors, neurosteroids modulate both synaptic and extrasynaptic receptors, which may explain their distinct clinical profile
Neurosteroids in depression
Studies investigating the composition of neurosteroids in depressed patients are rather rare. First clinical studies reported reduced levels of 3α-reduced steroids in corticospinal fluid and plasma of depressed patients and a subsequent increase following SSRI treatment [22,23,24]. These findings are in line with molecular data showing that SSRIs or mirtazapine may increase concentrations of 3α-reduced steroids through shifting the activity of the 3α-hydroxysteroid oxidoreductase towards the reductive direction (Fig. 1) [25, 26]. This mechanism may contribute to the antidepressant and anxiolytic effects of these compounds. Moreover, following medication with different antidepressants we found an increase in 3β-reduced steroids, which exert functional antagonistic properties [22,23,24]. To investigate whether these changes in neurosteroid composition more likely reflect clinical response patterns, we did a series of studies with nonpharmacological treatments of depression such as sleep deprivation [27], repetitive transcranial magnetic stimulation [28] and electroconvulsive therapy [29]. However, none of these treatments affected neurosteroid composition independent of treatment response. On the other hand, treatment with mirtazapine normalized the altered neurosteroid pattern in both responders and non-responders to treatment, suggesting a pharmacological effect via neurosteroidogenic enzymes [26]. Another study suggested alterations in neurosteroid composition also in perinatal depression [30]. Interestingly, in patients with premenstrual dysphoric disorder (PMDD), the sensitivity of exogenously administered allopregnanolone was shown to be dependent on the menstrual cycle [31]. Although 3α-reduced steroids are altered in depression, these changes are far less pronounced than in postpartum depression and conflicting results have been reported on the relationship between hormonal changes and symptom onset [32]. Although the reported findings of alterations in neurosteroid composition are subtle and only few studies are available, there is a good rationale for the therapeutic use of either exogenous or endogenous neurosteroids in affective disorders, which may provide a basis for novel treatment options.
Therapeutic effects of 3α-reduced steroids
Although 3α-reduced steroids and their behavioral properties have been known for decades, only recently industrial efforts have been undertaken to put these molecules forward as therapeutic agents. The first neurosteroid to receive approval for the treatment of postpartum depression by the FDA is brexanolone [33]. The formula of brexanolone is identical to that of the naturally occurring allopregnanolone (Fig. 1). It is a special preparation for intravenous application, that should be administered for 60 h [34, 35]. In these two studies, a rapid, clinically meaningful antidepressant effect was observed that persisted throughout the study period of 30 days. The most common adverse effects were dizziness and headache. These findings were replicated in the HUMMINGBIRD clinical program with rapid improvement of depressive, anxiety and insomnia symptoms [36]. A post-marketing survey revealed no serious safety concerns for brexanolone, with sedation being the most serious side effect [37]. From a pathophysiological perspective the administration of 3α-reduced steroids such as brexanolone may alleviate affective symptoms in postpartum depression [11]. After parturition, progesterone and its 3α-reduced metabolites show an enormous decline, which may cause affective symptoms and may be alleviated by exogenous administration of 3α-reduced steroids [5]. Thus, neurosteroid replacement may be directly related to the pathophysiology of postpartum depression. However, a practical inconvenience of brexanolone is the 60-hour intravenous infusion. An oral medication was needed and was introduced with zuranolone (Fig. 1), which solves the problem of administration. In 2023, zuranolone was approved by the FDA for the treatment of postpartum depression [38, 39]. It was shown that zuranolone produced a reduction in depressive symptoms within 15 days of treatment that lasted until day 45 [39, 40]. In addition, zuranolone improved sleep patterns in a model of insomnia in healthy volunteers [11, 41]. In these studies, side effects of zuranolone were similar to those of brexanolone (headache, dizziness, and somnolence). The approval of two 3a-reduced steroids for postpartum depression represents a major breakthrough in the treatment of this mental disorder.
A series of studies have been conducted to further study the efficacy of zuranolone in major depressive disorder. Zuranolone significantly improved depressive symptoms within 15 days of treatment when compared to placebo [42] and increased patient-reported quality of life within this time frame [43]. In addition, a recent meta-analysis pointed towards a beneficial effect of zuranolone within two weeks of treatment [44]. Several follow-up studies have been published recently. A study with 50 mg zuranolone was able to reproduce the superiority over placebo on day 15 in reducing the Hamilton Depression Scale (HAM-D) score with the first signs of onset already on day 3 [45]. However, in the so-called MOUNTAIN study, zuranolone did not meet its primary endpoint at a dose of 30 mg on day 15 of treatment [46]. In the SHORELINE study, an open-label phase 3 trial, repeated treatment courses were offered as needed, with responders requiring fewer than two treatment courses during one year of follow-up [47]. Another recent meta-analysis suggested antidepressant and anxiolytic efficacy of zuranolone with an optimal dose of 30 mg, with an increased risk of side effects with increasing the dose [48]. It is noteworthy, however, that in contrast to postpartum depression, zuranolone has not been approved for major depressive disorder. Concerns have been raised about limited efficacy, incompatibility with breastfeeding, putative impairment of psychomotor function, and potential for abuse [49]. Other issues that need to be evaluated include how long the effects of zuranolone can be maintained after treatment is discontinued, whether zuranolone can really work as an interval therapy requiring only short treatment intervals of 14 days, thereby changing the course of the disease without the need for maintenance or relapse medications, and whether zuranolone can be administered for a longer period of time in relation to side effects. So far, headache, somnolence and dizziness are the most prominent side effects within a two-week treatment. The side effect profile over longer treatment periods remains to be determined. It will be of great interest to see what the future potential of zuranolone in the treatment of major depressive disorder will be.
