
Abstract
Colloidal particles play a pivotal role in numerous applications across various disciplines, many of which necessitate precise control over particle size and size distribution. Seeded growth reactions have been established as effective methods for reproducibly accessing tailor-made particles. However, conventional batch-wise syntheses only yield discrete particle sizes. With the increasing focus on complex structures in current research, there is a demand for innovative and adaptable techniques to produce colloidal particles with precise sizes and size distributions. The Controlled Emulsion Extraction Process (CrEEP) is capable of addressing this challenge. Here, we present in detail how this synthesis works and demonstrate its reliability and versatility. Our approach exploits the time-dependent particle growth and enables accessing dispersions of controlled particle size distributions. We highlight these possibilities through a variation of the monomer feed and feed composition, resulting in gradual changes in both size and glass transition temperature, respectively. Beyond its application to polymer particles, CrEEP can be seamlessly extended to other seeded-growth mechanisms, such as the silica Stöber synthesis. Consequently, the Controlled Extraction Stöber Process (CrESP) similarly yields a size gradient, showcasing the generality of this synthetic advancement.
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Colloids play a crucial role in various scientific and civil fields. Colloidal particles are widely used for coatings [1], sensor application [2], catalysis [3], photonics [4, 5], plasmonics [6], passive cooling [7, 8], energy management [9], and in health treatment [10]. In many of these applications, the properties are closely related to the particle size and the size distribution. For example, drug delivery directly depends on different-sized nanoparticles depending on their target [11]. Here, the size influences the targeting and accumulation ability and the penetration in the body. Very small particles, \(<{5}~\textrm{nm}\) will be quickly metabolized, whereas larger sizes prevent particles from penetrating tissue [12]. Moreover, applications such as photonic sensors, displays, or other optical devices either depend on a particular size [13] or a narrow particle size distribution [14].
A widely used approach to synthesize narrowly dispersed nanoparticles with great control of the particle diameter is seeded growth methods. Established methods have been reported for silica [15,16,17] and polymer particles [18, 19] or quantum dots [20]. These methods lead to nanoparticles with narrow size distributions and are also used on an industrial level to fabricate dispersions for specific applications. While the composition of each nanoparticle can be controlled owing to the batch-wise synthesis, only discrete particle sizes have been obtained upon completion of the reaction [21]. More complex mesostructures can be achieved by introducing a hierarchy through two or more particle sizes. Size features are obtained by co-crystallization or layer stacking of binary or multinary particle sets [22,23,24]. The disparity of these features is limited by the particle size variability between the individual batch syntheses. However, increasing the number of adjusted batch syntheses to lower the size differences is infeasible, as hundreds of individual experiments would be needed. Additionally, the intrinsic particle size distribution, even of narrowly dispersed batches, prohibits the unambiguous distinction of batches with only a few nanometers in size difference. In the literature, gradual changes in size are only realized post-synthesis [25,26,27,28], or after assembly [29]. Consequently, novel synthetic strategies are required to access advanced particle dispersions that pave the way to complex and gradual mesostructures.
Recently, Schöttle et al. extended the well-known continuous emulsifier-free emulsion polymerization to obtain narrowly dispersed particle fractions with a continuous size gradient [18, 19, 30]. In the so-called controlled emulsion extraction process (CrEEP), fractions of the latex suspension are stored in a thin tube, separated by an air bubble exploiting the slow and time-dependent growth of polymeric seed particles. Monomer feed and fractionated extraction of the particle suspension were conducted with multiple syringe pumps, resulting in limitations regarding volume, reliability, and practicality.
In this work, we elaborate on the full potential of the CrEEP synthesis. We introduce technical improvements to the synthesis setup. In particular, replacing syringe pumps with multichannel peristaltic pumps provides a higher degree of reliability and versatility. Beyond the initial goal of CrEEP, creating a size gradient, we are also able to extensively control the process, which leads to the possibility of actively selecting distinct particle sizes. Finally, CrEEP synthesis can be applied to other seeded growth reactions. We demonstrate this versatility by a variation of the monomer feed ratio, which results in particles of gradually changing size and glass transition temperatures. Beyond that, CrEEP can also be applied to sol–gel syntheses such as Stöber synthesis [31], which we then call the controlled extraction Stöber process (CrESP).
