
Abstract
Aryl hydrocarbon receptor (AhR) is a ligand-dependent transcription factor that binds to structurally diverse synthetic and naturally occurring chemicals including dioxins, flavonoids, tryptophan photoproducts, and Malassezia metabolites. Upon binding to its ligands, cytoplasmic AhR translocates to the nucleus, heterodimerizes with aryl hydrocarbon receptor nuclear translocator (ARNT), and mediates numerous biological and toxicological effects by inducing the transcription of various AhR-responsive genes. AhR ligation controls oxidation/antioxidation, epidermal barrier function, photo-induced response, melanogenesis, and innate immunity. This review summarizes recent advances in the understanding of the regulatory mechanisms of skin homeostasis mediated by the AhR/ARNT system.
Similar content being viewed by others

Introduction
The skin is a highly sophisticated sensory organ covering the surface of the body. The sensing of external physiological and chemical stimuli plays key roles in self-defense and homeostasis. Aryl hydrocarbon receptor (AhR, also called dioxin receptor) is a chemical receptor that responds to exogenous and endogenous chemicals by inducing/repressing the expression of several genes with toxic or protective effects in a wide range of species and tissues [8]. The best-characterized high-affinity ligands for AhR include several ubiquitous hydrophobic environmental contaminants, such as halogenated and nonhalogenated polycyclic aromatic hydrocarbons (e.g., dioxins and benzo[a]pyrene) [8, 26]. Recent studies have also demonstrated that AhR can bind and be activated by structurally diverse chemicals, such as various phytochemicals [34, 52], Malassezia metabolites [45], and photo-induced chemicals [9, 93, 94] with a wide range of affinities. As keratinocytes, sebocytes, fibroblasts, melanocytes, endothelial cells, Langerhans cells, and other immune cells possess AhR [23, 30, 31, 34, 82, 84], the physiological and pathological processes of skin homeostasis and differentiation are variably affected by the ligand-dependent activation of the AhR signal transduction pathway.
AhR/ARNT signaling
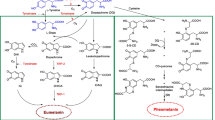
Aryl hydrocarbon receptor is a basic helix-loop-helix/Per-ARNT-Sim (bHLH-PAS)-containing transcription factor essential for adaptive responses to xenobiotics by inducing xenobiotic-metabolizing enzymes such as cytochrome P450 1A1 (CYP1A1) [51]. Most AhR ligands such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and benzo[a]pyrene are very hydrophobic; these ligands enter target cells via diffusion and bind to cytosolic AhR, which exists in an inactive or latent state as a multiprotein complex containing heat shock protein 90 (hsp90), immunophilin-like XAP2, the co-chaperone protein p23, and pp60src (Fig. 1) [1]. Upon ligand binding, AhR is presumed to undergo a conformational change that exposes its N-terminal nuclear localization sequence, facilitating the nuclear translocation of the AhR–ligand complex. The translocated HSP90-bound AhR subsequently dissociates from the HSP90 complex by binding to a structurally related nuclear protein, aryl hydrocarbon receptor nuclear translocator (ARNT) [22]. AhR–ARNT dimerization facilitates the conversion and transformation of the ligand–AhR–ARNT complex into its high-affinity DNA-binding form [22, 75]. Meanwhile, the dissociated pp60src activates epidermal growth factor receptor (EGFR) and induces the internalization and nuclear translocation of EGFR (Fig. 1) [1, 39].

Schematic representation of the AhR/ARNT signaling system. Aryl hydrocarbon receptor (AhR) resides in the cytoplasm as a protein complex with hsp90, XAP2, and p23. Various external and internal ligands like dioxins, dietary flavonoids, Malassezia metabolites, and ultraviolet light-induced metabolites bind to and activate AhR. Upon ligand binding, ligand–AhR protein complex translocates into the nucleus, where AhR nuclear translocator (ARNT) binds to it, releasing hsp90, XAP2, p23, and pp60src. The ligand–AhR–ARNT complex binds to the xenobiotic-responsive element (XRE) and induces the transcription of responsive genes such as cyp1A1. During the process of metabolism of ligands (e.g., dioxins) by CYP1A1, a large number of reactive oxygen species (ROS) are produced. This ROS generation is closely related to various cellular responses, such as cytokine production and DNA damage. Meanwhile, the dissociated pp60src activates epidermal growth factor receptor (EGFR) and induces its internalization and nuclear translocation. Moreover, the AhR signaling induces the transcription of AhR repressor (AhRR). This induced AhRR forms a heterodimer with ARNT, which competes with AhR/ARNT heterodimer to bind to the XRE sequence, consequently inhibiting AhR transcriptional activity
The binding of the heterodimeric ligand–AhR–ARNT complex to its specific DNA recognition site, namely, the xenobiotic-responsive element (XRE) or dioxin-responsive element, upregulates the transcription of responsive genes such as cyp1a1. CYP1A1 is a member of a multigene family of xenobiotic-metabolizing enzymes [8, 26, 51]. Besides its physiological role in the detoxification of polycyclic aromatic compounds, the activity of this enzyme can be deleterious because it generates mutagenic metabolites and reactive oxygen species (ROS). In addition to the upregulation of CYP1A1 and consequent ROS production, a high-affinity AhR ligand, TCDD, causes a broad spectrum of biochemical and toxicological effects, such as teratogenesis, immunosuppression due to thymic involution, and tumor promotion. Extensive studies on the function of AhR using AhR-deficient mice have demonstrated that AhR is responsible for most, if not all, of the toxic effects caused by TCDD [51].
Moreover, the AhR signaling pathway is downregulated via feedback inhibition, particularly via the activity of the AhR repressor (AhRR) [50, 51]. The AhRR promoter has a functional XRE sequence, and its gene expression is enhanced upon the ligand activation of AhR. The induced AhRR forms a heterodimer with ARNT, which competes with AhR/ARNT heterodimer to bind to the XRE sequence, consequently inhibiting AhR transcriptional activity. However, the feedback competition of AhRR/ARNT in AhR/ARNT signaling is a complicated and poorly understood regulatory system (Fig. 1) [51].
Role of AhR/ARNT in oxidative stress
The cells lining the outer and inner surfaces of the body, such as keratinocytes and airway epithelial cells, express AhR/ARNT complex [4, 84]. Benzo[a]pyrene actively induces the nuclear translocation of AhR and subsequent CYP1A1 and ROS generation, leading to DNA damage (i.e., 8-hydroxydeoxyguanosine production) and interleukin 8 (IL-8) production in keratinocytes, as well as mucin (MUC5AC) production in airway epithelial cells [4, 84]. AhR knockdown by specific siRNA abrogates this series of reactions, indicating their dependence on AhR. These results are concordant with the fact that the carcinogenic and inflammatogenic activities of benzo[a]pyrene and TCDD are abolished in AhR-null mice [51, 73]. Using an ex vivo skin organ culture system, Costa et al. [7] confirmed that benzo[a]pyrene actually upregulates CYP1A1 expression, ROS production, and subsequent protein peroxidation. As benzo[a]pyrene is one of the major harmful ingredients of tobacco smoke, AhR-mediated IL-8 production may explain why tobacco smoking exacerbates IL-8-related inflammatory skin diseases such as psoriasis and palmoplantar pustulosis [2, 10, 84].
