
Abstract
Suicide is a leading cause of death worldwide. Suicide ideation (SI) is a known risk factor for suicide behaviour (SB). The current psychobiology and genetic predisposition to SI and SB are poorly defined. Despite convincing relevance of a genetic background for SI, there is no current implementable knowledge about the genetic makeup that identifies subjects at risk for it. One of the possible reasons for the absence of a clear-cut evidence is the polygenetic nature of SI along with the very large sample sizes that are needed to observe significant genetic association result. The CATIE sample was instrumental to the analysis. SI was retrieved as measured by the Calgary test. Clinical possible covariates were identified by a nested regression model. A principal component analysis helped in defining the possible genetic stratification factors. A GWAS analysis, polygenic risk score associated with a random forest analysis and a molecular pathway analysis were undertaken to identify the genetic contribution to SI. As a result, 741 Schizophrenic individuals from the CATIE were available for the genetic analysis, including 166,325 SNPs after quality control and pruning. No GWAS significant result was found. The random forest analysis conducted by combining the polygenic risk score and several clinical variables resulted in a possibly overfitting model (OOB error rate < 1%). The molecular pathway analysis revealed several molecular pathways possibly involved in SI, of which those involved in microglia functioning were of particular interest. A medium-small sample of SKZ individuals was analyzed to shed a light on the genetic of SI. As an expected result from the underpowered sample, no GWAS positive result was retrieved, but the molecular pathway analysis indicated a possible role of microglia and neurodevelopment in SI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Suicide ideation (SI) and behavior (SB) are a world-wide concern. About 800.000 individuals per year globally commit suicide. The standardized suicide rate for males and females were respectively 12 and 6/100.000 in 2019. Suicide is the second leading cause of death in younger individuals [1]. It has been estimated that the costs related to SB were in 2014 as high as 5.53 billion dollars in lost economic income with an average cost for a single suicide event estimated to be as high as 803,000 dollars [2].
The current approach to the identification and prevention of SB consists in at least three different kinds of interventions, including school-level prevention [3], brief psychological and supportive interventions [4] and follow-up interventions [5]. Exposure to suicide or suicide attempts increases the risk of suicidal behavior [6]. One of the strongest clinical predictors of SB is the non-suicidal self-injury [7]. SI and SB are favored by predisposing risk factors such as loneliness, hopelessness, demoralization, economics factors, cultural factors, or social isolation due to belonging to a social minority [8, 9].
At the moment of writing there is no implementable biologic tool to predict suicide risk in specific populations, but it is reported that the absolute risk is higher in psychiatric populations, with a suicide risk during the first year of follow-up of 1.4% compared with 0.23% in non-psychiatric populations. Evaluating the risk of SB in individual affected by major psychiatric disorders is essential to prevent this occurrence [10, 11]. In particular, one group of individuals that have higher risk for SI and SB are those suffering from Schizophrenia (SKZ). SKZ is a chronic mental disorder with a prevalence of about 1% in general population. It is characterized by positive symptoms, including hallucinations, delusions and disorders of thought that may lead to aggressive or risky behavior; negative symptoms defined by impaired speech, tendence to isolation, abulia and emotional flattening; and cognitive impairment with deficits in verbal and working memory and attention disorders [12]. Regarding the pathogenesis, SKZ has been described as a neurodevelopmental disorder in which alterations of central nervous system can appear around or before birth, as well as around the onset of psychosis. A central role has been attributed to the disruption of the neural pruning, which could lead to an excessive loss of synapses and consequently to the emergence of the illness [13].
The SB rate in SKZ ranges from 1.49% (completers) [14], to 20.3% (attempters) [15]. The RR is as high as 9.76 (completers) [16].
As for SKZ [17], both SI and SB have a genetic basis, reviewed in here [18] and here [19]. It has been reported that the genetic contribution to the development of SI and SB would range from 30 to 50% [1]. In particular, classical genetic studies reported a RR of 8.38 for biological relatives of individuals who committed suicide. Despite this, risk genes for SI and SB in SKZ have not been consistently identified [9]. The last GWAS analyses revealed although a genetic predisposition that can be determined via the polygenic risk score (PRS): SB and SI probably have a multigenic nature [20]. SKZ also have a genetic origin, this confirmed by the first twin studies reviewed here [21]and here [22] and by the last GWAS investigations in this field, see for example here [23] and here [24].
The present contribution aims at the identification of the genetic risk for suicide in individuals with SKZ. The hypothesis under analysis is that there is a combined genetic background for SI and SB in SKZ individuals. This line of research is not new, see Table 1 for an overview about a selection of the most relevant studies in the field. In the present contribution we undertake a combined approach that includes both a classic GWAS approach, a molecular pathway analysis, a polygenic risk score (PRS) and a machine learning approach to test the hypothesis that a specific set of molecular pathways or a specific genetic background may inform the risk for SI in SKZ. Due to the limits of the database under analysis it is only SI, which is taken into consideration, and not SB. SI has been reported to be a risk factor for SB: about 1 in 10 of the subjects with SI will experience SB [25].