Methods
Materials
We used water of ultrapure quality. Methyl methacrylate (MMA, 99 %, n-butyl acrylate (nBA, 99 %), 4-Styrenesulfonic acid sodium salt hydrate (NaSS) and potassium persulfate (KPS, 99.99 %), tetraethyl orthosilicate (TEOS, 99 %) were purchased from Sigma Aldrich. MMA and nBA were destabilized over Alox B prior to use, all other chemicals were used as received. The ammonia solution (35 %) was purchased from Fisher Chemicals and ethanol (EtOH 99.8 %) was obtained from VWR Chemicals.
Controlled emulsion extraction process (CrEEP)
Seed particles were synthesized in a 100mL three-necked flask equipped with a reflux condenser and septa. With 48 mL water pre-filled, the system was degassed under a constant argon stream for 60 min at 80 °C and stirred at 650 rpm. With 5 min equilibration time between each step, 1) 1.7 mL MMA/nBA mixture (90/10 vol%), 2) 2 mg NaSS in 1 mL H2O, 3) 40 mg KPS in 1 mL H2O were added. The seed synthesis was allowed to run for 30 min before starting the semi-batch process. For the extraction process, a silicon tube (2 m, \(\text{ID}=2~\textrm{mm}\)), filled with water, was connected to the Reglo ICC 4-channel peristaltic pump (Ismatec) through a peristaltic tube (\(\text {ID}=1.14~\textrm{mm}\)). The other end was inserted into the reaction mixture in the flask through a septum. A cannula, attached to another peristaltic tube (\(\text {ID}=1.14~\textrm{mm}\)) was bent a1nd inserted into the silicon tube in the flask to allow air fractionation. A cannula for monomer addition was inserted through a septum and connected to a peristaltic tube (\(\text {ID}=1.52~\textrm{mm}\)), which was inserted into a monomer reservoir. The pump started automatically after seed particle synthesis. For the growth, 2.9 mL of the same monomer mixture as before was added with 2.9 mLh-2. Simultaneously, 5 mL were extracted with 5 mLh-2, while 16 \(\mu\)L air was pumped into the silicon tube every 30 s with 31 mLh-2. The \(T_\text {g}\) gradient was achieved by adding an nBA monomer reservoir. We started with 20 mL 90/10 vol% MMA/nBA mixture. From the nBA reservoir, nBA was pumped (peristaltic tube \(\text {ID}=1.52~\textrm{mm}\)) to the other reservoir (16 mL, 16 mLh-2). During the extraction pause, the air was pumped at a constant rate of 5 mLh-2. Subsequently, the air channel returned to standard operation.
Controlled extraction Stöber process (CrESP)
Seed particles were synthesized by loading 45.6 mL EtOH and 10.7 mL water into a 100 mL three-necked flask equipped with a gas bubbler and septa at 60 °C and 300 rpm stirring speed. After an equilibration time of 5 min 17.25 mL ammonia solution and a preheated mixture of 5.7 mL TEOS and 22.8 mL EtOH were added. The reaction was then allowed to proceed overnight. Purification was carried out by centrifugation for 10 min at 10 krpm in a high-speed centrifuge (Sigma 3–30KS, Osterode am Harz, Germany). This was repeated two more times after the particles were redispersed in water.