In addition to oxidative stress, recent studies have demonstrated that the AhR/ARNT system mediates antioxidative and protective signaling in response to different ligands, such as flavonoids, herbal medicines, and azoles (Fig. 2) [20, 21, 28, 59, 83]. For example, ketoconazole binds and induces the nuclear translocation of AhR without producing ROS. Instead, it activates nuclear factor-erythroid 2-related factor-2 (Nrf2) and subsequently NAD(P)H:quinone oxidoreductase 1 (Nqo1), which are key molecules that protect cells from ROS-induced oxidative damage [20, 28, 83]. Ketoconazole actually inhibits benzo[a]pyrene- and tumor necrosis factor-alpha (TNF-α)-induced ROS and IL-8 production, which is abolished by AhR or Nrf2 knockdown, but not by AhRR knockdown [83]. Similar findings have been obtained with traditional herbal remedies such as Bidens pilosa extract [34]. Both benzo[a]pyrene and TNF-α also induce marked ROS production in endothelial cells. B. pilosa extract potently inhibits ROS production by upregulating Nrf2 and Nqo1, which are abrogated by knockdown of AhR or Nrf2 [34]. The tea flavonoid epigallocatechin gallate upregulates Nrf2 and Nqo1 expression while downregulating AhR and CYP1A1 expression [21]. Quercetin, one of the flavonoids, efficiently induces AhR activation and CYP1A1 production [53]. However, it potently inhibits ultraviolet B (UVB)-induced ROS production [95]. In addition, quercetin also induces AhRR mRNA upregulation [59]. Benzo[a]pyrene-induced ROS production is AhR-dependent since it is inhibited by siRNA specific for AhR [84]; however, ketoconazole- and quercetin-mediated AhR activation occurs without ROS production [83, 95]. Therefore, the AhR-related production of ROS is likely to be evoked in a ligand-dependent manner. These complicated results indicate that the AhR/ARNT system acts as a master switch for up and downregulating oxidative stress by modulating diverse genes (e.g., those of AhR, AhRR, CYP1A1, Nrf2, and Nqo1). However, the precise mechanisms by which various phytochemicals and environmental pollutants differentially affect the AhR/ARNT system remain largely unknown.

AhR ligation induces not only oxidative stress but also antioxidative response in a ligand-dependent manner. Environmental pollutants such as benzo[a]pyrene and TCDD bind to AhR and induce ROS production, DNA damage, and inflammatory cytokine production. In contrast, ketoconazole and certain flavonoids bind to AhR, resulting in the activation of Nrf2 and subsequent induction of antioxidative enzymes such as Nqo1. These antioxidative enzymes inhibit ROS production, DNA damage, and inflammatory cytokine production. Thus, AhR acts as a master switch for oxidation and antioxidation
Role of AhR/ARNT in epidermal barrier function
Coal tar comprises at least 10,000 high-molecular-weight hydrocarbon and aromatic compounds, which may target the AhR/ARNT system. Topical coal tar remedies have been widely used to treat inflammatory skin diseases for at least two millennia [49]. Using organotypic skin models with primary keratinocytes from atopic dermatitis patients and controls, van den Bogaard et al. [86] demonstrated that coal tar activates AhR, resulting in the induction of epidermal differentiation (i.e., upregulation of filaggrin, loricrin, and hornerin expression), and thickens the cornified layer. Furthermore, AhR knockdown by siRNA completely abrogates this effect. In atopic dermatitis patients, coal tar completely restores the expression of major skin barrier proteins including filaggrin. Coal tar also diminishes spongiosis, apoptosis, and CCL26 expression in organotypic skin stimulated by the Th2 cytokines IL-4 and IL-13 via the dephosphorylation of STAT6; this is most likely due to the AhR-regulated activation of the Nrf2 antioxidation pathway [86]. Many studies have shown that AhR mediates the upregulation of epidermal differentiation [42, 43, 67, 77]. TCDD increases the quantity of cornified envelopes in monolayer cultures and organotypic cultures of keratinocytes [43]. TCDD also enhances filaggrin, involucrin, transglutaminase, and IL-1β expression [42, 63, 77]. In addition, TCDD exposure significantly augments the mRNA expression of other epidermal differentiation complex genes [38]: repetin, hornerin, late cornified envelope (LCE) 3E, LCE3A, LCE2B, LCE2A, LCE1C, small proline-rich protein (SPRR) 1A, SPRR2A, SPRR2B, S100A9, S100A12, and S100A7 [77]. Accordingly, the targeted ablation of ARNT in mouse epidermis results in profound defects in desquamation and epidermal barrier function, particularly decreased filaggrin and SPRR2A expression [18]. It is quite interesting that the increase in cornified envelope proteins such as SPRRs decrease oxidative stress by quenching excess ROS [88, 89]. Recent work by Kennedy et al. [33] has also revealed that TCDD increases the expression of 40 % of the genes of the epidermal differentiation complex found on chromosome 1q21, such as hornerin, filaggrin, SPRR2B, SPRR4, and LCE3A. In addition, TCDD increases the expression of 75 % of the genes required for de novo ceramide biosynthesis, leading to the overproduction of ceramides 1, 2, 3, 4, 5, 6, 7, and 9 without affecting the levels of cholesterol and free fatty acids. Moreover, the cornified envelope formation induced by TCDD is blocked in the presence of antioxidative agents, quercetin, catalase, or N-acetyl-l-cysteine, indicating an important role for ROS production in the TCDD-induced acceleration of epidermal terminal differentiation [33].
Exposure to extremely high concentrations of dioxins induces chloracne in humans, as was demonstrated in the Yusho and Seveso industrial accidents in Japan and Italy, respectively (Fig. 3) [3, 14]. The pathology of chloracne is characterized by hyperkeratinization of the interfollicular squamous epithelium, hyperproliferation and hyperkeratinization of hair follicle cells, and a metaplastic response of the sebaceous glands [29, 61, 63, 68, 76, 85]. Highly lipophilic dioxins appear to accumulate in and are excreted via sebaceous glands and sebum [25, 74], which may efficiently excrete dioxins from the intoxicated body [47]. TCDD also affects the differentiation of sebaceous gland cells, probably by switching human sebocytes toward keratinocyte-like differentiation [29]. Although the precise mechanism behind chloracne is not understood, sustained AhR hyperactivation and exaggerated hyperkeratinization of pilosebaceous units may be the cause of this devastating toxicity.