Materials and methods
Sample
The sample under analysis is the NIMH CATIE sample (NIMH contract NO1 MH90001). SKZ patients were enrolled between 1/2001 and 12/2004. CATIE was a multi-phase randomized controlled trial of antipsychotic medications involving 1460 persons with SCZ followed for up to 18 months. The main focus of the CATIE investigation was to test the tolerability of a number of antipsychotic treatments in the “real world” conditions. 51% of CATIE participants donated a DNA sample. This sub-sample is the core of the present investigation. A diagnosis of SKZ was conducted according to the Structured Clinical Interview for DSM-IV (SCID).
Inclusion criteria were: (1) a diagnosis of SCZ, (2) age 18–67 years, (3) clinical decision that oral medication was appropriate, (4) adequate decisional capacity, and (5) provision of written informed consent. Exclusion criteria were: (1) a diagnosis of schizoaffective disorder, (2) mental retardation, or (3) another cognitive disorder, (4) a history of serious adverse reactions to the proposed treatments, (5) only one psychotic episode, (6) a history of treatment resistance, (7) being pregnant or breastfeeding, (8) or a serious and unstable medical condition.
DNA sampling & cell line establishment
Peripheral venous blood samples were sent to the Rutgers University Cell and DNA Repository (RUCDR) where cell lines were established via EBV transformation. RUCDR employs stringent quality control procedures and the success rate for immortalization exceeds 99%. Sample DNA concentrations were quantified and normalized via the use of Picogreen dsDNA Quantitation Kits (Molecular Probes, Eugene, OR).
GWAS sampling and quality control
Affymetrix 500 K “A” chipset and a custom 164 K chip created by Perlegen to provide further coverage for the original sample. For the present analysis, the genetic data available from the HIMH database were downloaded after permission. SNPs (Single Nucleotide Polymorphism) were excluded for allele frequency < 0.01 and low genotype call rate. Deviations from the Hardy–Weinberg equilibrium were accepted under a P-threshold of 0.0001. Pruning was implemented according to standard –indep-pairwise 50 5 0.5 criteria (window size in SNPs (e.g., 50), the number of SNPs to shift the window at each step (e.g., 5), the VIF threshold. The VIF is 1/(1-R^2)). Lambda values served to exclude inflation factors. To avoid genetic stratification events a principal component analysis was conducted and the first 6 components were used as covariates. Covariates were also created from the clinical study of the sample, to avoid spurious genetic associations. For each SNP the statistics of association were generated and used to conduct the analysis of quality (to calculate lambda values), to run the basic GWAS analysis along with the calculation of the PRS and to conduct the molecular pathway analysis. PRS was calculated as the sum of the weighted effect size (or beta) calculated for each SNP. The effect size (or beta) gives the strength of the statistical association of every SNP with the phenotype under analysis. This association is weighted for the number of copies of the specific SNP in each individual.
Genetic association tests and molecular pathway analysis
Plink [26] served for the genetic association test. Single tests for association were generated for every SNP under a logistic regression model. SNPs associated with the investigated phenotype were ranked according to the p of association. SNPs showing a significant (nominal significance, not GWAS significance) (P < 0.05) association with the phenotype under analysis were selected. The genes harboring such variations were identified and were used as the input for the molecular pathway analysis. The molecular pathway analysis was conducted using the R software suit [27] through the packages Bioconductor [28] and ReactomePA [29]. The Reactome [30] is a manually curated database that includes chemical reactions, biological processes, and molecular pathways. SNPs that were used for the molecular pathway analysis were not preselected. Exonic, intronic, and SNPs from regulatory regions were entered into the analysis, allowing for a coverage of the molecular pathways as large as possible. The test for enrichment is corrected for multiple testing, considering the total number of molecular pathways that are tested for enrichment. Both the Bonferroni test (referred to as Padjust in the Results section) for multiple testing and the false discovery rate (referred to as q value in the Results section) were implemented in the analysis. The analysis was carried out in a Linux system in Bash language and the computations were performed through access at the Aarhus University superPC (https://genome.au.dk/). Polygenic risk score (PRS) was calculated and was instrumental to inform the machine learning model.
Machine learning
A random forest (RF) method was applied to test the predictive power of a combined genetic and clinical profile. RF was chosen because it has a non-parametric capacity and can be implemented to solve the classification of two-category issues, which is the case on the present investigation. Multiple decision trees are created and combined in a single model, creating the best fitting classification based by the decision points of each decision tree. The sample was divided in a training set (70%) and a testing set (30%). Accuracy, Specificity and Sensitivity (Confusion Matrix) were used to describe the model.
Definition of the principal outcome
The outcome under analysis is SI as measured by the Calgary scale [31], item number 8. Suicide ideation is explored at each interview and ranked according to the level of intensity as “Absent”, “Mild”, “Moderate” and “Severe”. The single individuals were classified as “cases” (did experience suicide ideation during the trial) and “controls” (did not experience suicide ideation during the trial).
Analysis of covariates
The following clinical covariates were taken into consideration: age, gender, race, employment, marital status, years of education, years of treatment, age at onset and medicine. The clinical variables that were significantly associated with the outcome were included in the genetic analysis as possible stratification factors. The intensity of psychotic symptoms was also analyzed in a nested regression model as a possible confounding factor. The possible genetic stratification was analyzed with the principal component analysis in plink and the first 6 components were included in the genetic analysis.