For the extraction process, a 100 mL three-necked flask equipped with a gas bubbler and septa was loaded with 68.4 mL EtOH, 6.39 mL water and 4.49 mL of a 3.9 wt% seed dispersion at 60 °C and 400 rpm stirring speed. A polyethylene tube (2 m, \(\text {ID}=2~\textrm{mm}\)) filled with a 6.4: 1 EtOH / water mixture was connected to the Reglo ICC 4-channel peristaltic pump (Ismatec) through a peristaltic tube (\(\text {ID}=1.14~\textrm{mm}\)). The other end was inserted into the reaction mixture in the flask through a septum. A cannula, attached to another peristaltic tube (\(\text {ID}=1.14~\textrm{mm}\)) was bent and inserted into the silicon tube in the flask to allow air fractionation. A cannula for the addition of the precursor was inserted through a septum and connected to a peristaltic tube (\(\text {ID}=1.14~\textrm{mm}\)), which was inserted into a reservoir with an EtOH/TEOS mixture (80/20 vol%). For the growth, 5 mL of the precursor mixture was added with 2.5 mLh-2. Simultaneously, 5 mL were extracted with 2.5 mLh-2, while 0.05 \(\mu\)L air was pumped into the silicon tube every 60 s with 80 mLh-2.
Scanning electron microscopy
Images were taken with a Zeiss Ultra plus (Carl Zeiss AG, Oberkochen, Germany) at 2 to 5 kV with either secondary electron or in-lens detection. The samples have been sputtered with a thin layer of platinum. Particle size analysis was performed with Matlab-based evaluation software. Before plotting, outliers were detected using the 1.5 interquartile distance method. Ondulations in the violin plots can be attributed to the pixel size of the SEM micrographs.
Confocal laser scanning microscopy
2D color images and topographic reconstructions were taken with a laser scanning confocal microscope (Olympus LEXT OLS5000, Tokyo, Japan) in reflection geometry with a white light source and a 405 nm laser. Images were obtained with \(20\times\) lens with NA. 0.45.
Dynamic light scattering
The hydrodynamic diameters (z-average) and size distributions were obtained by a ZetaSizer Nano ZS (Malvern Instruments, Malvern, United Kingdom) at backscattering geometry (178\(^{\circ }\)). For the silica particles, the diameters were retrieved from the peak maximum of the size distribution instead of the z-average.
Microspectroscopy
Microspectroscopy spectra of drop-casted particle suspensions were measured on an Olympus IX73 inverted microscope with a \(40\times\) lens with NA. 0.6. All spectra were collected in transmission geometry with a halogen light source. The detection was carried out with a Zeiss CCD. The spot size was determined by an LED pilot light, which projects the sensor’s field of view onto the sample by tracing back the optical pathway of the microscope (A.S. & Co. GmbH, München, Germany).
Diffuse reflectance spectroscopy
Diffuse reflectance UV–vis spectra in a wavelength-range from 250 to 2500 nm were collected with a Cary 5000 spectrometer (Agilent Technologies, Santa Clara, CA USA) equipped with an integrating sphere (Labsphere Inc., North Sutton, NH USA). The samples were adhered to carbon adhesive tape on an aluminum SEM stab and mounted to the reflectance port. A Spectralon diffuse white standard (Labsphere Inc., North Sutton, NH USA) was used as a reference.
Atomic force microscopy
Atomic force microscopy was performed with a Bruker Dimension Icon equipped with a Nanoscope V controller (Bruker, Billerica, MA USA). The samples were imaged by PeakForce©-QNM mode using a ScanAsyst-Air cantilever (\(f_\text {nom}=70~\textrm{kHz}\), \(k_\text {nom}=0.4~{\textrm{Nm}^{-1}}\), Bruker, Billerica, MA USA). Temperature control of the samples was obtained by a temperature stage equipped with a thermal applications controller (Bruker, Billerica, MA USA). After at least 5 min of thermal equilibration at 25 °C, the sample was imaged. The images were evaluated by means of the Nanoscope Analysis program (Bruker, Billerica, MA USA).
Nuclear magnetic resonance spectroscopy
The \(^1\)H-NMR spectra were acquired using a Bruker Avance-III HD FT-NMR spectrometer equipped with a 5mm CryoProbe™ Prodigy BBO 500 S2, that operates at a \(\text {B}_0\) field of 11.7T (\(v_0(^1\text {H})=500.1~\textrm{MHz}\)). The samples were dissolved in CDCl3 and placed into conventional 5 mm NMR tubes. The measurements were carried out with single pulse (SP) excitation, a recycle delay of 1 s, and 256 repetitions. The pulse length was set to 11.5 \(\mu\)s corresponding to a tip angle of 30\(^{\circ }\). The spectra were referenced to TMS using chloroform as a secondary reference. For the quantitative determination of the monomer ratio in the MMA/nBA-copolymer, the signal at 3.5 to 3.7 ppm (MMA, O–CH3) and the signal at 3.8 to 4.1 ppm (nBA, –CH2–) were integrated and compared.