Structure and function of TSPO
An alternative to the administration of exogenous 3a-reduced steroids is the promotion of endogenous neurosteroidogenesis. Much research has focused on the translocator protein 18 kDa (TSPO), suggesting it as a promising candidate for endogenous steroid formation. The following section on the structure and function of TSPO is taken in part from a recent review by our group in the same journal and presented again here for reasons of clarity [11]. The translocator protein 18 kDa (TSPO) is a 169 amino acid comprising protein of the outer mitochondrial membrane (OMM) [13, 14], which is associated with other proteins residing in the OMM such as voltage-dependent anion channel (VDAC), but also with cytosolic proteins, e.g., the steroidogenic acute regulatory protein (StAR) and proteins of the inner mitochondrial membrane (IMM), such as the adenine nucleotide transporter (ANT) [13, 14]. TSPO mediates numerous biological functions such as mitochondrial cholesterol transport, porphyrin transport and heme synthesis, apoptosis, cell proliferation, and transport of ions and metabolites [13, 14]. Although TSPO is particularly abundant in steroid producing tissues it can be found substantially also in the brain, liver, heart, and the immune system. The broad range of expression and its pleiotropic functional properties render TSPO an interesting target for many disease areas [13, 14, 50]. Currently, the exact role of TSPO for steroidogenesis has been challenged by studies in various knock-out mice [51,52,53], where steroidogenesis remains unaffected by TSPO knockout.
Nevertheless, TSPO ligands exert numerous functions including anti-inflammatory properties, which on the one hand offer a much broader pharmacological profile but on the other hand should be considered when developing TSPO ligands for therapeutic purposes both in terms of clinical and side effects. Recently, some TSPO ligands, such as the benzodiazepine diazepam (which also has some affinity for TSPO [14]), have been shown to activate synaptic pruning, leading to increased synaptic C1q deposition, removal of excitatory synapses, microglial phagocytosis of synaptic proteins, and decline in cognitive function [5, 54]. Such findings of impaired cognitive function, which may be due to a loss of hippocampal and cortical excitatory synapses, may provide an explanation for why certain benzodiazepines, such as diazepam, may cause cognitive impairment in humans. On the other hand, a recent study showed that the TSPO ligand XBD173 prevented the amyloid β-induced neurotoxicity and dendritic spine loss and even exerted precognitive effects in a mouse model of Alzheimer’s disease [55]. Thus, the question of whether TSPO ligands may have beneficial or even detrimental effects on cognition and in neurodegenerative disorders warrants further investigation.
TSPO ligands in the treatment of anxiety and depression
TSPO expression and activity [11, 13, 14, 56, 57] play an important role for psychiatric disorders and their treatment. A variety of studies have investigated the expression of TSPO in stress-related disorders [5, 11, 50]. These studies investigated either the expression of TSPO mRNA in peripheral mononuclear cells, the binding characteristics of the TSPO ligand PK11195 to platelet membranes, or protein expression in thrombocytes [58,59,60]. Meanwhile, various PET studies reported increased TSPO expression in depression [61, 62] and obsessive-compulsive disorder [63]. Recently, it has been suggested that TSPO PET imaging may even predict the clinical response to celecoxib treatment in major depression [64], thereby highlighting the potential of TSPO ligands as personalized approaches both in diagnostics and for selecting treatment procedures in relation to their outcome. In patients suffering from PTSD, it has been shown that higher C-reactive protein (CRP) levels are associated with lower prefrontal-limbic TSPO availability and PTSD severity [65]. Moreover, in neurodegenerative disorders, such as Alzheimer´s disease but also in depression with cognitive impairment, upregulation of TSPO labeling has been reported in PET studies [62, 66]. However, TSPO expression should not unequivocally be considered as a marker of neuroinflammation, since also neuronal activation may increase TSPO levels [67]. These findings suggest that TSPO ligands exert antidepressant effects through their anti-inflammatory properties. Moreover, TSPO gene variants, such as the rs6971 Ala147Thr polymorphism, which affects ligand binding and cholesterol uptake, should be considered in clinical studies when assessing TSPO binding or function. For example, both bipolar disorder and diurnal cortisol rhythm in bipolar disorder have been linked to this TSPO polymorphism [68, 69].
Various TSPO ligands may display anxiolytic or putative antidepressant properties in rodents [11, 14, 70]. In a translational study, our group showed that the selective TSPO ligand XBD173 enhanced GABAergic neurotransmission in brain slices via the induction of neurosteroidogenesis and effectively reduced the number of pharmacologically induced panic attacks in rodents in the absence of sedation [70]. Moreover, XBD173 displayed anti-panic and anxiolytic efficacy in humans using an experimental anxiety paradigm involving a challenge with cholecystokinin tetrapeptide (CCK-4). Whereas the benzodiazepine alprazolam caused sedation and withdrawal symptoms after only 7 days of treatment, these were absent in the XBD173-treated subjects. Recent molecular studies using global or neuronal TSPO knockout mice further dissected the role of neuronal TSPO for modulating anxiety- and depression-related behavior and the effects of XBD173 [57]. A recent animal study suggested that the TSPO ligand AC-5216 (XBD173) may exert rapid antidepressant and memory enhancing effects [71]. The capability of TSPO ligands to exert anxiolytic and antidepressant effects has also been demonstrated for other novel TSPO ligands such as YL-IPAo8 in a rat model of postpartum depression [72] or the antagonistic ligand ONO-2952 [73]. Therefore, TSPO represents a promising target for the development of fast-acting anxiolytics and antidepressants with a favorable side effect profile.
Currently, the only clinically available TSPO ligand is etifoxine (Fig. 2), which is approved in France. Etifoxine has a dual mode of action, as it targets TSPO but also directly α2 and α3 containing GABAA receptors [74]. Initial clinical studies with etifoxine have provided first evidence for a clinical anxiolytic effect, which showed comparable efficacy to the benzodiazepine lorazepam in patients suffering from adjustment disorders with anxiety [75]. The anxiolytic effects of etifoxine comparable to clonazepam have recently been confirmed in a randomized controlled double-blind clinical trial in patients with anxiety disorders [76]. At the cellular level, we have characterized the effects of etifoxine in comparison to benzodiazepines regarding neurosteroidogenesis [56]. Moreover, our group recently described differential effects of etifoxine and alprazolam on hypothalamic-pituitary-adrenal (HPA) axis activity in the Trier Social Stress Test in Virtual Reality (VR-TSST) [77]. Furthermore, we recently showed that etifoxine but not alprazolam reduced the fear-potentiated startle in comparison to placebo in an experimental threat paradigm in healthy volunteers [78]. However, the brain circuits underlying the anxiolytic and/or anti-stress effects of TSPO ligands have not been identified so far. Therefore, we conducted a double blinded within-design study with healthy male volunteers to address this issue. In this study, we could also show differential effects of etifoxine and alprazolam on GABAergic function as assessed by double-pulse transcranial magnetic stimulation (TMS) [79] and subtle effects on microbiome composition [80]. To address the question of whether etifoxine may affect peripheral steroid concentrations, steroid profiles were obtained in plasma in a subset of study participants which are reported below.