Result.
Sociodemographic variables
741 individuals from the CATIE study had both the genetic information and the clinical information to complete the present analysis. 274 individuals reported a positive SI at least once during the trial, while 467 individuals did not report it. The sociodemographic characteristics of the sample are reported in Table 2. Briefly, 196 females and 545 males were included, the mean age at the moment of enrolling was 40.89 ± 11.05 years. The mean age at onset and the mean duration of treatment were respectively 14.3 ± 10.83 years and 16.69 ± 11.2 years. Multiple ethnic groups were included in the analyses. White individuals were the most represented group (n = 489), followed by Black or African-American (n = 221). Most part of the enrolled individuals did not have a job (n = 627) at the moment of enrolling, the most part of them had though achieved a GED/High school diploma (n = 252) or at least some college (n = 178) level of education. The most part of the analyzed individuals were never married (n = 441). The covariate analysis for the sociodemographic variables showed that “RACE” as defined in the database was significantly associated with SI: the most relevant result was retrieved for the “Black” as defined in the database population. Individuals with SI in the “Black” group were largely less represented with respect to those without SI (0,36% vs 33%). This variable was then included in the covariate analysis for the genetic association tests. A nested mixed effect regression analysis was instrumental to test the impact of medicines (“DRUGTERM2” in the database) on suicidal ideation in the database. As reported in Table 2, “RACE” and “MARITAL”, indicating respectively the ethnicity and the marital status, were used as the clinical covariates for the genetic analysis.
Genetic and clinical variables
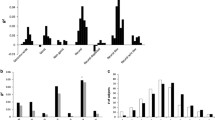
Of the initial 486,935 SNPs available in the genetic database, 166,325 passed the quality analysis because of low allelic representation in the database or because of unbalance in the HWE test. Of the initial database, 322,851 SNPs were excluded after the pruning analysis. The genetic database was not imputated because of the low quality of the imputation analysis in this specific dataset. Pairwise identity-by-state distance clustering allowed for the introduction of the first six dimensions of the pairwise identity-by-state distance structure as covariates for the genetic analysis. The analysis of genetic and clinical covariates resulted in a lambda value as large as 1.01, indicating no inflation factor in the final genetic analysis. The QQ-plot showing the distribution of the observed VS expected P values and the Manhattan plot are represented in Fig. 1.

GWAS analysis. A QQ plot analysis showing no inflation factor after correction for the clinical and genetic covariates along with the Manhattan plot of the GWAS analysis showing no SNPs reaching the GWAS significance level
None of the SNPs investigated achieved a genome-wide significant association with the outcomes under analysis. The following molecular pathways were found to be significantly associated with the outcome under analysis after correction for multiple testing: (1) Protein–protein interactions at synapses; (2) Neurexins and neuroligins; (3) O-linked glycosylation; (4) Glucuronidation; (5) Neuronal System; (6) Defective B3GALTL causes PpS; (7) cGMP effects; (8) Collagen chain trimerization; (9) O-glycosylation of TSR domain-containing proteins; (10) Nitric oxide stimulates guanylate cyclase; (11) Non-integrin membrane-ECM interactions and (12) NCAM1 interactions. The result of the molecular pathway analysis is detailed in Table 3.
The machine learning approach to the analysis was conducted to the application of a random forest technique, which was deemed appropriate for the “case VS controls” phenotype under analysis. The initial database was split in 400 individuals as the training test, and 351 individuals for the test sample. As a result, a random forest of 20 tress was enough to obtain stable result from the OOB (out of Bag) result, with an OOB estimate error of 0.88%. The confusing matrix reported in Table 4 suggests that the model was highly predictive of the final classification in the test sample. The mtry parameter was set at 5, allowing for 5 variables to be included in the model at each node. The predictive variables used in the random forest analysis were the PRS as calculated in the training test, age, gender, ethnicity, marital status, years at presentation and years of education. The analysis of the single weight of the genetic and clinical variables in predicting the final classification was unbalanced towards the genetic component of the analysis.
Discussion
Suicide ideation, known also as suicidal thoughts, is a phrase used to depict preoccupations and whishes linked to the idea of killing oneself [25]. There is at the moment of writing no consensus on a gold standard to assess SI [32] and the psychodiagnostics tools commonly used to assess it are not effective in identifying people at imminent risk of suicide [33, 34]. SI is considered a better predictor of lifetime risk for suicide than imminent risk [35] and can be distinguished in "active suicidal ideation”, characterized by specific suicidal ideas, with plans aiming to get death [36]; and “passive suicidal ideation”, described as a general desire do die, without any specific plan or mean to give oneself death [37]. There is evidence reporting that up to 75% of patients who died by suicide denied SI in the last month before acting suicide[35, 38].