Differential scanning calorimetry
Differential scanning calorimetry was measured using a TA Instruments Discovery DSC 2500 (TA Instruments, New Castle, DE USA). Measurements were conducted between -20 to 180 °C with 20 Km\(^{-1}\). DSC curves are evaluated with the built-in TRIOS software from TA instruments.
Color-loss evaluation
The glass slide with 24 drop-casted fractions was placed onto a black-coated heat plate (PZ 28-2, Harry Gestigkeit GmbH, Düsseldorf, Germany) The temperatures were tracked every 0.5 s with the digital multimeter DAQ6510 (Keithley Tektronix, Solon, OH USA), equipped with a Type T thermocouple. The video was recorded with a smartphone (Fairphone 3+) at 30 fps in full HD (\(1920{}\times 1080~\textrm{pixel}\)) at an angle of 10\(^{\circ }\) and at a distance of 10 cm in MP4 format under constant ISO. The setup was illuminated by a full-spectrum lamp (Walimax pro LED Niova 600 Plus Daylight) with a light diffuser at an angle of 10\(^{\circ }\) and a distance of 30 cm. For video analysis, the Python package OpenCV library was used. Every 30th frame (corresponding to one-second time steps) was extracted and divided into 24 rectangles covering each drop-casted fraction. Subsequently, the mean color value was calculated over the drop-casted area, and a Savitzky-Golay filter from the SciPy library was used to fit the time-resolved color-loss data. The color-loss on-set temperatures were determined from the first derivatives.
Results and discussion
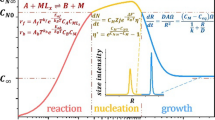
Seeded growth syntheses for a variety of nanoparticles [30, 32, 33], nanorods [34], and nanofibers [35, 36] are well described in the literature. The relevance of the synthesis of dispersions with defined particle size distributions lies in the separation of the nucleation and growth stages. This allows temporal control over the particle size [37, 38]. It starts with the formation of a nuclei, either a micell, in the case of emulsion polymerization, or primary particles in the Stöber process. The surfactant-free emulsion polymerization relies on monomer droplets in the water phase. Since the initiator is water-soluble and the monomer can diffuse and dissolve to some extent in the aqueous phase, polymerization can take place in the aqueous medium. Oligomers are formed, grow, and build nuclei micelles. Due to agglomeration, the particles grow until they are colloidally stable [39]. Similar steps occur in the Stöber process. SiOEt4 (TEOS) will initially hydrolyze in a basic environment and perform a condensation reaction to form the first nuclei. Two pathways for subsequent particle growth are outlined in the literature [15, 38]. The monomer addition model describes an initial nucleation phase resulting from a high TEOS concentration. Following this rapid nucleation phase, the concentration drops below a certain nucleation threshold. From this point onward, particle size increases solely due to surface growth. In contrast, the controlled aggregation model suggests a growth mechanism based on the aggregation of nuclei and primary particles until they achieve colloidal stability. Nucleation continues throughout the reaction until TEOS concentration decreases below the nucleation threshold. Subsequently, surface growth dominates.

