
Abstract
Purpose
Disturbance of cochlear microcirculation is discussed as final common pathway of various inner ear diseases. Hyperfibrinogenemia causing increased plasma viscosity is a possible factor for a critical reduction of cochlear blood flow that might lead to sudden sensorineural hearing loss (SSHL). The aim was to determine the efficacy and safety of drug-induced defibrinogenation by ancrod for SSHL.
Methods
Double-blind, randomized, placebo-controlled, multicenter, parallel group, phase II (proof-of-concept) study (planned enrollment: 99 patients). Patients received an infusion of ancrod or placebo (day 1) followed by subcutaneous administrations (day 2, 4, 6). Primary outcome was the change in pure tone audiogram air conduction average until day 8.
Results
The study was terminated early due to slow recruiting (31 enrolled patients: 22 ancrod, 9 placebo). A significant improvement of hearing loss was registered in both groups (ancrod: − 14.3 dB ± 20.4 dB, − 39.9% ± 50.4%; placebo: − 22.3 dB ± 13.7 dB, − 59.1% ± 38.0%). A statistically significant group-difference was not detected (p = 0.374). Placebo response of 33.3% complete and 85.7% at least partial recovery was observed. Plasma fibrinogen levels were reduced significantly by ancrod (baseline: 325.2 mg/dL, day 2: 107.2 mg/dL). Ancrod was tolerated well, no adverse drug reaction was of severe intensity, no serious adverse events occurred.
Conclusion
Ancrod reduced fibrinogen levels that support its mechanism of action. The safety profile can be rated positively. Since the planned number of patients could not be enrolled, no efficacy conclusion can be drawn. The high rate of placebo response challenges clinical trials for SSHL and needs to be considered in future investigations.
Trial registrations
This study was registered in the EU Clinical Trials Register, EudraCT-No. 2012-000066-37 at 2012-07-02.
Similar content being viewed by others

Introduction
Sudden sensorineural hearing loss (SSHL) is a frequent inner ear disorder with a wide-ranging incidence (USA: 5-27/100,000; Germany: 160/100,000) probably due to a high rate of spontaneous recovery before seeking medical attention [1,2,3]. Corticosteroids constitute the standard therapy, even though level of evidence is low [4]. Etiology remains unclear in the majority of cases, 71% are ultimately classified as idiopathic. Moreover, the underlying pathomechanism is still not completely understood, proposed theories include infectious causes, autoimmune disorders, or microcirculatory disturbances [5]. Impairment of the cochlear microcirculation was discussed to be relevant due to its sudden and single sided occurrence similar to a central retina vein occlusion of obvious vascular origin [6]. A vascular origin has been postulated as either the primary cause or part of a multifactorial genesis. Moreover, impaired cochlear microcirculation may be the final common pathogenic pathway of other etiological factors. Hyperfibrinogenemia was identified as a risk factor for SSHL [7]. Fibrinogen, a large glycoprotein influences rheologic properties by increasing viscosity and promoting aggregation of erythrocytes and thrombocytes [7,8,9,10]. An increased blood viscosity in SSHL-patients was not only confirmed as a potential etiopathological factor, but also discussed as a promising therapeutic target [7, 11]. Reducing plasma-fibrinogen by fibrinogen/LDL-apheresis was demonstrated beneficial in clinical studies [12,13,14]. Inner ear microcirculation cannot be investigated in the living human. A guinea pig animal model demonstrated a reduction of cochlear blood flow caused by hyperfibrinogenemia leading to hearing loss. Conversely, drug-induced reduction of elevated fibrinogen levels by ancrod caused an increase in cochlear blood flow and recovery of acute hearing loss [15, 16]. The snake venom thrombin-like enzyme ancrod cleaves fibrinogen without activating factor XIII, generating soluble fibrin polymers rapidly digested by plasmin and thus eliminated from the circulation via the reticuloendothelial system [17, 18].
The aim of this randomized placebo-controlled trial was to evaluate the efficacy of drug-induced defibrinogenation by ancrod as primary treatment in patients with SSHL.