SCZ patients with SI have over a six-fold increase of suicide [39], while people who had at least one lifetime psychotic event has double odds of experiencing SI, triple odds of a future suicide attempt, and four times the odds of dying by suicide [11]. Given those data, the importance of identifying biological and genetic biomarkers emerges. Our study tried to reach this aim using a classic GWAS approach, a molecular pathway analysis and modelling the polygenic risk score with the clinical predictors in a model through machine learning. No SNP alone reached a GWAS significance in identifying the genetic risk for SI. This result may depend on the underpowered sample, which does not allow to statistically distinguish the noise signal from the true association findings. This negative association result is also explained by the probable biologic complex and most likely polygenic nature of SI. To tackle the latter hypothesis a molecular pathway analysis was undertaken, along with a random forest analysis (machine learning), that considered both the PRS – which includes all the SNPs associated with the phenotype under analysis –. As a main result, a likely overfitting random forest model was retrieved from the analysis, with an error rate in the test sample as low as < 1%. This finding is to be taken with extreme cautiousness, since overfitting is a known possible bias of random forests [40] and it is unlikely that the model under analysis reaches such high level of accuracy. On the other hand, the molecular pathway analysis revealed some significant and interesting result. Table 3 and Fig. 2 report the result of the molecular pathway analysis. Some molecular pathways that resulted to be significantly enriched in variations associated with the outcome under analysis are closely related to the neurological function such as the “Protein–protein interactions at synapses”, “Neuronal system” or “Neurexins and neuroligins”. It is relevant to stress that the second pathway, that related to neurexins and neuroligins was previously found to be associate with cognitive functions [41, 42]. A recent meta-analysis conducted by Qingqin and colleagues [20], consistently reported that one SNP located in the neuroligin 1 (NLGN1) gene was significantly associated with suicide death and suicidal behavior in a sample combining 3765 cases and 6572 controls. The relevance of genetic variants located in the neurorexin 1 towards the suicide risk, was also recently reported by William and colleagues [43]. Negative association findings are also reported [44], this being possibly related to the power of the studies (small sample sizes) reporting negative association result. Overall, it can be underlined that the molecular pathway analysis as conducted in the present investigation is a hypothesis – free strategy to interrogate the whole genome while taking advantage of the current knowledge about the known molecular cascades. It is of note, that out of a hypothesis free approach, three of the molecular pathways resulting to be enriched in SNPs associated with SI are related to the function of neurons. This finding can help to defend the robustness of our investigation.

Molecular pathway analysis
Pathways related to glycosylation and glucuronidation were also reported to be significantly enriched in SNPs associated with the phenotype under analysis. These pathways are involved in a fan of different biologic events, and it is difficult to retrieve their specific role in determining the suicide risk. One pathway classically related to glycosylation was recently reported to play a role in the genetics of depression: B3GALTL (Beta 3-Glucosyltransferase) was found to be associated with suicide behavior in a proteome-wide association study on depression. The integration of data from 500,199 individuals with genome-wide data and 376 human brain proteomes identified 19 genes associated – and being causal of – depression. One of the proteins that were found to be associated with depression was the product of the B3GALTL. B3GALTL gene product codes for a protein that is implicated in synaptogenesis [45]. Quite interestingly, B3GALTL is expressed in the brain by neurons and astrocytes (proteinatlas.org). It is also expressed in T cells. This finding is then of particular relevance: Microglia are the primary immune system cells in the central nervous system, and they function like peripheral macrophages, releasing a multitude of pro-inflammatory cytokines and chemokines. Post-mortem examination of suicide patients shows an increased density of microglia in anterior cingulate cortex, dorsolateral prefrontal cortex, and mediodorsal thalamus regions [46]. An increased microglial activation is identified by PET in patients with SI [47]. Microglial cells affect the tryptophan-kynurenine pathway, increasing the production of neurotoxic metabolites such as quinolinic acid, a NMDA agonist [46] and creating an imbalance in the glutamatergic system. Glutamate neurotransmission is the background of a normal cognitive and emotional processing, so its disruption can lead to increased impulsivity, depressed mood, and suicidality [48]. Consistently with this finding, another important molecule involved in neuroinflammation is the 18-kDa Translocator Protein (TSPO), whose expression was significantly increased in patients with SI, most robustly in the regions of the anterior cingulate cortex [47]. Refer to Table 5, for previous relevant studies about schizophrenia, neuroinflammation and SI.
A pathway related to the Nitric oxide (NO) function was also found to be enriched in SNPs associated with the outcome under analysis. NO has many functions in the human body [49]. It regulates vascular tone and blood flow by activating soluble guanylate cyclase (sGC) in the vascular smooth muscle, and it controls mitochondrial O2 consumption by inhibiting cytochrome c oxidase. Of note, NO is also used as a regulator of the metabolic state of neurons. Our result is consistent with previous findings in literature. Baltazar-Gaytan reported that reduced NO levels were found in the anterior-pituitary region of young suicide completers [50]. The relevance of this pathway as a possible mediator between early trauma and suicide behavior [51].
Neurodevelopment may be the physiologic process that that is affected in subjects experiencing SI as it is hypothesized that neuroplasticity may be a key event in many psychiatric disorders [52]. This is consistent with our findings were the molecular pathways associated with neuronal migration and proliferation were associated with SI.