Schematic setup for controlled extraction syntheses (CrEEP and CrESP). i) A three-necked flask is equipped with synthesis-specific equipment such as a reflux condenser or gas bubbler. Cannulas for monomer input, air fractionation, and inert gas are inserted through septa. A silicon tube for CrEEP (or a PE tube for CrESP) is also inserted through a septum and placed into the reaction mixture. ii) The cannula connected to the air channel is bent to fit into the extraction tube. For CrEEP, the fractionation happens in a 30 s interval, whereas CrESP requires 60 s. iii) The heart of the setup is the peristaltic pump with several channels to address monomer feed, air feed, extraction, and possible change of the monomer ratio. iv) The monomer/precursor reservoirs can be purged with inert gas. They can be connected via the pump, allowing for changes in the feed composition

Characterization of particle sizes of the time-resolved fractionation. a Light microscopy images (\(20\times\) lens) of drop-casted fractions taken in reflection geometry. The seed particles at 0 min reaction time show a dark blue color. With ongoing reaction time, we see a clear change in color. At 60 min, the particles reflect yellow light. The SEM micrographs also show an increase in particle size. The scale bar equals 750 nm. b We analyze the particle size with µ-vis transmission spectroscopy (cf. Fig. 8). From the stop band position we calculated the particle size (cf. Eq. 1). Each fifth fraction was measured. Accordingly, the particles grow linearly in size from 175 to 245 nm during the addition of monomer. Only a few nanometer size differences appear between the fractions. c The particle size of each fifth fraction was determined in dynamic light scattering. Here, the hydrodynamic diameter \(\text {D}_\text {H}\) gradually increases from 208 to 300 nm. Furthermore, the PDI stays mostly below a value of 0.05, indicating a high monodispersity of the particles
We will give detailed insights into the capabilities of our controlled extraction syntheses setup (Fig. 1). Therefore, we address the four sections i) synthesis, ii) fractionation, iii) peristaltic pump, and iv) monomer supply individually. For synthesis (section i), a three-necked flask is equipped with the necessary accessories for the aspired synthesis, such as reflux condensers or gas bubblers. Cannulas allow for an inert gas flow and semi-batch approaches such as continuous monomer/precursor addition. For extraction and fractionation (section ii), a thin silicone or polyethylene tube is inserted into the reaction mixture through a septum. As the reaction proceeds, particles can be continuously extracted and stored in the tube, retaining the time-dependent size and composition throughout the synthesis. To prevent particle diffusion and obtain individual particle fractions, air is periodically injected into the extraction tube via a bent cannula. The extraction and air injection, as well as the monomer addition, are controlled by a multichannel peristaltic pump (section iii). The peristaltic pump allows for convenient computer-aided control and gives access to complex pump protocols. The particular feature of a peristaltic pump to convey liquids from a container not only eliminates the discrete volume restrictions of syringe pumps but also offers a more consistent pressure throughout the air injection volume, leading to more uniform bubbles. Moreover, the monomer can be purged with inert gas, and a continuous change in the monomer/precursor composition (section iv) is possible. The presented universal setup was modified to target the needs of individual synthesis mechanisms and the desired gradual changes. The different variations of the setup used in this publication are shown in Fig. 6.
A straightforward application of the synthesis setup is the production of size gradients of polymer particles (Fig. 2). A continuous feed of monomer combined with extraction and fractionation yields 110 fractions with an increasing particle size. The coloration of selected drop-casted samples is a result of the structural order displayed in the corresponding scanning electron microscopy (SEM) measurements (Fig. 2a). A quantitative analysis of \(>1000\) particles per fraction from SEM images confirms the growth of the particles during the reaction. Their median diameter increase from 160 to 240 nm (Fig. 7). As self-assembly is only achieved by particles of a narrow size distribution, the color of the images attests to the monodispersity of the extracted fractions [22]. The redshift of the coloration corroborates the increasing particle diameter. The coloration of the drop-casted particle suspension arises from a periodic change in the refractive index. The wavelength of the constructively reflected light is directly proportional to the periodicity of the refractive index variation. In the case of particle assemblies, this is proportional to the particle size [40, 41]. For a more rigorous size evaluation, every fifth fraction was drop casted, and the obtained colloidal crystals (CC) were measured via visible light microspectroscopy (µ-vis) in transmission geometry. The stop bands (Fig. 8) of the spectra were used to calculate the particle size using Bragg-Snell’s law [42]:
with the wavelength \(\lambda _c\) at the peak position, the diameter of the spheres D, the dielectric filling ratio f and the refractive indices of the spheres \(n_\text {sphere}\) and air \(n_\text {air}\). The received diameters confirm the increasing particle size, as they show linear growth throughout the 60 min reaction time from 175 to 245 nm (Fig. 2b). This linear trend is also verified by dynamic light scattering (DLS, Fig. 2c). The offset to the previous measurements is a result of the determined hydrodynamic diameter \(D_\text {H}\) that contains the hydration shell. The polydispersity index received from the DLS instrument affirms the narrow particle size distribution of each fraction.