Severe chloracne in Yusho patients (oral intoxication of a high concentration of 2,3,4,7,8-pentachlorodibenzofuran)
AhR-null mice appear normal at birth, but their growth is slightly slower than that of wild-type mice during the first few weeks of life. Thereafter, they catch up and no difference is apparent in animals over 12 weeks of age [73]. Tauchi et al. [80] generated transgenic mice expressing the constitutively active form of AhR in keratinocytes. At birth, these transgenic mice were normal, but severe skin lesions developed postnatally, along with marked scratching. Prominent epidermal acanthosis and hyperkeratosis were evident, with severe dermal infiltration of lymphocytes and polymorphonuclear cells in the lesional skin. Th2-skewed immune deviation was evident in the skin lesions and spleen in the transgenic mice, with an elevated circulating IgE level [80]. Interestingly, keratinocyte-specific ARNT-deficient mice generated by a K5-Cre-loxP system exhibit severe skin barrier dysfunction and die because of rapid dehydration [79]. The keratinocyte-specific ARNT disruption results in significant changes in the amount and composition of ceramides, but not cholesterol and fatty acids. The most prominent changes in the ceramide composition of the ARNT -null epidermis are observed for ceramide 2 and ceramide 5. The 4-sphingenine that these ceramides normally contain is largely replaced by sphinganine due to the impaired transcription of dihydroceramide desaturase isozyme, Des-2 [79]. Another type of epidermal ARNT -null mice generated by Geng et al. using a K14-Cre-loxP system also exhibits the upregulation of genes of the epidermal differentiation complex (S100A8, S100A9, S100A10, SPRR1, and SPRR 2) and the alteration of ceramides. In addition, the ARNT-null epidermis exhibits the upregulation of secretory leukocyte protease inhibitor (Slpi), which inhibits stratum corneum chymotryptic enzyme (kallikrein 7), leading to hyperkeratosis due to impaired corneodesmosome degradation and delayed detachment of corneocytes [18]. These studies highlight the importance of the AhR/ARNT system in keratinocyte terminal differentiation.
Although ROS production has been shown to be a prerequisite in the TCDD-induced upregulation of keratinocyte terminal differentiation [33], excessive antioxidant activity also hampers the epidermal barrier function [69, 71]. K5-Cre-Nrf2 transgenic mice generated by Schäfer et al. [69] express high levels of constitutively active Nrf2 in the epidermis together with the overexpression of Nqo1 and other antioxidative enzymes. Unexpectedly, their skin is dry with hair loss and scaling. In terms of their histology, epidermal acanthosis and hyperkeratosis are evident, with sebaceous gland enlargement and hair follicle abnormality. Like ARNT-null keratinocytes [18], the Nrf2-transgenic keratinocytes exhibit the upregulated expression of Slpi, SPRR2d, and SPRR2 h. The upregulated Slpi again inhibits kallikrein 7 activity, leading to impaired detachment of corneocytes, which results in hyperkeratosis [69]. The prolonged Nrf2 activation in K5-Cre-Nrf2 transgenic mice also causes sebaceous gland enlargement and seborrhea due to upregulation of epigen, a recently identified ligand for EGFR. Upon aging, the upregulation of Slpi, SPRR2d, and epigen in the pilosebaceous unit results in infundibular acanthosis, hyperkeratosis, and cyst formation mimicking chloracne [71]. Therefore, Slpi, SPRR2d, and epigen are crucial target molecules of Nrf2 in modifying epidermal and pilosebaceous differentiation. Kelch-like ECH-associated protein-1 (Keap1) is an Nrf2 repressor protein. In keeping with the findings in Nrf2-transgenic mice, Keap1-null mutation induces constitutive Nrf2 activation leading to hyperkeratosis [90]. With regard to the effect of EGFR signaling on the AhR/ARNT system, EGFR signaling blocks TCDD-induced CYP1A1 production as well as filaggrin upregulation [78]. Meanwhile, EGFR signaling is capable of activating the Nrf2/Nqo1 system [62]. However, there remain a plethora of unanswered questions in terms of the crosstalk among AhR, Nrf2, and EGFR signaling.
Role of AhR/ARNT in photobiology, melanogenesis, and immunodermatology
The critical roles of ROS and the AhR/ARNT system in photobiology have been elegantly reviewed by Schäfer et al. [70] and Krutmann et al. [37]. Besides environmental contaminants and dietary constituents [52, 57], many endogenous compounds including various indoles, heme, and arachidonic acid metabolites are AhR agonists; moreover, tryptophan is the precursor of many of the most active ligands for AhR [57]. Fritsche et al. [12] first demonstrated that UVB irradiation induces the intracellular tryptophan photoproduct, 6-formylindolo[3,2-b]carbazole (FICZ), which eventually induces the nuclear translocation of AhR. Furthermore, AhR-knockout mice exhibit compromised UVB responsiveness. Thus, AhR signaling is an integral part of the UVB stress response [12]. FICZ, which is formed upon the exposure of tryptophan solutions, cell culture media, or cells to UV radiation, binds to AhR with greater affinity than TCDD [57]. UVB is the most efficient means of generating FICZ from tryptophan [57, 60]. FICZ formation increases 40- to 400-fold in the presence of the photosensitizer riboflavin (vitamin B2), especially upon exposure to UVA and visible light; this is because riboflavin absorbs light efficiently at these longer wavelengths [57, 60]. This relatively easy conversion of tryptophan to FICZ in the presence of riboflavin and light suggests that the same process could occur in the skin. Thus, the formation of FICZ may explain the reported UV-dependent activation of CYP1 enzymes in human skin [93]. Indeed, FICZ metabolites are detected in human urine [93].
6-Formylindolo[3,2-b]carbazole is a high-affinity ligand for AhR; its K d value is 0.07 nM, which is two or more orders of magnitude less than that of low-affinity ligands such as prostaglandin and lipoxin derivatives [56, 57, 60]. FICZ upregulates the expression of AhR-responsive genes (e.g., CYP1A1) in an efficient but transient manner; this is because FICZ is rapidly metabolized by CYP1A1 in a feedback mechanism [92–94]. This sequence of events, which is typical of autoregulatory loops in biological signaling, suggests that FICZ may be an important physiological ligand for AhR. UVB as well as FICZ does indeed activate the AhR/ARNT system and upregulates the gene expression of CYP1A1 and matrix metalloproteinase 1, which are abolished in the presence of AhR antagonists [81]. Both FICZ and UVB activate EGFR and its downstream signaling extracellular signal-regulated kinases 1 and 2 and cyclooxygenase-2 (COX-2) [1, 12]. The induction of CYP1A1 and COX-2 mRNAs in the skin of mice exposed to UVB was blunted in AhR-deficient mice [12]. Moreover, it has been shown that AhR signaling may play an antiapoptotic role in UVB-exposed skin [11], strongly indicating that AhR signaling does in fact contribute to photocarcinogenesis, as proposed by Agostinis et al. [1]. However, we have to keep in mind that FICZ is a tiny fraction of tryptophan photoproducts and the outline proposed above has yet to be confirmed empirically. The role of FICZ in cutaneous photobiology remains largely unclear.