Materials and methods
Participants
The study was approved by the responsible Ethics Committees of the participating centers (approval number of the leading Ethics Committee of the University Medical Center Göttingen 14/7/12). It was funded and the investigational medical product (IMP) was provided by the sponsor of this clinical trial, the Nordmark Pharma GmbH. The study was performed in accordance with the Declaration of Helsinki, version 10/2008. The complete study protocol and statistical analysis plan is available in Supplemental Material 1. All participants gave written informed consent. This study was registered in the EU Clinical Trials Register, EudraCT-No. 2012-000066-37 at 2012-07-02.
Inclusion criteria comprised male or female patients aged 18–70 years presenting with acute (< 7d after onset), untreated unilateral idiopathic SSHL ≥ 30 dB in at least 2 consecutive frequencies or ≥ 20 dB in 3 consecutive frequencies and not greater than 90 dB based upon evaluation of 8 frequencies, 0.125, 0.25, 0.5, 1, 2, 4, 6, and 8 kHz, compared to the contralateral ear. Symmetric hearing according to patient’s recollection before onset was required. Amongst others, exclusion criteria were any pre-treatment of SSHL-related hearing loss within the preceding 30 days, e.g. with steroids, or current medication that interferes with coagulation, like anticoagulants or antiplatelet drugs. All in- and exclusion criteria are listed in detail in Supplemental Material 1, page 31–32.
Study design
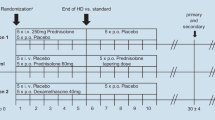
This was a randomized, double-blind, multi-center, placebo-controlled, parallel-group phase II proof-of-concept study on efficacy, safety, and tolerability comparing ancrod with placebo (2:1 randomization) as primary treatment in patients with unilateral SSHL. The study was initiated in 19 German and 5 Czech sites. Thereof, in 8 sites patients were enrolled. Treatment and assessments were performed in an outpatient setting including 7 visits and a study duration for each patient of 90 days.
The primary outcome measure was the change in pure tone audiogram air conduction thresholds in the affected ear from screening/day 1 until day 8. The pure tone average (PTA) was calculated as the arithmetic mean of air conduction thresholds at affected consecutive frequencies within 0.125, 0.25, 0.5, 1, 2, 4, 6, and 8 kHz. A non-affected frequency within two affected frequencies was included. Secondary outcome measures included: change in speech audiometry, fibrinogen concentration, biomarkers (TNF-α, CD38+ , CD40+ cells), patient and physician assessment of change in hearing impairment, and tinnitus severity.
Assessments and interventions
Screening, enrollment, and randomization needed to be performed within a 36 h period. Audiometric tests at time of screening and after treatment (day 8) included pure tone and speech audiometry (specific national test per country to determine the dB hearing level (dB HL) value where 50% of single words will be understood; Germany: German language Freiburg Monosyllabic Test). Hearing impairment, and the occurrence and the degree of tinnitus was moreover assessed by means of a 11-item numeric rating scale (NRS) from 0 to 10. Laboratory tests before study drug administration included plasma fibrinogen concentration.
Patients received the initial infusion of the IMP at day 1 (0.167 IU ancrod/kg bw/h or placebo). The dose was adjusted based on the screening fibrinogen concentration: 2 h infusion if screening fibrinogen concentration was ≥ 180 and ≤ 360 mg/dl, corresponding to a total dose of 0.33 IU/kg bw; 3 h infusion if screening fibrinogen concentration was > 360 mg/dl, corresponding to a total dose of 0.50 IU/kg bw. On day 2, again fibrinogen concentration was assessed to decide on proceeding with defibrinogenation treatment. If the fibrinogen concentration was < 50 mg/dl, the patient was taken off treatment. If it was ≥ 50 mg/dl, treatment proceeded with subcutaneous administration of either ancrod (1 IU/kg bw) or placebo on day 2, 4, and 6. This regimen was shown to be effective in defibrinogenation and was accompanied with the lowest incidence of bleeding complications [19].
On day 8 the treatment effect was evaluated by above mentioned tests and during follow-up at day 30 and 90. Blood samples were taken for coagulation diagnostics, biomarkers, and neutralizing anti-ancrod antibodies at defined time points throughout the clinical trial.