Creep allows for adjusting the particle size distribution by adjusting the extraction process. We demonstrate this with the implementation of an extraction pause of 20 min during the particle growth. A pause is best realized by substituting the dispersion fractions by air. This ensures an otherwise unchanged extraction and fractionation procedure. The particle sizes determined by µ-vis (Fig. 9) show that the implementation of a pause does not disturb the overall evolution of the particle size, as the sizes are in accordance with the continuous gradient discussed above. SEM and DLS measurements further confirm these results (Fig. 10). Monodispersity is ensured by a PDI below a value of 0.05 and by the appearance of a stop band.

Characterization of the extraction pause and diffuse reflectance UV–vis measurements of filtered particle mixtures. a µ-vis transmission spectroscopy of an extraction pause experiment. Here, the extraction is stopped between 20 min and 40 min leading to a gap in particle size of nearly 30nm (200 to 227 nm). b Diffuse reflectance UV–vis measurements of small (172± 0.3 nm) and large (245± 0.4 nm) particles, a binary mixture of both, a mixture of all fractions from a full range gradient (cf. Fig. 2) and the analog mixture of a gradient with an extraction pause. Above, the corresponding samples with the measurement spot size are displayed, highlighted by the red dashed circle
We want to highlight the relevance of particle size distributions for the case of light scattering. For that, we performed diffuse reflectance UV–vis measurements (Fig. 3b) of filtrated mixtures of the full-size gradient, the pause gradient, as well as a binary mixture of small (172 ± 0.3 nm) and large particles (245± 0.4 nm). Additionally, we provide the spectra for the filtered single neat particle suspensions, which resemble the seed and end fraction of the size gradient. The resulting single-component samples show distinct peaks at 350 nm and 525 nm, which is in accordance with the literature of colloidal glasses [30, 43]. All mixed particle size distributions fall between these two distinct cases. We observe marked differences but lack a clear trend regarding the particle size distribution. The binary mixture represents the utmost case of an extraction pause. It appears to be dominated by the large particle’s scattering properties (cf. Fig. 9c, d). The pause gradient, however, does not transition between the binary mixture and the full gradient. This highlights the complex interplay of light scattering with the present particle sizes and their arrangement in a colloidal glass. As demonstrated for these distinct three cases, the particle size distribution significantly controls the scattering behavior. Changes in the absolute reflectance are additionally governed by the thickness of the colloidal glass and its crack density (see photographs of Fig. 3b).
We now supplement the standard procedure with a gradual change in the monomer composition. We continuously added nBA to the monomer reservoir with an initial mixture of 92.3/7.7 mol% MMA/nBA (90/10 vol%) (Fig. 11a). The dead volume (1 mL) of the peristaltic tube causes a delay of 12 min until the monomer reservoir reaches the reaction mixture. At 60 min a monomer composition of 62/38 mol% inside the reaction mixture is obtained (Fig. 11b). NMR measurements of every 20th fraction are compared with the theoretical expectations. For that, the integrals of the O–CH3 of the MMA are set into relation to the prime –CH2– of the n-butyl group in nBA (Fig. 11c, d) [44]. The monomer ratio does not exactly match the expected values. We assume that not all monomer was consumed, leading to the observed offset (Fig. 4a). The continuous change in the composition does not affect the particle growth, as the sizes determined by µ-vis measurements are in close agreement with previous gradients (Figs. 4b, and 12a, b). Concomitantly, the increasing content of nBA built into the particles evokes a drop in the glass transition temperature (\(T_\text {g}\)) across the particle radius [45, 46]. As polymer particles with decreasing \(T_\text {g}\) face the problem of beam damage during SEM measurements (cf. Fig. 12c), we use atomic force microscopy (AFM) as another imaging technique for a complementary particle size evaluation (Fig. 4c). The evaluation of the topographical images confirmed the expected linear trend with the reaction time.