Normal murine melanocytes also express functional AhR [31]. Using standard UVB tanning protocols, Jux et al. [31] have demonstrated that AhR-deficient mice develop a significantly weaker tan than wild-type mice and that epidermal tyrosinase activity is decreased in AhR-deficient mice. However, tanning response and tyrosinase activity are normal in keratinocyte-specific AhR-conditional knockout mice, indicating that downregulation of the melanogenic response is a direct effect of UVB/AhR signaling on melanocytes [31]. In fact, AhR can modulate melanogenesis by controlling the expression of melanogenic genes in melanocytes. Luecke et al. have reported that exposing normal human melanocytes to TCDD activates the AhR signaling pathway, as well as the AhR-dependent induction of tyrosinase activity, with the elevation of total melanin content. Neither the induction of tyrosinase enzyme activity nor that of total melanin could be attributed to the enhanced cell proliferation of melanocytes; instead, they are due to the induction of tyrosinase and tyrosinase-related protein 2 gene expression [44]. Nakamura et al. [54] have demonstrated that tobacco smoke extract exerts similar melanogenic effects by activating AhR in melanocytes. In this context, Schallreuter et al. have demonstrated that AhR signaling is severely impaired in the lesional and nonlesional skin in cases of vitiligo, despite the presence of FICZ [72]. UVB phototherapy is the mainstay treatment for vitiligo. A recent study by Lan et al. [39] has revealed that excimer light (peak wavelength 308 nm) is more potent at inducing melanogenesis of cultured melanocytes than narrow-band UVB (peak wavelength 311 nm) because the former upregulates AhR–EGFR-dependent tyrosinase activity more efficiently than the latter.
Wang et al. [91] examined the functional AhR gene polymorphisms and suggested that the T allele of rs10249788, which is located in the promoter of the AhR gene, is associated with a protective effect against vitiligo in Han Chinese populations. Concordantly, our recent study clarified that a transcription factor, nuclear factor 1-C (NF1C), which suppresses AhR gene transcription, preferentially binds to the C allele over the T allele at rs10249788 [41]. Therefore, it is conceivable that subjects with the T allele at rs10249788 express higher levels of AhR and are more melanogenic than those with the C allele.
AhR-mediated melanogenesis may also explain the marked hyperpigmentation that occurred in victims of the Yusho industrial accident, who were exposed to extremely high levels of dioxin-related compounds (Fig. 4) [27, 53]. However, it should be mentioned that skin pigmentation was not always recognized in the victims of TCDD intoxication [19, 68]. Yusho patients may be different, as they were exposed to a wide variety of polyhalogenated polycyclic hydrocarbons, in particular polychlorinated biphenyls, which may simply produce a charcoal-like blackish skin color.

Hyperpigmentation in Yusho patients
Malassezia yeasts are unique in that they are virtually the sole eukaryote among the microbial flora of the skin [15]. Malassezia furfur produces malassezin and other indole derivatives by converting l-tryptophan in culture medium [35, 48]. Since l-tryptophan is present in sweat [24], Malassezia is expected to produce these compounds on the surface of human skin in vivo. Indeed, human skin extracts from seborrheic dermatitis lesions infected by Malassezia contain indirubin, FICZ, indolo[3,2-b]carbazole (ICZ), and malassezin [16, 17]. Interestingly, these molecules are active AhR agonists and induce CYP1A1 production [45]. In the presence of malassezin, human melanocytes undergo apoptosis; this may explain why pityriasis versicolor induces depigmented macules [36]. Thus, how AhR discriminates similar but oppositely agonistic ligations by TCDD (i.e., in melanogenesis) and malassezin (i.e., in melanocyte apoptosis) is an intriguing issue [36, 54].
In the immune system, AhR appears to play a crucial role in T-helper 17 (Th17) and regulatory T (Treg) cells [13, 46, 64–66, 87]. AhR-deficient mice can develop Th17 cells, but fail to respond to AhR ligands to enhance Th17 cell development [87]. AhR activation during the induction of experimental autoimmune encephalomyelitis accelerates the onset and increases the pathology of this condition in wild-type mice, but not in AhR-deficient mice [87]. The development of Treg cells is reciprocally related to that of Th17 cells. The agonistic ligation of AhR by TCDD induces functional Treg cells, suppressing experimental autoimmune encephalomyelitis. On the other hand, AhR activation by FICZ interferes with Treg development, boosts Th17 cell differentiation, and increases the severity of experimental autoimmune encephalomyelitis in mice [65].
Aryl hydrocarbon receptor-mediated Treg cell induction appears to operate in UV-induced immunosuppression. Navid et al. [55] have demonstrated that the AhR antagonist 3-methoxy-4-nitroflavone reduces UV-mediated immunosuppression as well as the induction of Treg cells in murine contact hypersensitivity. Conversely, AhR activation by the agonist 4-n-nonylphenol suppresses the induction of contact hypersensitivity and induces antigen-specific Treg cells similarly to UV radiation. This has been further confirmed in AhR-knockout mice, which exhibit significantly reduced UV radiation- and 4-n-nonylphenol-induced immunosuppression [55].
Kynurenine is an important immunoinhibitory metabolite of tryptophan and is generated by indoleamine 2,3-dioxygenase (IDO) [58]. AhR activation by TCDD is required to induce IDO expression in dendritic cells. In the presence of lipopolysaccharide or CpG, bone marrow-derived dendritic cells skew the differentiation of naïve T cells toward Treg cells rather than Th17 cells, however, the capacity of developing Treg cells is deteriorated in AhR-deficient dendritic cells. The restoration of the Treg-inducible function in AhR-deficient dendritic cells by exogenous kynurenine indicates that AhR/IDO/kynurenine induction is important for the generation of tolerogenic dendritic cells under lipopolysaccharide or CpG stimulation [58].
Epidermal Langerhans cells also express AhR [30]. When cultured, AhR-deficient Langerhans cells show insufficient maturation and decreased expression of costimulatory molecules such as CD40, CD80, and CD24a. In keeping with this notion, AhR-deficient mice exhibit significantly weaker contact hypersensitivity to hapten [30]. The maturation of Langerhans cells is stimulated by granulocyte–macrophage colony-stimulating factor produced from surrounding keratinocytes. However, this production of granulocyte–macrophage colony-stimulating factor is significantly decreased in AhR-deficient keratinocytes [30]. Another immunocompetent bone marrow-derived cell type in the murine epidermis is the γδ T cells (dendritic epidermal T cells; DETCs) [32]. The proliferation and distribution of DETCs are markedly impaired in AhR-null mice due to insufficient expression of c-Kit, which is a downstream target molecule of AhR [32].