Statistical analysis
The statistical analysis plan is available in Supplemental Material 1.
Based on 2-sided t tests it was calculated that 87 evaluable patients should be treated to confirm a treatment difference of 15 dB PTA value of active treatment versus placebo with a 2:1 randomization, an α-level of 5%, and a statistical power of 90%, assuming a standard deviation of 20%. To compensate for dropouts, 99 patients were planned to be enrolled. Due to slow recruitment the study was terminated early after enrollment of 37 patients and the collected data was analyzed.
The primary analysis is based on the intention-to-treat population (full analysis set; FAS); in addition, a per-protocol analysis (excluding patients not fulfilling the evaluability criteria; per-protocol set, PPS) was performed. Evaluability criteria were: SSHL as defined per inclusion criterion; valid PTA data for day 1 and 8; adhere reasonably well to the study protocol without major protocol deviations.
Quantitative efficacy variables (including the primary efficacy variable) were analyzed by means of an analysis of covariance (ANCOVA) based on the generalized linear model with treatment group (fixed factor) and the covariates baseline value and country (study centers were pooled according to an external factor: country). For the primary efficacy variable, confirmative testing was performed aiming to show superiority in favor of ancrod over placebo. Qualitative parameters were analyzed with Fisher's exact test (2 × 2-tables) or the Freeman-Halton test (tables larger than 2 × 2). A two-sided 5% significance level was applied. All other variables were compared between treatment groups in an explorative manner.
Subgroups were evaluated descriptively for the primary efficacy endpoint. Subgroup analyses and a step-down ANCOVA were applied to identify risk or influential factors.
Safety data were analyzed descriptively by treatment group. Absolute and relative frequencies were calculated for adverse events (AEs) by system organ class and preferred term. AE rates were compared between the treatment groups by Fisher’s exact test.
Statistical analyses were performed using the SAS® version 9.3 (Statistical Analysis System, SAS Institute, Cary, NC, USA).
Results
Patients, demographics, and baseline values
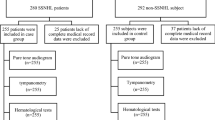
The progress of patients throughout the trial is reported by the consolidated standards of reporting trials diagram (Fig. 1). 620 patients were screened for eligibility from 08/2013 to 09/2018. The most frequent causes for exclusion were hearing loss not according to inclusion criteria (n = 82), pretreatment of SSHL (n = 71), and SSHL onset > 7d (n = 58). Eligibility assessment was accomplished within 36 h. During that period, 24 patients presented with spontaneous recovery the other day. Thirty-seven patients were enrolled into the trial. Of these, six patients were excluded after already having signed informed consent due to screening failure since they did not meet the in-/exclusion criteria (n = 4) or again presented with spontaneous recovery during enrollment period (n = 2). Thirty-one patients (83.8%) were randomized to study treatment. The study was terminated early as the planned 99 patients could not be enrolled in a reasonable time period.

Consolidated Standards of Reporting Trials Flow Diagram. aScreening failure was defined as a patient having signed the informed consent form, but discontinuing before being randomized
Twenty-two patients (71.0%) received ancrod and 9 (29.0%) placebo. These 31 cases were analyzed according to the intention-to-treat approach (FAS). For demographic data and baseline characteristics see Table 1. With regard to the per-protocol analysis, 20 cases were included in the PPS since 11 were excluded due to at least one major protocol deviation. These were: in-/exclusion criteria violation (n = 2), deviation in the audiological measurement regarding primary outcome measure (n = 4), study medication not administered as stipulated by study protocol (n = 8). Multiple reasons were possible per patient. In total, 9/22 (40.9%) ancrod and 2/9 (22.2%) placebo patients had at least one major protocol deviation.