Steroid plasma profile following treatment with etifoxine
The study population of 36 male participants between the ages of 18 and 55 years was screened by a physician for the absence of physical and psychiatric disorders by physical examination and the German version of the Mini-International Neuropsychiatric Interview (M.I.N.I.) [81, 82]. Study population and design are described in more detail elsewhere [79, 80]. The trial was registered at the European Clinical Trials Register (EudraCT number: 2018-002181-40) and at the German Register of Clinical Studies (DRKS-ID: DRKS00020267) and approved by the ethics committee of University of Regensburg and the German Federal Institute for Drugs and Medical Devices (BfArM). All participants gave their written informed consent. Throughout the experiment, participants were required to abstain from alcohol, driving, and the use of heavy machinery. The order of medication intake was pseudo-randomly assigned - placebo, alprazolam (1.5 mg/d in 3 doses of 0.5 mg) and etifoxine (150 mg/d in 3 doses of 50 mg) for 5 days each, with a washout period of at least 7 days between medications. Due to the complexity and expense of the steroid analysis, only placebo and etifoxine samples from 25 subjects were complete and could be analyzed. These were collected on day 5, 60 min after the 12:00 midday medication intake (placebo or etifoxine). Steroid profiles were determined by means of a highly specific and sensitive gas chromatography coupled to tandem mass spectrometry analysis as described previously [53]. A two-sided t-test was performed between etifoxine and placebo samples per neurosteroid using a bootstrapped (k = 10,000) null distribution generated from the study data.
Figure 3 (Fig. 3) shows peripheral plasma levels of pregnenolone (PREG), progesterone (PROG), 5α-dihydroprogesterone (5α-DHP), 3α,5α-tetrahydroprogesterone (3α,5α-THP, allopregnanolone), 5α-dihydrodeoxycorticosterone (5α-DHDOC), and 3α,5α-tetrahydrodeoxycorticosterone (3α,5α-THDOC) on a logarithmic scale (absolute values are provided in Table 1).

Steroid profile in plasma following administration of etifoxine and placebo in healthy male volunteers. Steroid concentrations in ng/ml are shown logarithmized on the y axis. Error bars indicate 95% confidence intervals. No significant effect of the etifoxine on plasma steroids was detectable. PREG: pregnenolone, PROG: progesterone, 5α-DHP: 5α-dihydrodroprogesterone, 3α,5α-THP: 3α,5α-tetrahydroprogesterone, 5α-DHDOC: 5α-dihydrodeoxycorticosterone, 3α,5α-THDOC: 3α,5α-tetrahydrodeoxycorticosterone
In this clinical study, no effects of etifoxine were detected in the plasma of male volunteers. This is in contrast to findings in rats, where etifoxine caused increases in progesterone and 3α-reduced steroids in both brain and plasma [83, 84] as well as in cellular systems [56]. Presumed differences in tissue-specific enzymatic machinery and between rats and humans preclude further conclusions. A part of the steroid pool, like allopregnanolone, is mainly formed in peripheral glands [85], whereas neuronal tissue provides enzymes such as the 5α-reductase and 3α-hydroxysteroid dehydrogenase to synthesize meaningful local concentrations of neurosteroids [85,86,87]. In rats, plasma levels are not fully correlated and much lower compared to their levels in neural tissue [88, 89]. Although these findings seem to weaken the role of peripheral plasma levels of neurosteroids as sensitive clinical biomarkers, measurable differences in plasma levels of specific neurosteroids could still be an important marker of pathological conditions with or without reflecting localized brain processes [90]. Ongoing clinical research in our group aims to study the sensitivity of peripheral steroid levels to therapeutic manipulation, e.g. by etifoxine treatment under pathological conditions [91].
Etifoxine and GABAkines in the treatment of depression
Because TSPO ligands may induce neurosteroidogenesis, they may represent an alternative approach to exogenously applied 3α-reduced steroids such as brexanolone or zuranolone in the treatment of postpartum depression or major depressive disorder. As outlined above, our group is currently performing a first proof of concept study assessing the potential of etifoxine as adjunct treatment in depression in relation to neuroimaging and microbiome parameters. Moreover, modifications of the etifoxine molecule, such as the GABAkine GRX-917 [92], may provide TSPO ligands with improved pharmacokinetic properties. It is noteworthy that side effects such as sedation, tolerance development and abuse liability, which are typical for benzodiazepines, have not been reported for TSPO ligands so far, which further supports their investigation as a novel treatment option.
Conclusion and outlook.
The neurosteroid field is known for decades. However, it has recently gained considerable interest due to industrial efforts, e.g., by SAGE/Biogen, to develop 3α-reduced steroids such as brexanolone and zuranolone as novel therapeutic agents for affective disorders. With respect to postpartum depression, it is quite clear that the dramatic drop in progesterone after childbirth leads to a corresponding drop in 3α-reduced steroids, which may contribute to the psychopathology of postpartum depression. As a clinical consequence, the administration of exogenous 3α-reduced steroids has a solid pathophysiological rationale. In fact, the portfolio of studies presented has led to the approval of brexanolone and zuranolone for the treatment of postpartum depression. The situation is less clear in major depressive disorder. Although subtle changes in neurosteroid composition have been reported in major depressive disorder and anxiety disorders, and antidepressants have been shown to interfere with neurosteroidogenic enzymes, the magnitude of alterations is only marginal compared to postpartum depression. Nevertheless, it is intriguing that the study portfolio presented by SAGE/Biogen suggests a rapid antidepressant potential of zuranolone also in major depressive disorder. However, zuranolone has not been approved by the FDA for major depressive disorder due to several concerns that need to be addressed. According to the authors, the following questions should be addressed: What will be the side-effect profile after a more prolonged administration beyond 14 days? Will there be abuse liability or tolerance effects? What will be the efficacy over a longer period of time? Will these agents find their place as a monotherapy or as adjuncts to standard antidepressants? Will interval therapy become a novel treatment regimen? Moreover, the induction of endogenous neurosteroidogenesis via TSPO ligands such as etifoxine or derivatives may be an interesting alternative approach. Will these two approaches provide similar clinical effects, or will there be a different clinical profile given the broad spectrum of action of TSPO ligands? In conclusion, it is encouraging that the progress made in the field of neurosteroids and TSPO has now opened the door to new treatment options. Hopefully, these will find their way as alternative strategies to existing pharmacological and non-pharmacological treatments for affective disorders.