Other studies
The AhR/ARNT system plays diverse and complex roles in tissue differentiation, immunoregulation, and carcinogenesis. As this system is fundamentally involved in various cellular responses, numerous papers have recently been published on it. The AhR/ARNT system is also important for the connection between food and health [40]. Exposure to AhR ligands through the human diet, even in the first weeks of life, is critical for the development of immune responses, as they control the maturation of innate lymphoid cells. Innate lymphoid cells drive immune responses against intestinal infections, and their generation is impaired in AhR-deficient mice. Mice fed a diet lacking natural AhR ligands suffer from deficient innate lymphoid cell generation and are prone to intestinal infection. The sole addition of the natural AhR ligand indole-3-carbinol to the diet restores both the generation of innate lymphoid cells and the immune response in an AhR-dependent manner. This highlights the importance of exposure to AhR agonists through the diet and their role in the maintenance of intestinal homeostasis [40]. Human blood actually contains a number of AhR ligands, such as resveratrol and indole-3-carbinol derived from vegetables, fruit, nuts, and herbs [5]. The complexity of regulatory mechanisms associated with the AhR/ARNT system has also been highlighted by a melanoma study by Contador-Troca et al. [6]; they found that AhR contributes to tumor-stroma interaction, that is, blocking melanoma growth and metastasis when expressed in tumor cells, but supporting melanoma when expressed in the stroma. Studies on skin surface sensing by AhR/ARNT should elucidate enigmatic host-environment interactions and may provide novel strategies for drug development.
References
Agostinis P, Garmyn M, Van Laethem A (2007) The aryl hydrocarbon receptor: an illuminating effector of the UVB response. Sci STKE 403:pe49
Akiyama T, Seishima M, Watanabe H, Nakatani A, Mori S, Kitajima Y (1995) The relationships of onset and exacerbation of pustulosis palmaris et plantaris to smoking and focal infections. J Dermatol 22(12):930–934
Caputo R, Monti M, Ermacora E, Carminati G, Gelmetti C, Gianotti R, Gianni E, Puccinelli V (1988) Cutaneous manifestations of tetrachlorodibenzo-p-dioxin in children and adolescents. Follow-up 10 years after the Seveso, Italy, accident. J Am Acad Dermatol 19(5 Pt. 1):812–819
Chiba T, Uchi H, Tsuji G, Gondo H, Moroi Y, Furue M (2011) Arylhydrocarbon receptor (AhR) activation in airway epithelial cells induces MUC5AC via reactive oxygen species (ROS) production. Pulm Pharmacol Ther 24(1):133–140. doi:10.1016/j.pupt.2010.08.002
Connor KT, Harris MA, Edwards MR, Budinsky RA, Clark GC, Chu AC, Finley BL, Rowlands JC (2008) AH receptor agonist activity in human blood measured with a cell-based bioassay: evidence for naturally occurring AH receptor ligands in vivo. J Expo Sci Environ Epidemiol 18(4):369–380
Contador-Troca M, Alvarez-Barrientos A, Barrasa E, Rico-Leo EM, Catalina-Fernández I, Menacho-Márquez M, Bustelo XR, García-Borrón JC, Gómez-Durán A, Sáenz-Santamaría J, Fernández-Salguero PM (2013) The dioxin receptor has tumor suppressor activity in melanoma growth and metastasis. Carcinogenesis 34(12):2683–2693. doi:10.1093/carcin/bgt248
Costa C, Catania S, De Pasquale R, Stancanelli R, Scribano GM, Melchini A (2010) Exposure of human skin to benzo[a]pyrene: role of CYP1A1 and aryl hydrocarbon receptor in oxidative stress generation. Toxicology 271(3):83–86. doi:10.1016/j.tox.2010.02.014
Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B (2011) Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol Sci 124(1):1–22. doi:10.1093/toxsci/kfr218
Diani-Moore S, Labitzke E, Brown R, Garvin A, Wong L, Rifkind AB (2006) Sunlight generates multiple tryptophan photoproducts eliciting high efficacy CYP1A induction in chick hepatocytes and in vivo. Toxicol Sci 90(1):96–110
Fortes C, Mastroeni S, Leffondré K, Sampogna F, Melchi F, Mazzotti E, Pasquini P, Abeni D (2005) Relationship between smoking and the clinical severity of psoriasis. Arch Dermatol 141(12):1580–1584
Frauenstein K, Sydlik U, Tigges J, Majora M, Wiek C, Hanenberg H, Abel J, Esser C, Fritsche E, Krutmann J, Haarmann-Stemmann T (2013) Evidence for a novel anti-apoptotic pathway in human keratinocytes involving the aryl hydrocarbon receptor, E2F1, and checkpoint kinase 1. Cell Death Differ 20(10):1425–1434. doi:10.1038/cdd.2013.102
Fritsche E, Schäfer C, Calles C, Bernsmann T, Bernshausen T, Wurm M, Hübenthal U, Cline JE, Hajimiragha H, Schroeder P, Klotz LO, Rannug A, Fürst P, Hanenberg H, Abel J, Krutmann J (2007) Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc Natl Acad Sci USA 104(21):8851–8856
Funatake CJ, Marshall NB, Steppan LB, Mourich DV, Kerkvliet NI (2005) Cutting edge: activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+ CD25+ cells with characteristics of regulatory T cells. J Immunol 175(7):4184–4188
Furue M, Uenotsuchi T, Urabe K, Ishikawa T, Kuwabara M (2005) Overview of Yusho. J Dermatol Sci Suppl (Suppl. 1):S3–S10
Gaitanis G, Magiatis P, Hantschke M, Bassukas ID, Velegraki A (2012) The Malassezia genus in skin and systemic diseases. Clin Microbiol Rev 25(1):106–141. doi:10.1128/CMR.00021-11
Gaitanis G, Magiatis P, Stathopoulou K, Bassukas ID, Alexopoulos EC, Velegraki A, Skaltsounis AL (2008) AhR ligands, malassezin, and indolo[3,2-b]carbazole are selectively produced by Malassezia furfur strains isolated from seborrheic dermatitis. J Invest Dermatol 128(7):1620–1625. doi:10.1038/sj.jid.5701252
Gaitanis G, Velegraki A, Magiatis P, Pappas P, Bassukas ID (2011) Could Malassezia yeasts be implicated in skin carcinogenesis through the production of aryl-hydrocarbon receptor ligands? Med Hypotheses 77(1):47–51. doi:10.1016/j.mehy.2011.03.020
Geng S, Mezentsev A, Kalachikov S, Raith K, Roop DR, Panteleyev AA (2006) Targeted ablation of Arnt in mouse epidermis results in profound defects in desquamation and epidermal barrier function. J Cell Sci 119(Pt. 23):4901–4912