Efficacy
The PTA before treatment was 35.6 ± 15.0 dB HL in the ancrod and 38.6 ± 10.4 dB HL in the placebo group (FAS). From baseline to day 8 (end of treatment), a treatment effect was observed in both groups. When adjusting for the covariates baseline PTA and country, the estimates for the treatment effect (least square means) achieved values of − 14.92 dB (CI [− 23.37, − 6.46]) for ancrod and -22.25 dB (CI [− 36.82, − 7.67]) for placebo with statistical significance (p = 0.05). The difference between the two study groups (ancrod-placebo) was estimated at 7.33 dB with 95% CI [− 9.41, 24.06]. A statistically significant difference could not be detected (p = 0.37). The corresponding analyses on the PPS is in line with these results (Table 2). Subgroup analyses did not identify any risk or influential factor on the study result (data not shown). The PTA over time of the intention-to-treat and PPS are shown in Table 3. Categorical change of SSHL from baseline to day 8 revealed a response rate of 50.0% (PPS: 66.7%) within ancrod and 85.7% (PPS: 85.7%) within placebo treated patients (placebo response).
Plasma fibrinogen concentration was reduced significantly by ancrod, whereas placebo treatment did not show a comparable effect (p < 0.0001). With regard to biomarkers (TNF-α, CD38+ , CD40+ cells), there was no relevant difference between the study groups and no correlation of a clinical effect of IMP (data not shown). Assessment of hearing impairment via NRS revealed a significant treatment effect from baseline to day 8 in both groups without any significant difference between the two study groups. Results of this assessment rated by the physician revealed comparable results (data not shown). The results of the secondary efficacy endpoints, including speech audiometry are depicted in Table 4.
Taken together, pure tone and speech audiometry, and assessments on hearing impairment and tinnitus showed a treatment effect in both study groups without revealing superiority of the ancrod treatment.
Safety
The frequency of observed AEs by patient was 15/22 (68.2%) in the ancrod and 5/9 (55.6%) in the placebo group with no significant difference (p = 0.683). The number of patients with AEs causally related to IMP is low: 6 ancrod (27.3%) and 2 (22.1%) placebo patients (p = 1.00). AEs causally related to IMP occurred at most for one patient in each treatment group. The AEs with a causal link to IMP and occurring in more than one patient is “Feeling hot” (n = 2), one in each study group. Moreover, due to its mode of action, a higher occurrence of bleedings was expected in the ancrod group. Accordingly, two cases of mild bleeding (hematoma, vaginal hemorrhage) were reported. These AEs were rated causally related to IMP. The patient with hematoma discontinued treatment with ancrod but completed the study. One patient in the placebo group discontinued the study due to non-serious AEs. A summary of AEs with absolute and relative frequencies by system organ class and preferred term is provided in the Supplemental Material 2.
No serious adverse event was reported. Just one AE was rated severe: this patient had received ancrod and the AE (unilateral deafness) was classified unrelated to effects of the study medication by the investigator. This was most likely progression of the disease under study, but was classified as AE. All other AEs were rated with mild or moderate intensity.
Except fibrinogen no clinically significant changes occurred in results of routine laboratory analyses, physical examination, and vital signs during treatment. One ancrod patient was tested positive for neutralizing anti-ancrod antibodies at screening and subsequently tested negative at day 8 and 30. At day 30, two patients were tested positive that had been tested negative at screening and day 8.
Discussion
This randomized controlled trial on drug-induced defibrinogenation for SSHL did not demonstrate a significant difference between ancrod and placebo with respect to the primary outcome measure. Unfortunately, no conclusion regarding efficacy can be drawn, since the study was terminated ahead of schedule.
The safety results show that ancrod was well tolerated. No serious adverse event was reported. Adverse drug reactions are within the expected range. Laboratory parameters show an expected decline in plasma fibrinogen in the ancrod group while remaining on a stable level in the placebo group. This observation supports ancrod’s mechanism of action and the dosing schedule. Ancrod was already marketed in Europe and Canada for indications like peripheral arterial occlusive disease, deep vein thrombosis, and prophylaxis for thromboembolism. It is easy to monitor by routine laboratory tests measuring plasma fibrinogen.