The present investigation is characterized by several limits. The CATIE study was not designed for the analysis of the phenotype under investigation, and it is underpowered to retrieve a GWAS significant result. In order to define the phenotype under analysis, an item of the Calgary scale was used and not a dedicated psychologic test, limiting the clinical information that can be retrieved by our analysis. No imputation was conducted due to the poor quality of the original database not granting valuable imputed data. The molecular pathway analysis is prone to false positive findings, despite the Bonferroni and FDR corrections. Moreover, it depends on the current annotation system, which is changing year after year. Table 6 (supplementary data) reports the genes that were found to be enriched in SNPs associated with the phenotype under analysis.
In the table, the gene ratio indicates the k/n value and the Bg ratio indicates the M/N value, where N is the total number of genes with annotation, M is the number of genes annotated in that distribution, n is the number of genes included in the analysis, and k is the number of genes annotated to the node. P adjust indicates the P level after Bonferroni correction for multitesting. q Value indicates the P level after false discovery rate correction for multitesting.
Confusion Matrix for the Ranfom Forest Analysis. OOB estimate of error rate was 0.88%. 20 trees with the analysis of up to 5 variables per node were used.
The molecular pathways containing significantly more SNPs associated with the phenotype under analysis than expected by chance are reported. Bar length represents the number of genes enriched in each pathway. The shadowing of the bars represents the P level after correction for multiple tests.
All the participants gave written, informed consent prior to entering the study, and the protocol was conducted in full compliance with the Declaration of Helsinki.
References
World Health Organization (2021) Suicide worldwide in 2019 Global Health Estimates’, Geneva
Doran CM, Kinchin I (2020) Economic and epidemiological impact of youth suicide in countries with the highest human development index. PLoS One. https://doi.org/10.1371/journal.pone.0232940
Walsh EH, McMahon J, Herring MP (2022) Research review: the effect of school-based suicide prevention on suicidal ideation and suicide attempts and the role of intervention and contextual factors among adolescents: a meta-analysis and meta-regression. J Child Psychol Psychiatry Alli Discip 63(8):836–845. https://doi.org/10.1111/jcpp.13598
Doupnik SK et al (2020) Association of suicide prevention interventions with subsequent suicide attempts, linkage to follow-up care, and depression symptoms for acute care settings: a systematic review and meta-analysis. JAMA Psychiatry 77(10):1021–1030. https://doi.org/10.1001/jamapsychiatry.2020.1586
Inagaki M et al (2015) Interventions to prevent repeat suicidal behavior in patients admitted to an emergency department for a suicide attempt: a meta-analysis’. J Affect Disord 175:66–78. https://doi.org/10.1016/j.jad.2014.12.048
Hill NTM et al (2020) Association of suicidal behavior with exposure to suicide and suicide attempt: a systematic review and multilevel meta-analysis. PLoS Med. https://doi.org/10.1371/JOURNAL.PMED.1003074
Ribeiro JD et al (2016) Self-injurious thoughts and behaviors as risk factors for future suicide ideation, attempts, and death: a meta-analysis of longitudinal studies. Psychol Med 46(2):225–236. https://doi.org/10.1017/S0033291715001804
Klonsky ED, May AM, Saffer BY (2016) Suicide, suicide attempts, and suicidal ideation. Annu Rev Clin Psychol 12:307–330. https://doi.org/10.1146/annurev-clinpsy-021815-093204
Fazel S, Runeson B (2020) Suicide. N Engl J Med 382(3):266–274. https://doi.org/10.1056/NEJMra1902944
Hubers AAM et al (2018) Suicidal ideation and subsequent completed suicide in both psychiatric and non-psychiatric populations: a meta-analysis. Epidemiol Psychiatr Sci 27(2):186–198. https://doi.org/10.1017/S2045796016001049
Yates K et al (2019) Association of psychotic experiences with subsequent risk of suicidal ideation, suicide attempts, and suicide deaths: a systematic review and meta-analysis of longitudinal population studies. JAMA Psychiatry 76(2):180–189. https://doi.org/10.1001/jamapsychiatry.2018.3514
Orzelska-Górka J, Mikulska J, Wiszniewska A, Biała G (2022) New atypical antipsychotics in the treatment of schizophrenia and depression. Int J Mol Sci. https://doi.org/10.3390/ijms231810624
Keshavan MS, Collin G, Guimond S, Kelly S, Prasad KM, Lizano P (2020) Neuroimaging in Schizophrenia. Neuroimag Clin North America 30(1):73–83. https://doi.org/10.1016/j.nic.2019.09.007