Geusau A, Abraham K, Geissler K, Sator MO, Stingl G, Tschachler E (2001) Severe 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) intoxication: clinical and laboratory effects. Environ Health Perspect 109(8):865–869
Haarmann-Stemmann T, Abel J, Fritsche E, Krutmann J (2012) The AhR–Nrf2 pathway in keratinocytes: on the road to chemoprevention? J Invest Dermatol 132(1):7–9. doi:10.1038/jid.2011.359
Han SG, Han SS, Toborek M, Hennig B (2012) EGCG protects endothelial cells against PCB 126-induced inflammation through inhibition of AhR and induction of Nrf2-regulated genes. Toxicol Appl Pharmacol 261(2):181–188. doi:10.1016/j.taap.2012.03.024
Hankinson O (1995) The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol 35:307–340
Hao N, Whitelaw ML (2013) The emerging roles of AhR in physiology and immunity. Biochem Pharmacol 86(5):561–570. doi:10.1016/j.bcp.2013.07.004
Hier SW, Cornbleet T, Bergeim O (1946) The amino acids of human sweat. J Biol Chem 166(1):327–333
Iida T, Hirakawa H, Matsueda T, Takenaka S, Yu ML, Guo YL (1999) Recent trend of polychlorinated dibenzo-p-dioxins and their related compounds in the blood and sebum of Yusho and Yu Cheng patients. Chemosphere 38(5):981–993
Ikuta T, Namiki T, Fujii-Kuriyama Y, Kawajiri K (2009) AhR protein trafficking and function in the skin. Biochem Pharmacol 77(4):588–596. doi:10.1016/j.bcp.2008.10.003
Imamura T, Matsumoto S, Akahane M, Kanagawa Y, Koike S, Tajima B, Matsuya S, Uchi H, Shibata S, Furue M (2009) Cutaneous symptoms such as acneform eruption and pigmentation are closely associated with blood levels of 2,3,4,7,8-penta-chlorodibenzofurans in Yusho patients, using data mining analysis. BMC Res Notes 2:27. doi:10.1186/1756-0500-2-27
Jaiswal AK (2004) Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med 36(10):1199–1207
Ju Q, Fimmel S, Hinz N, Stahlmann R, Xia L, Zouboulis CC (2011) 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters sebaceous gland cell differentiation in vitro. Exp Dermatol 20(4):320–325. doi:10.1111/j.1600-0625.2010.01204.x
Jux B, Kadow S, Esser C (2009) Langerhans cell maturation and contact hypersensitivity are impaired in aryl hydrocarbon receptor-null mice. J Immunol 182(11):6709–6717. doi:10.4049/jimmunol.0713344
Jux B, Kadow S, Luecke S, Rannug A, Krutmann J, Esser C (2011) The aryl hydrocarbon receptor mediates UVB radiation-induced skin tanning. J Invest Dermatol 131(1):203–210. doi:10.1038/jid.2010.269
Kadow S, Jux B, Zahner SP, Wingerath B, Chmill S, Clausen BE, Hengstler J, Esser C (2011) Aryl hydrocarbon receptor is critical for homeostasis of invariant gammadelta T cells in the murine epidermis. J Immunol 187(6):3104–3110. doi:10.4049/jimmunol.1100912
Kennedy LH, Sutter CH, Leon Carrion S, Tran QT, Bodreddigari S, Kensicki E, Mohney RP, Sutter TR (2013) 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated production of reactive oxygen species is an essential step in the mechanism of action to accelerate human keratinocyte differentiation. Toxicol Sci 132(1):235–249. doi:10.1093/toxsci/kfs325
Kohda F, Takahara M, Hachiya A, Takei K, Tsuji G, Yamamura K, Furue M (2013) Decrease of reactive oxygen species and reciprocal increase of nitric oxide in human dermal endothelial cells by Bidens pilosa extract: a possible explanation of its beneficial effect on livedo vasculopathy. J Dermatol Sci 72(1):75–77. doi:10.1016/j.jdermsci.2013.05.008
Krämer HJ, Kessler D, Hipler UC, Irlinger B, Hort W, Bödeker RH, Steglich W, Mayser P (2005) Pityriarubins, novel highly selective inhibitors of respiratory burst from cultures of the yeast Malassezia furfur: comparison with the bisindolylmaleimide arcyriarubin A. ChemBioChem 6(12):2290–2297
Krämer HJ, Podobinska M, Bartsch A, Battmann A, Thoma W, Bernd A, Kummer W, Irlinger B, Steglich W, Mayser P (2005) Malassezin, a novel agonist of the aryl hydrocarbon receptor from the yeast Malassezia furfur, induces apoptosis in primary human melanocytes. ChemBioChem 6(5):860–865
Krutmann J, Morita A, Chung JH (2012) Sun exposure: what molecular photodermatology tells us about its good and bad sides. J Invest Dermatol 132(32 Pt. 2):976–984. doi:10.1038/jid.2011.394
Kypriotou M, Huber M, Hohl D (2012) The human epidermal differentiation complex: cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp Dermatol 21(9):643–649. doi:10.1111/j.1600-0625.2012.01472.x
Lan CC, Yu HS, Lu JH, Wu CS, Lai HC (2013) Irradiance, but not fluence, plays a crucial role in UVB-induced immature pigment cell development: new insights for efficient UVB phototherapy. Pigment Cell Melanoma Res 26(3):367–376. doi:10.1111/pcmr.12077
Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, Wilhelm C, Veldhoen M (2011) Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147(3):629–640. doi:10.1016/j.cell.2011.09.025
Li D, Takao T, Tsunematsu R, Morokuma S, Fukushima K, Kobayashi H, Saito T, Furue M, Wake N, Asanoma K (2013) Inhibition of AHR transcription by NF1C is affected by a single-nucleotide polymorphism, and is involved in suppression of human uterine endometrial cancer. Oncogene 32(41):4950–4959. doi:10.1038/onc.2012.509
Loertscher JA, Lin TM, Peterson RE, Allen-Hoffmann BL (2002) In utero exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin causes accelerated terminal differentiation in fetal mouse skin. Toxicol Sci 68(2):465–472
Loertscher JA, Sattler CA, Allen-Hoffmann BL (2001) 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters the differentiation pattern of human keratinocytes in organotypic culture. Toxicol Appl Pharmacol 175(2):121–129
Luecke S, Backlund M, Jux B, Esser C, Krutmann J, Rannug A (2010) The aryl hydrocarbon receptor (AHR), a novel regulator of human melanogenesis. Pigment Cell Melanoma Res 23(6):828–833. doi:10.1111/j.1755-148X.2010.00762.x
Magiatis P, Pappas P, Gaitanis G, Mexia N, Melliou E, Galanou M, Vlachos C, Stathopoulou K, Skaltsounis AL, Marselos M, Velegraki A, Denison MS, Bassukas ID (2013) Malassezia yeasts produce a collection of exceptionally potent activators of the Ah (dioxin) receptor detected in diseased human skin. J Invest Dermatol 133(8):2023–2030. doi:10.1038/jid.2013.92