The observed reduction of plasma fibrinogen by ancrod is in line with in vivo guinea pig studies, in which this drug-induced reduction of elevated fibrinogen levels resulted in an increase of cochlear blood flow accompanied by recovery of acute hearing loss [15]. The effect of defibrinogenation therapy on cochlear microcirculation cannot be investigated in patients. However, the strategy of reducing plasma-fibrinogen as a therapeutic target in SSHL was supported by former studies investigating the treatment with the snake venom thrombin-like enzyme batroxobin compared to systemic corticosteroid application: Kubo et al. reported a higher recovery rate (57.3% versus 38.7%) and greater improvement of other symptoms closely related to recovery of the inner ear, such as tinnitus and aural fullness [20]. Suzuki et al. found the overall hearing outcomes similar to high-dose steroid therapy, but according to subgroup analysis they recommended steroids for patients with moderate and defibrinogenation therapy for those with severe hearing loss [21]. In line, post-hoc analysis of high-dose steroid versus batroxobin treatment led to the conclusion that defibrinogenation therapy should be chosen specifically for patients with profound hearing loss and initial high fibrinogen [22]. An open study, investigating batroxobin in combination with low-molecular dextran, vasodilators, and vitamins found a correlation between hearing recovery and onset of treatment [23]. This poses the question of a relevance of timely administration within the course of defibrinogenation treatment versus the effect of spontaneous recovery.
Drug-induced enzymatic defibrinogenation has the advantage of an easy, cost-effective, and widely accessible treatment. Previous studies investigated more elaborate extracorporeal techniques to prove the value of defibrinogenation in SSHL-treatment. A superior beneficial effect of fibrinogen/LDL-apheresis in comparison to the standard therapy of high-dose steroids was demonstrated in clinical studies [13, 14]. Moreover, non-responders of standard therapies significantly improved after receiving fibrinogen/LDL-apheresis as second-line therapy, even though the timeframe of spontaneous remissions was exceeded [12]. Likewise, rheopheresis, which reduces a defined spectrum of rheologically relevant high molecular weight proteins from plasma, including fibrinogen, showed to be effective in a case series of SSHL patients [24]. Additionally, a large study comparing this procedure to i.v. corticosteroids or hemodilution demonstrated an equal efficacy [25], and in cases of recurrent SSHL that was refractory to infusion therapy rheopheresis still achieved improvement [26]. Lastly, in an uncontrolled pilot study a single procedure of specific fibrinogen apheresis achieved complete remission of hearing loss in 60% and in another 20% at 4 weeks post-onset [27]. In summary, lowering plasma fibrinogen levels displays a promising therapeutic target and fibrinogen is of potential importance in the pathophysiology of SSHL.
However, conducting and interpreting clinical studies on SSHL is challenging, especially under the terms of inherent high rates of spontaneous recovery. Rates of complete recovery range from 35 to 68% [28, 29], even though these derive from studies which were performed 40 years ago. More recent studies report placebo response rates of 26% complete recovery [30, 31]. Rates of partial recovery were stated between 51 and 89% (placebo or untreated patients) [28, 29, 32, 33]. Spontaneous recovery may be more pronounced in mild-to-moderate affected SSHL patients than in patients with SSHL > 60 dB HL [31]. In line, a high placebo response of 33.3% complete and 85.7% at least partial recovery was observed in the present study. Moreover, 4.2% of the 620 screened patients recovered during the 36 h enrollment period. Considering the placebo-controlled design the present study might have had better prerequisites than other randomized trials that tested against e.g., the therapeutic standard, systemic steroids. These results challenge any study design regarding SSHL and consequently, a considerably stronger effect is required to reach a significant difference in a randomized controlled trial. This needs to be preconceived in future clinical trials.
The study was terminated early due to insufficient recruitment. This is probably caused by (1) demanding in-/exclusion criteria: After 5 years and eligibility screening of 620 patients with acute hearing loss, only 37 of the initially planned 99 patients were enrolled, of which 31 were randomized (5%). Furthermore, considering these highly selective criteria an adequate representation of “common” SSHL can be called into question. (2) Involvement of predominantly tertiary referral centers: The most frequently constituted exclusion criteria were “pretreated SSHL” (n = 71) and “onset > 7 days ago” (n = 58), which lead to presumption of potentially higher recruitment numbers, if rather primary and secondary care providers have been involved. (3) High complexity of study protocol and logistics: This is reflected by the observation of a high rate of major protocol deviations (35.5%) despite several investigator meetings and continuous monitoring. In order to increase inclusion numbers several efforts were made e.g., reports in local press, advertisement in public transport, online presence, Google AdWords.