References
Malhi GS, Mann JJ (2018) Depression. The Lancet 392:2299–2312. https://doi.org/10.1016/S0140-6736(18)31948-2
Cheng Q, Huang J, Xu L, Li Y, Li H, Shen Y, Zheng Q, Li L (2020) Analysis of Time-Course, Dose-Effect, and influencing factors of antidepressants in the treatment of Acute adult patients with Major Depression. Int J Neuropsychopharmacol 23:76–87. https://doi.org/10.1093/ijnp/pyz062
Kritzer M (2021) Ketamine for treatment of mood disorders and suicidality: a narrative review of recent progress. Ann Clin Psychiatry 33:4. https://doi.org/10.12788/acp.0048
Sáiz-Vázquez O, Gracia-García P, Ubillos-Landa S, Puente-Martínez A, Casado-Yusta S, Olaya B, Santabárbara J (2021) Depression as a risk factor for Alzheimer’s Disease: a systematic review of longitudinal Meta-analyses. J Clin Med 10:1809. https://doi.org/10.3390/jcm10091809
Rupprecht R, Wetzel CH, Dorostkar M, Herms J, Albert NL, Schwarzbach J, Schumacher M, Neumann ID (2022) Translocator protein (18 kDa) TSPO: a new diagnostic or therapeutic target for stress-related disorders? Mol Psychiatry 27:2918–2926. https://doi.org/10.1038/s41380-022-01561-3
Edinoff AN, Nix CA, Hollier J, Sagrera CE, Delacroix BM, Abubakar T, Cornett EM, Kaye AM, Kaye AD (2021) Benzodiazepines: uses, dangers, and clinical considerations. Neurol Int 13:594–607. https://doi.org/10.3390/neurolint13040059
Pariente A, De Gage SB, Moore N, Bégaud B (2016) The benzodiazepine–dementia disorders Link: current state of knowledge. CNS Drugs 30:1–7. https://doi.org/10.1007/s40263-015-0305-4
Barker M (2004) Persistence of cognitive effects after withdrawal from long-term benzodiazepine use: a meta-analysis. Arch Clin Neuropsychol 19:437–454. https://doi.org/10.1016/S0887-6177(03)00096-9
Furukawa TA, Streiner D, Young LT, Kinoshita Y (2001) Antidepressants plus benzodiazepines for major depression. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD001026
Ogawa Y, Takeshima N, Hayasaka Y, Tajika A, Watanabe N, Streiner D, Furukawa TA (2019) Antidepressants plus benzodiazepines for adults with major depression. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD001026.pub2
Rupprecht R, Pradhan AK, Kufner M, Brunner LM, Nothdurfter C, Wein S, Schwarzbach J, Puig X, Rupprecht C, Rammes G (2022) Neurosteroids and translocator protein 18 kDa (TSPO) in depression: implications for synaptic plasticity, cognition, and treatment options. Eur Arch Psychiatry Clin Neurosci 273:1477–1487. https://doi.org/10.1007/s00406-022-01532-3
Rupprecht R, Holsboer F (1999) Neuroactive steroids: mechanisms of action and neuropsychopharmacological perspectives. Trends Neurosci 22:410–416. https://doi.org/10.1016/S0166-2236(99)01399-5
Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapère J-J, Lindemann P, Norenberg MD, Nutt D, Weizman A, Zhang M-R, Gavish M (2006) Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci 27:402–409. https://doi.org/10.1016/j.tips.2006.06.005
Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M (2010) Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov 9:971–988. https://doi.org/10.1038/nrd3295
Chen Z-W, Bracamontes JR, Budelier MM, Germann AL, Shin DJ, Kathiresan K, Qian M-X, Manion B, Cheng WWL, Reichert DE, Akk G, Covey DF, Evers AS (2019) Multiple functional neurosteroid binding sites on GABAA receptors. PLOS Biol 17:e3000157. https://doi.org/10.1371/journal.pbio.3000157
Rudolph U, Crestani F, Benke D, Brünig I, Benson JA, Fritschy J-M, Martin JR, Bluethmann H, Möhler H (1999) Benzodiazepine actions mediated by specific γ-aminobutyric acidA receptor subtypes. Nature 401:796–800. https://doi.org/10.1038/44579
Luscher B, Maguire JL, Rudolph U, Sibille E (2023) GABAA receptors as targets for treating affective and cognitive symptoms of depression. Trends Pharmacol Sci 44:586–600. https://doi.org/10.1016/j.tips.2023.06.009
Locci A, Pinna G (2017) Neurosteroid biosynthesis down-regulation and changes in GABA A receptor subunit composition: a biomarker axis in stress‐induced cognitive and emotional impairment. Br J Pharmacol 174:3226–3241. https://doi.org/10.1111/bph.13843
Paul SM, Pinna G, Guidotti A (2020) Allopregnanolone: from molecular pathophysiology to therapeutics. A historical perspective. Neurobiol Stress 12:100215. https://doi.org/10.1016/j.ynstr.2020.100215
Althaus AL, Ackley MA, Belfort GM, Gee SM, Dai J, Nguyen DP, Kazdoba TM, Modgil A, Davies PA, Moss SJ, Salituro FG, Hoffmann E, Hammond RS, Robichaud AJ, Quirk MC, Doherty JJ (2020) Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator. Neuropharmacology 181:108333. https://doi.org/10.1016/j.neuropharm.2020.108333
Zorumski CF, Paul SM, Izumi Y, Covey DF, Mennerick S (2013) Neurosteroids, stress and depression: potential therapeutic opportunities. Neurosci Biobehav Rev 37:109–122. https://doi.org/10.1016/j.neubiorev.2012.10.005
Romeo E, Ströhle A, Spalletta G, Michele FD, Hermann B, Holsboer F, Pasini A, Rupprecht R (1998) Effects of antidepressant treatment on neuroactive steroids in Major Depression. Am J Psychiatry 155:910–913. https://doi.org/10.1176/ajp.155.7.910