Liang Y, Wu M, Zou Y, Wan X, Liu Y, Liu X (2023) Prevalence of suicide ideation, self-harm, and suicide among Chinese patients with schizophrenia: a systematic review and meta-analysis. Front Public Health. https://doi.org/10.3389/fpubh.2023.1097098
Álvarez A et al (2022) A systematic review and meta-analysis of suicidality in psychotic disorders: stratified analyses by psychotic subtypes, clinical setting and geographical region. Neurosci Biobehav Rev. https://doi.org/10.1016/j.neubiorev.2022.104964
Correll CU, Solmi M, Croatto G, Schneider LK, Rohani-Montez SC, Fairley L, Smith N, Bitter I, Gorwood P, Taipale H, Tiihonen J (2022) Mortality in people with schizophrenia: a systematic review and meta-analysis of relative risk and aggravating or attenuating factors. World Psychiatry 21(2):248–71
Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB (2015) Inflammation and immunity in schizophrenia: Implications for pathophysiology and treatment. Lancet Psychiatry 2(3):258–270. https://doi.org/10.1016/S2215-0366(14)00122-9
Roy A, Segal NL, Centerwall BS, Dennis Robinette C. Hospital, ‘Suicide in Twins Subsequent to collection of these 11 twin pairs, previously unpublished data about suicide in twins surfaced (collected by’. [Online]. Available: http://archpsyc.jamanetwork.com/
Roy A, Segal NL (2001) ‘Suicidal behavior in twins: a replication’. [Online]. Available: https://www.sciencedirect.com/journal/journal-of-affective-disorders
Li QS et al (2023) Genome-wide association study meta-analysis of suicide death and suicidal behavior. Mol Psychiatry 28(2):891–900. https://doi.org/10.1038/s41380-022-01828-9
Cardno AG, Gottesman II (2000) Twin studies of schizophrenia: from bow-and-arrow concordances to star wars mx and functional genomics. Am J Med Genet. https://doi.org/10.1002/(SICI)1096-8628(200021)
Legge SE, Santoro ML, Periyasamy S, Okewole A, Arsalan A, Kowalec K (2021) Genetic architecture of schizophrenia: a review of major advancements. Psychol Med 51(13):2168–2177. https://doi.org/10.1017/S0033291720005334
Gawde U et al (2022) ‘Multimorbidity landscape of schizophrenia: Insights from meta-analysis of genome wide association studies. Schizophrenia Res 243:214–216. https://doi.org/10.1016/j.schres.2022.03.013
Liu J et al (2021) Genome-wide association study followed by trans-ancestry meta-analysis identify 17 new risk loci for schizophrenia. BMC Med. https://doi.org/10.1186/s12916-021-02039-9
Harmer B, Lee S, Vi Duong TH, Saadabadi A. ‘Suicidal Ideation continuing education activity. [Online]. Available: https://www-ncbi-nlm-nih-gov.auh.aub.aau.dk/books/NBK565877/
Purcell S et al (2007) PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575. https://doi.org/10.1086/519795
Team RC (2020) A language and environment for statistical computing
Alexa A, Rahnenführer J (2023) Gene set enrichment analysis with topGO. [Online]. Available: https://www.mpi-inf.mpg.de/%e2%88%bcalexa
‘Package “Reactome PA”’, https://doi.org/10.18129/B9.bioc.ReactomePA
Gillespie M et al (2022) The reactome pathway knowledgebase 2022. Nucleic Acids Res 50(D1):D687–D692. https://doi.org/10.1093/nar/gkab1028
Addington J, Shah H, Liu L, Addington D (2014) Reliability and validity of the Calgary Depression Scale for Schizophrenia (CDSS) in youth at clinical high risk for psychosis. Schizophr Res 153(1–3):64–67. https://doi.org/10.1016/j.schres.2013.12.014
Berman AL, Silverman MM (2017) How to ask about suicide? A question in need of an empirical answer. Crisis 38(4):213–216. https://doi.org/10.1027/0227-5910/a000501
Glenn CR, Kleiman EM, Cha CB, Deming CA, Franklin JC, Nock MK (2018) Understanding suicide risk within the research domain criteria (RDoC) framework: a meta-analytic review. Depress Anxiety 35(1):65–88. https://doi.org/10.1002/da.22686
American Psychiatric Association (2015) The American Psychiatric Association practice guidelines for the psychiatric evaluation of adults. Am Psychiatric Assoc. https://doi.org/10.1176/appi.books.9780890426760
Berman AL (2018) Risk factors proximate to suicide and suicide risk assessment in the context of denied suicide ideation. Suicide Life Threat Behav 48(3):340–352. https://doi.org/10.1111/sltb.12351
McCullumsmith C (2015) ‘Laying the groundwork for standardized assessment of suicidal behavior. J Clin Psychiatry 76(10):e1333–e1335. https://doi.org/10.4088/JCP.14com09603
Baca-Garcia E et al (2011) Estimating risk for suicide attempt: Are we asking the right questions?: Passive suicidal ideation as a marker for suicidal behavior. J Affect Disord 134(1–3):327–332. https://doi.org/10.1016/j.jad.2011.06.026
Silverman MM, Berman AL (2014) Suicide risk assessment and risk formulation part I: a focus on suicide ideation in assessing suicide risk. Suicide Life Threat Behav 44(4):420–431. https://doi.org/10.1111/sltb.12065