Strengths and limitations
The strength of this study is the placebo-controlled design displaying the high placebo response rate. The major limitation is the early study termination due to patient recruitment difficulties that does not allow drawing conclusions on efficacy. Consequently, other study limitations are the demanding in- and exclusion criteria, and the complex study protocol and logistics that resulted in the involvement of predominantly tertiary referral centers. In these limitations, on the contrary, lies a strength of the study: the lessons learned allow investigators to draw conclusions for designing future study protocols on SSHL.
Conclusions
Ancrod reduced fibrinogen levels supporting its mechanism of action. The safety profile can be rated positive. The planned number of patients could not be enrolled; therefore, no efficacy conclusion can be drawn. A distinct improvement of SSHL occurred in both study arms. The high placebo response rate in SSHL challenges clinical trials and needs to be considered in future investigations. Conclusions about in- and exclusion criteria, high complexity of the study protocol and logistics, and involvement of primary and secondary care providers may provide benefit for future studies.
Availability of data and material
Underlying research materials, the full trial protocol, and raw data related to this work can be obtained on reasonable request from the corresponding author.
References
Alexander TH, Harris JP (2013) Incidence of sudden sensorineural hearing loss. Otol Neurotol 34(9):1586–1589. https://doi.org/10.1097/MAO.0000000000000222
Klemm E, Deutscher A, Mosges R (2009) A present investigation of the epidemiology in idiopathic sudden sensorineural hearing loss. Laryngorhinootologie 88(8):524–527. https://doi.org/10.1055/s-0028-1128133
Chandrasekhar SS, Tsai Do BS, Schwartz SR, Bontempo LJ, Faucett EA, Finestone SA, Hollingsworth DB, Kelley DM, Kmucha ST, Moonis G, Poling GL, Roberts JK, Stachler RJ, Zeitler DM, Corrigan MD, Nnacheta LC, Satterfield L (2019) Clinical practice guideline: sudden hearing loss (update). Otolaryngol Head Neck Surg 161(1):S1–S45. https://doi.org/10.1177/0194599819859885
Wei BP, Stathopoulos D, O’Leary S (2013) Steroids for idiopathic sudden sensorineural hearing loss. Cochrane Database Syst Rev 7:CD003998. https://doi.org/10.1002/14651858.CD003998.pub3
Chau JK, Lin JR, Atashband S, Irvine RA, Westerberg BD (2010) Systematic review of the evidence for the etiology of adult sudden sensorineural hearing loss. Laryngoscope 120(5):1011–1021. https://doi.org/10.1002/lary.20873
Glacet-Bernard A, Roquet W, Coste A, Peynegre R, Coscas G, Soubrane G (2001) Central retinal vein occlusion and sudden deafness: a possible common pathogenesis. Eur J Ophthalmol 11(2):197–199
Suckfull M, Wimmer C, Reichel O, Mees K, Schorn K (2002) Hyperfibrinogenemia as a risk factor for sudden hearing loss. Otol Neurotol 23(3):309–311
Fabry TL (1987) Mechanism of erythrocyte aggregation and sedimentation. Blood 70(5):1572–1576
Dintenfass L, Kammer S (1977) Plasma viscosity in 615 subjects. Effect of fibrinogen, globulin, and cholesterol in normals, peripheral vascular disease retinopathy, and melanoma. Biorheology 14(5–6):247–251
Budzynski AZ (1986) Fibrinogen and fibrin: biochemistry and pathophysiology. Crit Rev Oncol Hematol 6(2):97–146
Ohinata Y, Makimoto K, Kawakami M, Haginomori S, Araki M, Takahashi H (1994) Blood viscosity and plasma viscosity in patients with sudden deafness. Acta Otolaryngol 114(6):601–607
Canis M, Heigl F, Suckfuell M (2012) Fibrinogen/LDL apheresis is a promising rescue therapy for sudden sensorineural hearing loss. Clin Res Cardiol Suppl 7(Suppl 1):36–40. https://doi.org/10.1007/s11789-012-0044-8