Uzunova V, Sheline Y, Davis JM, Rasmusson A, Uzunov DP, Costa E, Guidotti A (1998) Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine. Proc Natl Acad Sci 95:3239–3244. https://doi.org/10.1073/pnas.95.6.3239
Ströhle A, Romeo E, Di Michele F, Pasini A, Hermann B, Gajewsky G, Holsboer F, Rupprecht R (2003) Induced panic attacks Shift γ-Aminobutyric acid type a receptor Modulatory Neuroactive Steroid Composition in patients with panic disorder: preliminary results. Arch Gen Psychiatry 60:161. https://doi.org/10.1001/archpsyc.60.2.161
Griffin LD, Mellon SH (1999) Selective serotonin reuptake inhibitors directly alter activity of neurosteroidogenic enzymes. Proc Natl Acad Sci 96:13512–13517. https://doi.org/10.1073/pnas.96.23.13512
Schüle C, Romeo E, Uzunov DP, Eser D, Di Michele F, Baghai TC, Pasini A, Schwarz M, Kempter H, Rupprecht R (2006) Influence of mirtazapine on plasma concentrations of neuroactive steroids in major depression and on 3α-hydroxysteroid dehydrogenase activity. Mol Psychiatry 11:261–272. https://doi.org/10.1038/sj.mp.4001782
Schüle C, di Michele F, Baghai T, Romeo E, Bernardi G, Zwanzger P, Padberg F, Pasini A, Rupprecht R (2003) Influence of sleep deprivation major depression on neuroactive steroids in major depression. Neuropsychopharmacology 28:577–581
Padberg F (2002) Plasma concentrations of neuroactive steroids before and after Repetitive Transcranial Magnetic Stimulation (rTMS) in Major Depression. Neuropsychopharmacology 27:874–878. https://doi.org/10.1016/S0893-133X(02)00355-X
Baghai TC, Di Michele F, Schüle C, Eser D, Zwanzger P, Pasini A, Romeo E, Rupprecht R (2005) Plasma concentrations of neuroactive steroids before and after Electroconvulsive Therapy in Major Depression. Neuropsychopharmacology 30:1181–1186. https://doi.org/10.1038/sj.npp.1300684
Deligiannidis KM, Kroll-Desrosiers AR, Tan Y, Dubuke ML, Shaffer SA (2020) Longitudinal proneuroactive and neuroactive steroid profiles in medication-free women with, without and at-risk for perinatal depression: a liquid chromatography-tandem mass spectrometry analysis. Psychoneuroendocrinology 121:104827. https://doi.org/10.1016/j.psyneuen.2020.104827
Timby E, Bäckström T, Nyberg S, Stenlund H, Wihlbäck A-CN, Bixo M (2016) Women with premenstrual dysphoric disorder have altered sensitivity to allopregnanolone over the menstrual cycle compared to controls—a pilot study. Psychopharmacology 233:2109–2117. https://doi.org/10.1007/s00213-016-4258-1
Walton N, Maguire J (2019) Allopregnanolone-based treatments for postpartum depression: Why/how do they work? Neurobiol Stress 11:100198. https://doi.org/10.1016/j.ynstr.2019.100198
Powell JG, Garland S, Preston K, Piszczatoski C (2020) Brexanolone (Zulresso): finally, an FDA-Approved treatment for Postpartum Depression. Ann Pharmacother 54:157–163. https://doi.org/10.1177/1060028019873320
Kanes S, Colquhoun H, Gunduz-Bruce H, Raines S, Arnold R, Schacterle A, Doherty J, Epperson CN, Deligiannidis KM, Riesenberg R, Hoffmann E, Rubinow D, Jonas J, Paul S, Meltzer-Brody S (2017) Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet 390:480–489. https://doi.org/10.1016/S0140-6736(17)31264-3
Meltzer-Brody S, Colquhoun H, Riesenberg R, Epperson CN, Deligiannidis KM, Rubinow DR, Li H, Sankoh AJ, Clemson C, Schacterle A, Jonas J, Kanes S (2018) Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet 392:1058–1070. https://doi.org/10.1016/S0140-6736(18)31551-4
Epperson CN, Rubinow DR, Meltzer-Brody S, Deligiannidis KM, Riesenberg R, Krystal AD, Bankole K, Huang M-Y, Li H, Brown C, Kanes SJ, Lasser R (2023) Effect of brexanolone on depressive symptoms, anxiety, and insomnia in women with postpartum depression: pooled analyses from 3 double-blind, randomized, placebo-controlled clinical trials in the HUMMINGBIRD clinical program. J Affect Disord 320:353–359. https://doi.org/10.1016/j.jad.2022.09.143
Garafola S, Shiferaw E, Dev V (2023) Safety of Brexanolone in adults with Postpartum Depression: Postmarketing Surveillance Data. Drugs - Real World Outcomes 10:351–356. https://doi.org/10.1007/s40801-023-00372-4
Heo Y-A (2023) Zuranolone: first approval. Drugs 83:1559–1567. https://doi.org/10.1007/s40265-023-01953-x
Barnes KN, Vogl CM, Nelson LA (2023) Zuranolone: the first FDA-Approved oral treatment option for Postpartum Depression. Ann Pharmacother 0:0. https://doi.org/10.1177/10600280231204953
Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, Doherty J, Jonas J, Li S, Sankoh AJ, Silber C, Campbell AD, Werneburg B, Kanes SJ, Lasser R (2021) Effect of Zuranolone vs Placebo in Postpartum Depression: a Randomized Clinical Trial. JAMA Psychiatry 78:951. https://doi.org/10.1001/jamapsychiatry.2021.1559
Bullock A, Gunduz-Bruce H, Zammit GK, Qin M, Li H, Sankoh AJ, Silber C, Kanes SJ, Jonas J, Doherty J (2022) A phase 1 double‐blind, placebo‐controlled study of zuranolone (SAGE‐217) in a phase advance model of insomnia in healthy adults. Hum Psychopharmacol Clin Exp 37:e2806. https://doi.org/10.1002/hup.2806
Gunduz-Bruce H, Silber C, Kaul I, Rothschild AJ, Riesenberg R, Sankoh AJ, Li H, Lasser R, Zorumski CF, Rubinow DR, Paul SM, Jonas J, Doherty JJ, Kanes SJ (2019) Trial of SAGE-217 in patients with major depressive disorder. N Engl J Med 381:903–911. https://doi.org/10.1056/NEJMoa1815981