Chapman CL, Mullin K, Ryan CJ, Kuffel A, Nielssen O, Large MM (2015) Meta-analysis of the association between suicidal ideation and later suicide among patients with either a schizophrenia spectrum psychosis or a mood disorder. Acta Psychiatr Scand 131(3):162–173. https://doi.org/10.1111/acps.12359
Hengl T, Nussbaum M, Wright MN, Heuvelink GBM, Gräler B (2018) Random forest as a generic framework for predictive modeling of spatial and spatio-temporal variables. PeerJ 8:2018. https://doi.org/10.7717/peerj.5518
Südhof TC (2008) ‘Neuroligins and neurexins link synaptic function to cognitive disease. Nature. https://doi.org/10.1038/nature07456
Dean C, Dresbach T (2006) Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci 29(1):21–29. https://doi.org/10.1016/j.tins.2005.11.003
William N et al (2021) Neurexin 1 variants as risk factors for suicide death. Mol Psychiatry 26(12):7436–7445. https://doi.org/10.1038/s41380-021-01190-2
Gaynor SC et al (2016) A targeted sequencing study of glutamatergic candidate genes in suicide attempters with bipolar disorder HHS public access. Am J Med Genet B Neuropsychiatr Genet 171(8):1080–1087. https://doi.org/10.1002/ajmg.b
Vasudevan D, Takeuchi H, Johar SS, Majerus E, Haltiwanger RS (2015) Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism. Curr Biol 25(3):286–295. https://doi.org/10.1016/j.cub.2014.11.049
Suzuki H et al (2019) Suicide and microglia: Recent findings and future perspectives based on human studies. Front Cell Neurosci 13:1–10. https://doi.org/10.3389/fncel.2019.00031
Holmes SE et al (2018) Elevated translocator protein in anterior cingulate in major depression and a role for inflammation in suicidal thinking: a positron emission tomography study. Biol Psychiatry 83(1):61–69. https://doi.org/10.1016/j.biopsych.2017.08.005
De Berardis D et al (2018) Eradicating suicide at its roots: Preclinical bases and clinical evidence of the efficacy of ketamine in the treatment of suicidal behaviors. Int J Mol Sci. https://doi.org/10.3390/ijms19102888
Liy PM, Puzi NNA, Jose S, Vidyadaran S (2021) Nitric oxide modulation in neuroinflammation and the role of mesenchymal stem cells. Exp Biol Med 246(22):2399–2406. https://doi.org/10.1177/1535370221997052
Baltazar-Gaytan E et al (2019) Increased cell number with reduced nitric oxide level and augmented superoxide dismutase activity in the anterior-pituitary region of young suicide completers. J Chem Neuroanat 96:7–15. https://doi.org/10.1016/j.jchemneu.2018.11.002
Lengvenyte A et al (2022) Combined effects of nitric oxide synthase 3 genetic variant and childhood emotional abuse on earlier onset of suicidal behaviours. Prog Neuropsychopharmacol Biol Psychiatry. https://doi.org/10.1016/j.pnpbp.2022.110617
Sweatt JD (2016) Neural plasticity and behavior – sixty years of conceptual advances. J Neurochem 139:179–199. https://doi.org/10.1111/jnc.13580
Saetre P et al (2010) The Tryptophan Hydroxylase 1 (TPH1) gene, schizophrenia susceptibility, and suicidal behavior: a multi-centre case-control study and meta-analysis. Am J Med Genet B Neuropsychiatr Genet 153(2):387–396. https://doi.org/10.1002/ajmg.b.30991
Fabbri C, Serretti A (2017) Role of 108 schizophrenia-associated loci in modulating psychopathological dimensions in schizophrenia and bipolar disorder. Am J Med Genet B Neuropsychiatr Genet 174(7):757–764. https://doi.org/10.1002/ajmg.b.32577
Bani-Fatemi A et al (2013) Analysis of CpG SNPs in 34 genes: association test with suicide attempt in schizophrenia. Schizophr Res 147(2–3):262–268. https://doi.org/10.1016/j.schres.2013.04.018
Finseth PI et al (2014) Association analysis between suicidal behaviour and candidate genes of bipolar disorder and schizophrenia. J Affect Disord 163:110–114. https://doi.org/10.1016/j.jad.2013.12.018
Lindholm Carlström E et al (2012) Association between a genetic variant in the serotonin transporter gene (SLC6A4) and suicidal behavior in patients with schizophrenia, 2012. [Online]. Available: https://behavioralandbrainfunctions.biomedcentral.com/articles/10.1186/1744-9081-8-24
Kowalczyk M et al (2020) Association of HSPA1B polymorphisms with paranoid schizophrenia in a polish population. Neuromolecular Med 22(1):159–169. https://doi.org/10.1007/s12017-019-08575-1
Suchanek-Raif R et al (2017) Association study of tumor necrosis factor receptor 1 (TNFR1) gene polymorphisms with schizophrenia in the polish population. Mediat Inflamm. https://doi.org/10.1155/2017/6016023
Wang JY, Jia CX, Lian Y, Sun SH, Lyu M, Wu A (2015) Association of the HTR2A 102T/C polymorphism with attempted suicide: a meta-analysis. Psychiatr Genet 25(4):168–177. https://doi.org/10.1097/YPG.0000000000000091
Giegling I et al (2008) Estrogen receptor gene 1 variants are not associated with suicidal behavior. Psychiatry Res 160(1):1–7. https://doi.org/10.1016/j.psychres.2007.05.007