Suckfull M (2002) Fibrinogen and LDL apheresis in treatment of sudden hearing loss: a randomised multicentre trial. Lancet 360(9348):1811–1817. https://doi.org/10.1016/S0140-6736(02)11768-5. (S0140-6736(02)11768-5[pii])
Bianchin G, Russi G, Romano N, Fioravanti P (2010) Treatment with HELP-apheresis in patients suffering from sudden sensorineural hearing loss: a prospective, randomized, controlled study. Laryngoscope 120(4):800–807. https://doi.org/10.1002/lary.20835
Weiss BG, Bertlich M, Bettag SA, Desinger H, Ihler F, Canis M (2017) Drug-induced defibrinogenation as new treatment approach of acute hearing loss in an animal model for inner ear vascular impairment. Otol Neurotol 38(5):648–654. https://doi.org/10.1097/MAO.0000000000001400
Ihler F, Strieth S, Pieri N, Gohring P, Canis M (2012) Acute hyperfibrinogenemia impairs cochlear blood flow and hearing function in guinea pigs in vivo. Int J Audiol 51(3):210–215. https://doi.org/10.3109/14992027.2011.622302
Bell WR Jr (1997) Defibrinogenating enzymes. Drugs 54(S3):18–30 (discussion 30-11)
Lenfors S, Gustafsson D (1996) New model for in vivo studies of pharmacological interventions with endogenous fibrinolysis: effects of thrombin inhibitors. Semin Thromb Hemost 22(4):335–342. https://doi.org/10.1055/s-2007-999028
Latallo ZS (1983) Retrospective study on complications and adverse effects of treatment with thrombin-like enzymes—a multicentre trial. Thromb Haemost 50(2):604–609
Kubo T, Matsunaga T, Asai H, Kawamoto K, Kusakari J, Nomura Y, Oda M, Yanagita N, Niwa H, Uemura T et al (1988) Efficacy of defibrinogenation and steroid therapies on sudden deafness. Arch Otolaryngol Head Neck Surg 114(6):649–652
Suzuki H, Furukawa M, Kumagai M, Takahashi E, Matsuura K, Katori Y, Shimomura A, Kobayashi T (2003) Defibrinogenation therapy for idiopathic sudden sensorineural hearing loss in comparison with high-dose steroid therapy. Acta Otolaryngol 123(1):46–50
Oya R, Horii A, Akazawa H, Osaki Y, Inohara H (2016) Prognostic predictors of sudden sensorineural hearing loss in defibrinogenation therapy. Acta Otolaryngol 136(3):271–276. https://doi.org/10.3109/00016489.2015.1104723
Shiraishi T, Kubo T, Okumura S, Naramura H, Nishimura M, Okusa M, Matsunaga T (1993) Hearing recovery in sudden deafness patients using a modified defibrinogenation therapy. Acta Otolaryngol Suppl 501:46–50
Balletshofer BM, Stock J, Rittig K, Lehn-Stefan A, Braun N, Burkart F, Plontke S, Klingel R, Haring HU (2005) Acute effect of rheopheresis on peripheral endothelial dysfunction in patients suffering from sudden hearing loss. Ther Apher Dial 9(5):385–390. https://doi.org/10.1111/j.1744-9987.2005.00316.x
Mosges R, Koberlein J, Heibges A, Erdtracht B, Klingel R, Lehmacher W, Group R-IS (2009) Rheopheresis for idiopathic sudden hearing loss: results from a large prospective, multicenter, randomized, controlled clinical trial. Eur Arch Otorhinolaryngol 266(7):943–953. https://doi.org/10.1007/s00405-008-0823-5
Uygun-Kiehne S, Straube R, Heibges A, Klingel R, Davids H (2010) Rheopheresis for recurrent sudden hearing loss: therapeutic options for patients refractory to infusion therapy. HNO 58(5):445–451. https://doi.org/10.1007/s00106-009-2004-2
Ullrich H, Kleinjung T, Steffens T, Jacob P, Schmitz G, Strutz J (2004) Improved treatment of sudden hearing loss by specific fibrinogen aphaeresis. J Clin Apheresis 19(2):71–78. https://doi.org/10.1002/jca.20001
Weinaug P (1984) Spontaneous remission in sudden deafness. HNO 32(8):346–351
Mattox DE, Simmons FB (1977) Natural history of sudden sensorineural hearing loss. Ann Otol Rhinol Laryngol 86(4 Pt 1):463–480. https://doi.org/10.1177/000348947708600406