Suthoff E, Kosinski M, Arnaud A, Hodgkins P, Gunduz-Bruce H, Lasser R, Silber C, Sankoh AJ, Li H, Werneburg B, Jonas J, Doherty J, Kanes SJ, Bonthapally V (2022) Patient-reported health-related quality of life from a randomized, placebo-controlled phase 2 trial of zuranolone in adults with major depressive disorder. J Affect Disord 308:19–26. https://doi.org/10.1016/j.jad.2022.03.068
Arnaud A, Suthoff E, Stenson K, Werneburg B, Hodgkins P, Bonthapally V, Jonas J, Meyer K, O’Day K (2021) Number needed to treat and Number needed to harm analysis of the zuranolone phase 2 clinical trial results in major depressive disorder. J Affect Disord 285:112–119. https://doi.org/10.1016/j.jad.2021.02.027
Clayton AH, Lasser R, Parikh SV, Iosifescu DV, Jung J, Kotecha M, Forrestal F, Jonas J, Kanes SJ, Doherty J (2023) Zuranolone for the treatment of adults with major depressive disorder: a Randomized, Placebo-Controlled phase 3 trial. Am J Psychiatry 180:676–684. https://doi.org/10.1176/appi.ajp.20220459
Clayton AH, Lasser R, Nandy I, Sankoh AJ, Jonas J, Kanes SJ (2023) Zuranolone in major depressive disorder: results from MOUNTAIN—A phase 3, Multicenter, Double-Blind, randomized, Placebo‐Controlled Trial. J Clin Psychiatry 84:22m14445. https://doi.org/10.4088/JCP.22m14445
Cutler AJ, Mattingly GW, Kornstein SG, Aaronson ST, Lasser R, Zhang H, Rana N, Brown C, Levin S, Miller C, Kotecha M, Forrestal F, Doherty J (2023) Long-term safety and efficacy of initial and repeat treatment courses with zuranolone in adult patients with major depressive disorder: interim results from the Open-Label, phase 3 SHORELINE Study. J Clin Psychiatry 85:23m14845. https://doi.org/10.4088/JCP.23m14845
Lin Y-W, Tu Y-K, Hung K-C, Liang C-S, Tseng P-T, Lin P-Y, Chia-Cheng Lai E, Hsu C-W (2023) Efficacy and safety of zuranolone in major depressive disorder: a meta-analysis of factor effect and dose-response analyses. eClinicalMedicine 66:102308. https://doi.org/10.1016/j.eclinm.2023.102308
Prasad V, Allely D (2024) Concerns that may limit the utility of Zuranolone. JAMA 331:105. https://doi.org/10.1001/jama.2023.26103
Rupprecht R, Rupprecht C, Di Benedetto B, Rammes G (2022) Neuroinflammation and psychiatric disorders: relevance of C1q, translocator protein (18 kDa) (TSPO), and neurosteroids. World J Biol Psychiatry 23:257–263. https://doi.org/10.1080/15622975.2021.1961503
Fan J, Campioli E, Midzak A, Culty M, Papadopoulos V (2015) Conditional steroidogenic cell-targeted deletion of TSPO unveils a crucial role in viability and hormone-dependent steroid formation. Proc Natl Acad Sci 112:7261–7266. https://doi.org/10.1073/pnas.1502670112
Selvaraj V, Stocco DM, Tu LN (2015) Minireview: Translocator Protein (TSPO) and steroidogenesis: a reappraisal. Mol Endocrinol 29:490–501. https://doi.org/10.1210/me.2015-1033
Liere P, Liu G-J, Pianos A, Middleton RJ, Banati RB, Akwa Y (2023) The Comprehensive Steroidome in Complete TSPO/PBR knockout mice under basal conditions. Int J Mol Sci 24:2474. https://doi.org/10.3390/ijms24032474
Shi Y, Cui M, Ochs K, Brendel M, Strübing FL, Briel N, Eckenweber F, Zou C, Banati RB, Liu G-J, Middleton RJ, Rupprecht R, Rudolph U, Zeilhofer HU, Rammes G, Herms J, Dorostkar MM (2022) Long-term diazepam treatment enhances microglial spine engulfment and impairs cognitive performance via the mitochondrial 18 kDa translocator protein (TSPO). Nat Neurosci 25:317–329. https://doi.org/10.1038/s41593-022-01013-9
Pradhan AK, Neumüller T, Klug C, Fuchs S, Schlegel M, Ballmann M, Tartler KJ, Pianos A, Garcia MS, Liere P, Schumacher M, Kreuzer M, Rupprecht R, Rammes G (2023) Chronic administration of XBD173 ameliorates cognitive deficits and neuropathology via 18 kDa translocator protein (TSPO) in a mouse model of Alzheimer’s disease. Transl Psychiatry 13:332. https://doi.org/10.1038/s41398-023-02630-z
Wolf L, Bauer A, Melchner D, Hallof-Buestrich H, Stoertebecker P, Haen E, Kreutz M, Sarubin N, Milenkovic V, Wetzel C, Rupprecht R, Nothdurfter C (2015) Enhancing Neurosteroid synthesis – relationship to the Pharmacology of Translocator protein (18 kDa) (TSPO) ligands and benzodiazepines. Pharmacopsychiatry 48:72–77. https://doi.org/10.1055/s-0034-1398507
Barron AM, Higuchi M, Hattori S, Kito S, Suhara T, Ji B (2021) Regulation of anxiety and depression by mitochondrial translocator protein-mediated steroidogenesis: the role of neurons. Mol Neurobiol 58:550–563. https://doi.org/10.1007/s12035-020-02136-5
Pini S, Martini C, Abelli M, Muti M, Gesi C, Montali M, Chelli B, Lucacchini A, Cassano GB (2005) Peripheral-type benzodiazepine receptor binding sites in platelets of patients with panic disorder associated to separation anxiety symptoms. Psychopharmacology 181:407–411. https://doi.org/10.1007/s00213-005-2247-x
Abelli M, Chelli B, Costa B, Lari L, Cardini A, Gesi C, Muti M, Lucacchini A, Martini C, Cassano GB, Pini S (2010) Reductions in platelet 18-kDa Translocator Protein Density Are Associated with adult separation anxiety in patients with bipolar disorder. Neuropsychobiology 62:98–103. https://doi.org/10.1159/000315440