Marshall NB, Vorachek WR, Steppan LB, Mourich DV, Kerkvliet NI (2008) Functional characterization and gene expression analysis of CD4+ CD25+ regulatory T cells generated in mice treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Immunol 181(4):2382–2391
Matsumoto S, Akahane M, Kanagawa Y, Kajiwara J, Todaka T, Yasukawa F, Uchi H, Furue M, Imamura T (2013) Individuals’ half-lives for 2,3,4,7,8-penta-chlorodibenzofuran (PeCDF) in blood: correlation with clinical manifestations and laboratory results in subjects with Yusho. Chemosphere 92(7):772–777. doi:10.1016/j.chemosphere.2013.04.005
Mayser P, Schäfer U, Krämer HJ, Irlinger B, Steglich W (2002) Pityriacitrin—an ultraviolet-absorbing indole alkaloid from the yeast Malassezia furfur. Arch Dermatol Res 294(3):131–134
McLean WH, Irvine AD (2013) Old King Coal—molecular mechanisms underlying an ancient treatment for atopic eczema. J Clin Invest 123(2):551–553. doi:10.1172/JCI67438
Mimura J, Ema M, Sogawa K, Fujii-Kuriyama Y (1999) Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev 13(1):20–25
Mimura J, Fujii-Kuriyama Y (2003) Functional role of AhR in the expression of toxic effects by TCDD. Biochim Biophys Acta 1619(3):263–268
Mohammadi-Bardbori A, Bengtsson J, Rannug U, Rannug A, Wincent E (2012) Quercetin, resveratrol, and curcumin are indirect activators of the aryl hydrocarbon receptor (AHR). Chem Res Toxicol 25(9):1878–1884. doi:10.1021/tx300169e
Nagayama J, Todaka T, Hirakawa H, Hori T, Kajiwara J, Yoshimura T, Furue M (2010) Polychlorinated dibenzofurans as a causal agent of fetal Yusho. Chemosphere 80(5):513–518. doi:10.1016/j.chemosphere.2010.04.062
Nakamura M, Ueda Y, Hayashi M, Kato H, Furuhashi T, Morita A (2013) Tobacco smoke-induced skin pigmentation is mediated by the aryl hydrocarbon receptor. Exp Dermatol 22(8):556–558. doi:10.1111/exd.12170
Navid F, Bruhs A, Schuller W, Fritsche E, Krutmann J, Schwarz T, Schwarz A (2013) The aryl hydrocarbon receptor is involved in UVR-induced immunosuppression. J Invest Dermatol 133(12):2763–2770. doi:10.1038/jid.2013.221
Nebert DW, Karp CL (2008) Endogenous functions of the aryl hydrocarbon receptor (AHR): intersection of cytochrome P450 1 (CYP1)-metabolized eicosanoids and AHR biology. J Biol Chem 283(52):36061–36065. doi:10.1074/jbc.R800053200
Nguyen LP, Bradfield CA (2008) The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol 21(1):102–116
Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T (2010) Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci USA 107(46):19961–19966. doi:10.1073/pnas.1014465107
Niestroy J, Barbara A, Herbst K, Rode S, van Liempt M, Roos P (2011) Single and concerted effects of benzo[a]pyrene and flavonoids on the AhR and Nrf2-pathway in the human colon carcinoma cell line Caco-2. Toxicol In Vitro 25(3):671–683. doi:10.1016/j.tiv.2011.01.008
Öberg M, Bergander L, Hakansson H, Rannug U, Rannug A (2005) Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol Sci 85(2):935–943
Panteleyev AA, Bickers DR (2006) Dioxin-induced chloracne—reconstructing the cellular and molecular mechanisms of a classic environmental disease. Exp Dermatol 15(9):705–730
Papaiahgari S, Yerrapureddy A, Hassoun PM, Garcia JG, Birukov KG, Reddy SP (2007) EGFR-activated signaling and actin remodeling regulate cyclic stretch-induced NRF2-ARE activation. Am J Respir Cell Mol Biol 36(3):304–312
Pastor MA, Carrasco L, Izquierdo MJ, Fariña MC, Martín L, Renedo G, Requena L (2002) Chloracne: histopathologic findings in one case. J Cutan Pathol 29(4):193–199
Pot C (2012) Aryl hydrocarbon receptor controls regulatory CD4+ T cell function. Swiss Med Wkly 142:w13592. doi:10.4414/smw.2012.13592
Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL (2008) Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453(7191):65–71. doi:10.1038/nature06880
Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, Weiner HL (2010) An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 107(48):20768–20773. doi:10.1073/pnas.1009201107
Ray SS, Swanson HI (2003) Alteration of keratinocyte differentiation and senescence by the tumor promoter dioxin. Toxicol Appl Pharmacol 192(2):131–145
Saurat JH, Kaya G, Saxer-Sekulic N, Pardo B, Becker M, Fontao L, Mottu F, Carraux P, Pham XC, Barde C, Fontao F, Zennegg M, Schmid P, Schaad O, Descombes P, Sorg O (2012) The cutaneous lesions of dioxin exposure: lessons from the poisoning of Victor Yushchenko. Toxicol Sci 125(1):310–317. doi:10.1093/toxsci/kfr223
Schäfer M, Farwanah H, Willrodt AH, Huebner AJ, Sandhoff K, Roop D, Hohl D, Bloch W, Werner S (2012) Nrf2 links epidermal barrier function with antioxidant defense. EMBO Mol Med 4(5):364–379. doi:10.1002/emmm.201200219
Schäfer M, Werner S (2011) The cornified envelope: a first line of defense against reactive oxygen species. J Invest Dermatol 131(7):1409–1411. doi:10.1038/jid.2011.119
Schäfer M, Willrodt AH, Kurinna S, Link AS, Farwanah H, Geusau A, Gruber F, Sorg O, Huebner AJ, Roop DR, Sandhoff K, Saurat JH, Tschachler E, Schneider MR, Langbein L, Bloch W, Beer HD, Werner S (2014) Activation of Nrf2 in keratinocytes causes chloracne (MADISH)-like skin disease in mice. EMBO Mol Med (Epub ahead of print)
Schallreuter KU, Salem MA, Gibbons NC, Maitland DJ, Marsch E, Elwary SM, Healey AR (2012) Blunted epidermal l-tryptophan metabolism in vitiligo affects immune response and ROS scavenging by Fenton chemistry, part 2: Epidermal H2O2/ONOO(–)-mediated stress in vitiligo hampers indoleamine 2,3-dioxygenase and aryl hydrocarbon receptor-mediated immune response signaling. FASEB J 26(6):2471–2485. doi:10.1096/fj.11-201897
Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, Fujii-Kuriyama Y, Ishikawa T (2000) Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci USA 97(2):779–782
Sorg O, Zennegg M, Schmid P, Fedosyuk R, Valikhnovskyi R, Gaide O, Kniazevych V, Saurat JH (2009) 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko: identification and measurement of TCDD metabolites. Lancet 374(9696):1179–1185. doi:10.1016/S0140-6736(09)60912-0