Nosrati-Zarenoe R, Hultcrantz E (2012) Corticosteroid treatment of idiopathic sudden sensorineural hearing loss: randomized triple-blind placebo-controlled trial. Otol Neurotol 33(4):523–531. https://doi.org/10.1097/MAO.0b013e31824b78da
Suckfuell M, Lisowska G, Domka W, Kabacinska A, Morawski K, Bodlaj R, Klimak P, Kostrica R, Meyer T (2014) Efficacy and safety of AM-111 in the treatment of acute sensorineural hearing loss: a double-blind, randomized, placebo-controlled phase II study. Otol Neurotol 35(8):1317–1326. https://doi.org/10.1097/MAO.0000000000000466
Desloovere C, Lorz M, Klima A (1989) Sudden sensorineural hearing loss influence of hemodynamical and hemorheological factors on spontaneous recovery and therapy results. Acta Otorhinolaryngol Belg 43(1):31–37
Desloovere C, Meyer-Breiting E, von Ilberg C (1988) Randomized double-blind study of therapy of sudden deafness: initial results. HNO 36(10):417–422
Acknowledgements
The Authors thank the sponsor of this clinical trial the Nordmark Pharma GmbH represented by Dr. Jörn Tonne for funding and supply of drugs, as well as his team of Clinical Development, especially Dr. Kristin Forßmann followed by Dr. Winrich Rauschning. For collaboration with conception of the clinical study we thank Dr. Klaus Rübsamen. Moreover, we acknowledge Dr. Martina Jordan and Dr. Gudrun Cimander (ClinSupport GmbH) for excellent support and monitoring during the entire study, Alexander Gissler of ProjectPharm for support with regulatory activities and monitoring of the study sites located in Czech Republic, and Dr. Christoph Ortland of Forschungsdock GmbH for support of project management activities. With regard to data management and biostatistics activities, we acknowledge the Medizinisches Wirtschaftsinstitut GmbH, represented by Dr. Dirk Osterkorn followed by X-act Cologne Clinical Research GmbH, among others Dieter Wohltmann and Andreas Bachinger.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was funded by the sponsor of this clinical trial, the Nordmark Pharma GmbH.
Author information
Authors and Affiliations
Contributions
BGW and MC collected data from the centers in Germany, reviewed data from all sites and provided interpretive analysis. BGW analyzed data and prepared the manuscript. JLS, SB, SS, BO, MB, TL, FI, and MC collected data from the centers in Germany. TF and JM collected data from the centers in Czech Republic. All authors discussed the results and implications and commented on the manuscript at all stages.
Corresponding author
Ethics declarations
Conflict of interest
The author BGW has received research funding by the Nordmark Pharma GmbH. This study was funded by the sponsor of this clinical trial, the Nordmark Pharma GmbH.
Ethical approval
The study was approved by the responsible Ethics Committees of the participating centers (approval number of the leading Ethics Committee of the University Medical Center Göttingen 14/7/12). The study was performed in accordance with the Declaration of Helsinki, version 10/2008.
Consent to participate
All participants included in the study gave written informed consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
405_2023_7896_MOESM2_ESM.pdf
Supplementary file2 Adverse events with absolute and relative frequencies by system organ class and preferred term (PDF 120 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weiss, B.G., Spiegel, J.L., Becker, S. et al. Randomized, placebo-controlled study on efficacy, safety and tolerability of drug-induced defibrinogenation for sudden sensorineural hearing loss: the lessons learned. Eur Arch Otorhinolaryngol 280, 4009–4018 (2023). https://doi.org/10.1007/s00405-023-07896-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00405-023-07896-z