Sarubin N, Baghai T, Lima-Ojeda J, Melchner D, Hallof-Buestrich H, Wolf L, Hilbert S, Milenkovic V, Wetzel C, Rupprecht R, Nothdurfter C (2016) Translocator Protein (TSPO) expression in platelets of depressed patients decreases during antidepressant therapy. Pharmacopsychiatry 49:204–209. https://doi.org/10.1055/s-0042-107795
Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, Suridjan I, Kennedy JL, Rekkas PV, Houle S, Meyer JH (2015) Role of Translocator Protein Density, a marker of Neuroinflammation, in the Brain during Major depressive episodes. JAMA Psychiatry 72:268. https://doi.org/10.1001/jamapsychiatry.2014.2427
Li H, Sagar AP, Kéri S (2018) Microglial markers in the frontal cortex are related to cognitive dysfunctions in major depressive disorder. J Affect Disord 241:305–310. https://doi.org/10.1016/j.jad.2018.08.021
Attwells S, Setiawan E, Wilson AA, Rusjan PM, Mizrahi R, Miler L, Xu C, Richter MA, Kahn A, Kish SJ, Houle S, Ravindran L, Meyer JH (2017) Inflammation in the neurocircuitry of obsessive-compulsive disorder. JAMA Psychiatry 74:833. https://doi.org/10.1001/jamapsychiatry.2017.1567
Attwells S, Setiawan E, Rusjan PM, Xu C, Hutton C, Rafiei D, Varughese B, Kahn A, Kish SJ, Vasdev N, Houle S, Meyer JH (2020) Translocator protein distribution volume predicts reduction of symptoms during open-label trial of Celecoxib in Major Depressive Disorder. Biol Psychiatry 88:649–656. https://doi.org/10.1016/j.biopsych.2020.03.007
Bhatt S, Hilmer AT, Girgenti MJ, Rusowicz A, Kapinos M, Nabuls N (2020) PTSD is associated with neuroimmune suppression: evidence from PET imaging and postmortem transctriptional studies. Nat Commun 11:2360. https://doi.org/10.1038/s41467-020-15930-5
Tournier BB, Tsartsalis S, Ceyzeriat K, Garibotto V, Millet P (2020) In vivo TSPO signal and neuroinflammation in Alzheimer´s disease. Cells 9:1941. https://doi.org/10.3390/cells9091941
Notter T, Schalbetter SM, Clifton NE, Mattei D, Richetto J, Thomas K, Meyer U, Hall J (2021) Neuronal activity increases translocator protein (TSPO) levels. Mol Psychiatry 26:2025–2037. https://doi.org/10.1038/s41380-020-0745-1
Colasanti A, Owen DR, Grozeva D, Rabiner EA, Matthews PM, Craddock N, Young AH (2013) Bipolar disorder is associated with the rs6971 polymorphism in the gene encoding 18 kDa translocator protein (TSPO). Psychoneuroendocrinology 38:2826–2829. https://doi.org/10.1016/j.psyneuen.2013.07.007
Prossin AR, Chandler M, Ryan KA, Saunders EF, Kamali M, Papadopoulos V, Zöllner S, Dantzer R, McInnis MG (2018) Functional TSPO polymorphism predicts variance in the diurnal cortisol rhythm in bipolar disorder. Psychoneuroendocrinology 89:194–202. https://doi.org/10.1016/j.psyneuen.2018.01.013
Rupprecht R, Rammes G, Eser D, Baghai TC, Schüle C, Nothdurfter C, Troxler T, Gentsch C, Kalkman HO, Chaperon F, Uzunov V, McAllister KH, Bertaina-Anglade V, La Rochelle CD, Tuerck D, Floesser A, Kiese B, Schumacher M, Landgraf R, Holsboer F, Kucher K (2009) Translocator Protein (18 kD) as target for Anxiolytics without Benzodiazepine-Like Side effects. Science 325:490–493. https://doi.org/10.1126/science.1175055
Shang C, Yao RM, Guo Y, Ding ZC, Sun LJ, Ran YH (2020) Translocator protein-mediated fast-onset antidepressant-like and memory-enhancing effects in chronically stressed mice. J Psychopharmacol 34:441–451. https://doi.org/10.1177/0269881119896304
Ren P, Ma L, Wang J-Y, Guo H, Sun L, Gao M-L, Liu Y-Z, Ma Y-Q, Li Y-F, Guo W-Z (2020) Anxiolytic and anti-depressive like effects of Translocator protein (18 kDa) Ligand YL-IPA08 in a rat model of Postpartum Depression. Neurochem Res 45:1746–1757. https://doi.org/10.1007/s11064-020-03036-9
Nozaki K, Ito H, Ohgidani M, Yamawaki Y, Sahin EH, Kitajima T, Katsumata S, Yamawaki S, Kato TA, Aizawa H (2020) Antidepressant effect of the translocator protein antagonist ONO-2952 on mouse behaviors under chronic social defeat stress. Neuropharmacology 162:107835. https://doi.org/10.1016/j.neuropharm.2019.107835
Mattei C, Taly A, Soualah Z, Saulais O, Henrion D, Guérineau NC, Verleye M, Legros C (2019) Involvement of the GABAA receptor α subunit in the mode of action of etifoxine. Pharmacol Res 145:104250. https://doi.org/10.1016/j.phrs.2019.04.034
Nguyen N, Fakra E, Pradel V, Jouve E, Alquier C, Guern ME (2006) Efficacy of etifoxine compared to lorazepam monotherapy in the treatment of patients with adjustment disorders with anxiety: a double-blind controlled study in general practice. Hum Psychopharmacol 21:139–149. https://doi.org/10.1002/hup.757
Vicente B, Saldicia S, Hormazabal N, Bustos C, Rubi P (2020) Etifoxine is non-inferior than clonazepam for the reduction of anxiety disorders: a randomized, double blind, non-inferiority trial. Psychopharmacology 237:3357–3367. https://doi.org/10.1007/s00213-020-05617-6
Bahr LM, Maurer F, Weigl J, Weber K, Melchner D, Dörfelt A (2021) Dissociation of endocrine responses to the Trier social stress test in virtual reality (VR-TSST) by the benzodiazepine alprazolam and the translocator protein 18 kDa (TSPO) ligand etifoxine. Psychoneuroendocrinology 124:105100. https://doi.org/10.1016/j.psyneuen.2020.105100