De Luca V, Hlousek D, Likhodi O, Van Tol HHM, Kennedy JL, Wong AHC (2006) The interaction between TPH2 promoter haplotypes and clinical-demographic risk factors in suicide victims with major psychoses. Genes Brain Behav 5(1):107–110. https://doi.org/10.1111/j.1601-183X.2005.00182.x
Wang Y et al (2010) Genetic polymorphisms in the SCN8A gene are associated with suicidal behavior in psychiatric disorders in the Chinese population. World J Biol Psychiatry 11(8):956–963. https://doi.org/10.3109/15622971003801936
Fanous AH et al (2009) Genetic variation in the serotonin 2A receptor and suicidal Ideation in a sample of 270 irish high-density schizophrenia families. Am J Med Genet B Neuropsychiatr Genet 150(3):411–417. https://doi.org/10.1002/ajmg.b.30833
Mullins N et al (2019) GWAS of suicide attempt in psychiatric disorders and association with major depression polygenic risk scores. Am J Psychiatry 176(8):651–660. https://doi.org/10.1176/appi.ajp.2019.18080957
Bani-Fatemi A, Graff A, Zai C, Strauss J, De Luca V (2016) GWAS analysis of suicide attempt in schizophrenia: Main genetic effect and interaction with early life trauma. Neurosci Lett 622:102–106. https://doi.org/10.1016/j.neulet.2016.04.043
Zai CC et al (2015) Investigation of the genetic interaction between BDNF and DRD3 genes in suicidical behaviour in psychiatric disorders. World J Biol Psychiatry 16(3):171–179. https://doi.org/10.3109/15622975.2014.953011
Karanović J et al (2015) Joint effect of ADARB1 gene, HTR2C gene and stressful life events on suicide attempt risk in patients with major psychiatric disorders. World J Biol Psychiatry 16(4):261–271. https://doi.org/10.3109/15622975.2014.1000374
Dada O et al (2021) Mediating effect of genome-wide DNA methylation on suicidal ideation induced by perceived stress. Psychiatr Genet 31(5):168–176. https://doi.org/10.1097/YPG.0000000000000281
Bani-Fatemi A, Strauss J, Zai C, Wong AHC, De Luca V (2017) Multiple tissue methylation analysis of HTR2A exon i in suicidal behavior. Psychiatr Genet 27(6):219–224. https://doi.org/10.1097/YPG.0000000000000183
De Luca V et al (2011) Parent of origin effect and differential allelic expression of BDNF Val66Met in suicidal behaviour. World J Biol Psychiatry 12(1):42–47. https://doi.org/10.3109/15622975.2010.506928
Sokolowski M, Wasserman J, Wasserman D (2016) Rare CNVs in suicide attempt include schizophrenia-associated loci and neurodevelopmental genes: a pilot genome-wide and family-based study. PLoS One. https://doi.org/10.1371/journal.pone.0168531
Feldcamp LA, Souza RP, Romano-Silva M, Kennedy JL, Wong AHC (2008) Reduced prefrontal cortex DARPP-32 mRNA in completed suicide victims with schizophrenia. Schizophr Res 103(1–3):192–200. https://doi.org/10.1016/j.schres.2008.05.014
Li J, Yoshikawa A, Meltzer HY (2017) Replication of rs300774, a genetic biomarker near ACP1, associated with suicide attempts in patients with schizophrenia: relation to brain cholesterol biosynthesis. J Psychiatr Res 94:54–61. https://doi.org/10.1016/j.jpsychires.2017.06.005
Aytac HM, Ozdilli K, Tuncel FC, Pehlivan M, Pehlivan S (2022) Tumor necrosis factor-alpha (TNF-α) −238 G/A polymorphism is associated with the treatment resistance and attempted suicide in schizophrenia. Immunol Invest 51(2):368–380. https://doi.org/10.1080/08820139.2020.1832115
Shinko Y et al (2020) Chemokine alterations in the postmortem brains of suicide completers. J Psychiatr Res 120:29–33. https://doi.org/10.1016/j.jpsychires.2019.10.008
Ermakov EA, Mednova IA, Boiko AS, Buneva VN, Ivanova SA (2023) Chemokine dysregulation and neuroinflammation in schizophrenia: a systematic review. Int J Mol Sci. https://doi.org/10.3390/ijms24032215
Purves-Tyson TD, Brown AM, Weissleder C, Rothmond DA, Shannon Weickert C (2021) Reductions in midbrain GABAergic and dopamine neuron markers are linked in schizophrenia. Mol Brain. https://doi.org/10.1186/s13041-021-00805-7
Rahimian R, Wakid M, O’Leary LA, Mechawar N (2021) The emerging tale of microglia in psychiatric disorders. Neurosci Biobehav Rev 131:1–29. https://doi.org/10.1016/j.neubiorev.2021.09.023
Trépanier MO, Hopperton KE, Mizrahi R, Mechawar N, Bazinet RP (2016) Postmortem evidence of cerebral inflammation in schizophrenia: a systematic review. Mol Psychiatry 21(8):1009–1026. https://doi.org/10.1038/mp.2016.90
Brisch R et al (2022) The role of microglia in neuropsychiatric disorders and suicide. Eur Arch Psychiatry Clin Neurosci 272(6):929–945. https://doi.org/10.1007/s00406-021-01334-z
Funding
Open access funding provided by Aalborg University Hospital.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Turiaco, F., Iannuzzo, F., Bruno, A. et al. Genetics of suicide ideation. A role for inflammation and neuroplasticity?. Eur Arch Psychiatry Clin Neurosci (2024). https://doi.org/10.1007/s00406-024-01836-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00406-024-01836-6