Soshilov A, Denison MS (2008) Role of the Per/Arnt/Sim domains in ligand-dependent transformation of the aryl hydrocarbon receptor. J Biol Chem 283(47):32995–33005. doi:10.1074/jbc.M802414200
Suskind RR (1985) Chloracne, “the hallmark of dioxin intoxication.” Scand J Work Environ Health 11(3 Spec. No.):165-71
Sutter CH, Bodreddigari S, Campion C, Wible RS, Sutter TR (2011) 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases the expression of genes in the human epidermal differentiation complex and accelerates epidermal barrier formation. Toxicol Sci 124(1):128–137. doi:10.1093/toxsci/kfr205
Sutter CH, Yin H, Li Y, Mammen JS, Bodreddigari S, Stevens G, Cole JA, Sutter TR (2009) EGF receptor signaling blocks aryl hydrocarbon receptor-mediated transcription and cell differentiation in human epidermal keratinocytes. Proc Natl Acad Sci USA 106(11):4266–4271. doi:10.1073/pnas.0900874106
Takagi S, Tojo H, Tomita S, Sano S, Itami S, Hara M, Inoue S, Horie K, Kondoh G, Hosokawa K, Gonzalez FJ, Takeda J (2003) Alteration of the 4-sphingenine scaffolds of ceramides in keratinocyte-specific Arnt-deficient mice affects skin barrier function. J Clin Invest 112(9):1372–1382
Tauchi M, Hida A, Negishi T, Katsuoka F, Noda S, Mimura J, Hosoya T, Yanaka A, Aburatani H, Fujii-Kuriyama Y, Motohashi H, Yamamoto M (2005) Constitutive expression of aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol Cell Biol 25(21):9360–9368
Tigges J, Haarmann-Stemmann T, Vogel CF, Grindel A, Hubenthal U, Brenden H, Grether-Beck S, Vielhaber G, Johncock W, Krutmann J, Fritsche E (2014) The new aryl hydrocarbon receptor antagonist E/Z-2-benzylindene-5,6-dimethoxy-3,3-dimethylindan-1-one protects against UVB-induced signal transduction. J Invest Dermatol 134(2):556–559. doi:10.1038/jid.2013.362
Tigges J, Weighardt H, Wolff S, Götz C, Förster I, Kohne Z, Huebenthal U, Merk HF, Abel J, Haarmann-Stemmann T, Krutmann J, Fritsche E (2013) Aryl hydrocarbon receptor repressor (AhRR) function revisited: repression of CYP1 activity in human skin fibroblasts is not related to AhRR expression. J Invest Dermatol 133(1):87–96. doi:10.1038/jid.2012.259
Tsuji G, Takahara M, Uchi H, Matsuda T, Chiba T, Takeuchi S, Yasukawa F, Moroi Y, Furue M (2012) Identification of ketoconazole as an AhR–Nrf2 activator in cultured human keratinocytes: the basis of its anti-inflammatory effect. J Invest Dermatol 132(1):59–68. doi:10.1038/jid.2011.194
Tsuji G, Takahara M, Uchi H, Takeuchi S, Mitoma C, Moroi Y, Furue M (2011) An environmental contaminant, benzo(a)pyrene, induces oxidative stress-mediated interleukin-8 production in human keratinocytes via the aryl hydrocarbon receptor signaling pathway. J Dermatol Sci 62(1):42–49. doi:10.1016/j.jdermsci.2010.10.017
Uchi H, Yasukawa F, Kiryu H, Hashimoto K, Furue M (2012) Infundibular cyst with seborrheic verruca-like cyst walls in a patient with Yusho disease exposed to dioxins. Eur J Dermatol 22(5):687–688. doi:10.1684/ejd.2012.1805
van den Bogaard EH, Bergboer JG, Vonk-Bergers M, van Vlijmen-Willems IM, Hato SV, van der Valk PG, Schröder JM, Joosten I, Zeeuwen PL, Schalkwijk J (2013) Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J Clin Invest 123(2):917–927. doi:10.1172/JCI65642
Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B (2008) The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453(7191):106–109. doi:10.1038/nature06881
Vermeij WP, Alia A, Backendorf C (2011) ROS quenching potential of the epidermal cornified cell envelope. J Invest Dermatol 131(7):1435–1441. doi:10.1038/jid.2010.433
Vermeij WP, Backendorf C (2010) Skin cornification proteins provide global link between ROS detoxification and cell migration during wound healing. PLoS ONE 5(8):e11957. doi:10.1371/journal.pone.0011957
Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, Harada T, Engel JD, Yamamoto M (2003) Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 35(3):238–245
Wang XW, Li K, Guo S, Qiang HN, Liu L, Song P, Wei C, Yi XL, Jian Z, Li Q, Li CY, Gao TW (2012) The association of functional polymorphisms in the aryl hydrocarbon receptor (AHR) gene with the risk of vitiligo in Han Chinese populations. Br J Dermatol 166(5):1081–1087. doi:10.1111/j.1365-2133.2011.10798.x
Wei YD, Bergander L, Rannug U, Rannug A (2000) Regulation of CYP1A1 transcription via the metabolism of the tryptophan-derived 6-formylindolo[3,2-b]carbazole. Arch Biochem Biophys 383(1):99–107
Wincent E, Amini N, Luecke S, Glatt H, Bergman J, Crescenzi C, Rannug A, Rannug U (2009) The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J Biol Chem 284(5):2690–2696. doi:10.1074/jbc.M808321200
Wincent E, Bengtsson J, Mohammadi Bardbori A, Alsberg T, Luecke S, Rannug U, Rannug A (2012) Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proc Natl Acad Sci USA 109(12):4479–4484. doi:10.1073/pnas.1118467109
Yin Y, Li W, Son YO, Sun L, Lu J, Kim D, Wang X, Yao H, Wang L, Pratheeshkumar P, Hitron AJ, Luo J, Gao N, Shi X, Zhang Z (2013) Quercitrin protects skin from UVB-induced oxidative damage. Toxicol Appl Pharmacol 269(2):89–99. doi:10.1016/j.taap.2013.03.015
Related articles recently published in Archives of Dermatological Research (selected by the journal’s editorial staff)
Watabe Y, Tomioka M, Watabe A, Aihara M, Shimba S, Inoue H (2013) The clock gene brain and muscle Arnt-like protein-1 (BMAL1) is involved in hair growth. Arch Dermatol Res 305:755–761
Acknowledgments
This work was partly supported by grants from The Ministry of Health, Labour and Welfare; The Ministry of Education, Culture, Sports, Science and Technology; and the Environment Technology Development Fund of the Ministry of the Environment, Japan.
Conflict of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Furue, M., Takahara, M., Nakahara, T. et al. Role of AhR/ARNT system in skin homeostasis. Arch Dermatol Res 306, 769–779 (2014). https://doi.org/10.1007/s00403-014-1481-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00403-014-1481-7