Brunner LM, Maurer F, Weber K, Weigl J, Milenkovic VM, Rupprecht R, Nothdurfter C, Mühlberger A (2022) Differential effects of the translocator protein 18 kDa (TSPO) ligand etifoxine and the benzodiazepine alprazolam on startle response to predictable threat in a NPU-threat task after acute and short-term treatment. Psychopharmacology 1:1–12. https://doi.org/10.1007/S00213-022-06111-X/FIGURES/3
Riebel M, Von Pappenheim B, Kanig C, Nothdurfter C, Wetter TC, Rupprecht R, Schwarzbach J (2023) GABAergic effects of Etifoxine and Alprazolam assessed by double pulse TMS. Pharmacopsychiatry 56:154–161. https://doi.org/10.1055/a-2078-4823
Manook A, Baghai TC, Riebel M, Nothdurfter C, Schwarzbach JV, Gessner A, Rupprecht R, Hiergeist A (2023) Short-term effects of etifoxine on human gut microbiome in healthy men. Front Neurosci 17:1188847. https://doi.org/10.3389/fnins.2023.1188847
Lecrubier Y, Sheehan D, Weiller E, Amorim P, Bonora I, Sheehan KH, Janavs J, Dunbar G (1997) The Mini International Neuropsychiatric interview (MINI). A short diagnostic structured interview: reliability and validity according to the CIDI. Eur Psychiatry 12:224–231. https://doi.org/10.1016/S0924-9338(97)83296-8
Ackenheil M, Stotz-Ingenlath G, Dietz-Bauer R, Vossen A (1999) MINI mini international neuropsychiatric interview, German version 5.0. 0 DSM IV. Munich Psychiatr Univ Clin
Verleye M, Akwa Y, Liere P, Ladurelle N, Pianos A, Eychenne B, Schumacher M, Gillardin J-M (2005) The anxiolytic etifoxine activates the peripheral benzodiazepine receptor and increases the neurosteroid levels in rat brain. Pharmacol Biochem Behav 82:712–720. https://doi.org/10.1016/j.pbb.2005.11.013
Liere P, Pianos A, Oudinet J-P, Schumacher M, Akwa Y (2017) Differential effects of the 18-kDa translocator protein (TSPO) ligand etifoxine on steroidogenesis in rat brain, plasma and steroidogenic glands: pharmacodynamic studies. Psychoneuroendocrinology 83:122–134. https://doi.org/10.1016/j.psyneuen.2017.05.022
Kancheva L, Hill M, Včeláková H, Vrbíková J, Pelikánová T, Stárka L (2007) The identification and simultaneous quantification of neuroactive androstane steroids and their polar conjugates in the serum of adult men, using gas chromatography–mass spectrometry. Steroids 72:792–801. https://doi.org/10.1016/j.steroids.2007.06.006
Baulieu E (1997) Neurosteroids: of the nervous system, by the nervous system, for the nervous system. Recent Prog Horm Res 52:1–32 PMID: 9238846
Stoffel-Wagner B (2003) Neurosteroid biosynthesis in the human brain and its clinical implications. Ann N Y Acad Sci 1007:64–78. https://doi.org/10.1196/annals.1286.007
Caruso D, Scurati S, Maschi O, De Angelis L, Roglio I, Giatti S, Garcia-Segura LM, Melcangi RC (2008) Evaluation of neuroactive steroid levels by liquid chromatography–tandem mass spectrometry in central and peripheral nervous system: Effect of diabetes. Neurochem Int 52:560–568. https://doi.org/10.1016/j.neuint.2007.06.004
Caruso D, Pesaresi M, Abbiati F, Calabrese D, Giatti S, Garcia-Segura LM, Melcangi RC (2013) Comparison of plasma and cerebrospinal fluid levels of neuroactive steroids with their brain, spinal cord and peripheral nerve levels in male and female rats. Psychoneuroendocrinology 38:2278–2290. https://doi.org/10.1016/j.psyneuen.2013.04.016
Kancheva R, Hill M, Novák Z, Chrastina J, Kancheva L, Stárka L (2011) Neuroactive steroids in periphery and cerebrospinal fluid. Neuroscience 191:22–27. https://doi.org/10.1016/j.neuroscience.2011.05.054
Brunner L-M, Riebel M, Wein S, Koller M, Zeman F, Huppertz G, Emmer T, Eberhardt Y, Schwarzbach J, Rupprecht R, Nothdurfter C (2024) The translocator protein 18 kDa ligand etifoxine in the treatment of depressive disorders—a double-blind, randomized, placebo-controlled proof-of-concept study. Trials 25:274. https://doi.org/10.1186/s13063-024-08120-x
Witkin JM, Lippa A, Smith JL, Jin X, Ping X, Biggerstaff A (2022) The imidazodiazepine, KRM-II-81: an example of a newly emerging generations of GABAkines for neurological and psychiatric disorders. Phamacol Biochem Behav 213:173321. https://doi.org/10.1016/j.pbb.2021.173321
Acknowledgements
This work has been supported by the German Research Foundation (Deutsche Forschungsgemeinschaft) (DFG), project number 422179811 to RR, CN and JS.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical standards
All clinical studies have been approved by the local ethics committee and have been performed in accordanve with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Conflict of interest
The other authors declare that they have no conflict of interest.
Competing interests
RR has received consultancy honoraria from SAGE/Biogen and GABA Therapeutics.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Riebel, M., Brunner, LM., Nothdurfter, C. et al. Neurosteroids and translocator protein 18 kDa (TSPO) ligands as novel treatment options in depression. Eur Arch Psychiatry Clin Neurosci (2024). https://doi.org/10.1007/s00406-024-01843-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00406-024-01843-7