Characterization of particles with a continuous variation of the monomer feed. a Comparison of expected monomer fractions calculated from the reaction mixture and experimental monomer fractions from NMR spectroscopy. Both show a similar trend for how the nBA content changes during synthesis. NMR spectroscopy reveals a change from 92.6/7.4 % to 85.9/14.1 % (MMA/nBA). b µ-vis transmission spectroscopy measurements of every fifth fraction. During the synthesis, the volume fraction of MMA and nBA is gradually changed from 90/10 vol% to 50/50 vol% (MMA/nBA). Yet, the spectroscopy data shows a size range similar to that of previous gradients. c Particle sizes extracted from AFM measurements. d Video of drop-casted samples on a heated stage (cf. video in SI). One frame per second is extracted from the video to analyze the color loss. A time matrix for selected fractions is shown in e. With increasing reaction time, color loss starts at lower temperatures. f When every image is evaluated, we get precise curves of that temperature-dependent color. g The on-set temperatures of the curves in f show a trend toward lower temperatures, which is in agreement with DSC. h DSC measurements of fractionated particle suspensions. From the DSC curves (Fig. 13), the on-set temperature, the \(T_\text {g}\), and the end-set temperature are evaluated. All three show a trend to lower temperatures with the reaction time, e.g., the initial temperature is reduced from initially 97.3 to 53.4 °C
The increase from 184 to 265 nm in particle size is in the same range as the results of µ-vis, as well as DLS and SEM (Fig. 12d, e).
We use the loss of structural coloration of the CCs above \(T_\text {g}\) for an optical readout of the \(T_\text {g}\) gradient induced by the changing monomer ratio [2, 47]. Every fifth fraction was drop-casted onto a glass slide and placed on a heat plate. We recorded the heating of the sample at a constant angle (cf. supporting video) and extracted an image of the individual fractions every second (Fig. 4d, e). Subsequently, we calculated the mean color value in the drop-casted area and fitted the time-resolved color loss data with a Savitzky-Golay-Filter (Fig. 4f).

Characterization of CrESP gradient silica particles. a SEM analysis of selected fractions. The particle sizes increase with the reaction time. b Evaluated SEM images confirm the growth from initially 384 to 500 nm. c DLS measurements of the silica particles during a live reaction reveal higher absolute sizes but show a similar gradual increase. The diameters were retrieved from the peak maximum of the size distribution. The PDI increases with reaction time as a result of secondary nucleation. The dashed green line indicates a value of 0.05
We then used the first derivative to determine the color loss on-set temperatures (Fig. 4g). The evaluated data show a beginning color loss of the seed particles at 83 °C, which gradually declines to 62 °C of the last fraction. The plateau from 0 to 12 min reaction time is caused by the tube´s dead volume, as described earlier. The extension of the plateau to approximately 20 min is a result of the growth mechanism of particles in continuous emulsifier-free emulsion polymerization. The radial growth of the particles results in a core with the initial monomer ratio and a multilayered shell, with increasing nBA content [19]. Each layer can be assumed to have an individual \(T_\text {g}\), leading to a superposition in the macroscopic particle. As a result, the core characteristics prevail over the thin plasticized multilayer shell. The latter is still too thin to fill the voids of the CC and suppress the structural coloration. Differential scanning calorimetry (DSC) provides a more precise analysis of the thermal properties of the fractions. In the heat flow (cf. Fig. 13), we see different rates of decreasing on-set, glass transition, and end-set temperatures. The broadening of the \(T_\text {g}\) resembles the \(T_\text {g}\) behavior of gradient co-polymers, already published in the literature [48, 49]. Since the \(T_\text {g}\) is calculated using the midpoint of the onset and the endset temperature, it will follow the onset. However, the end-set stays nearly the same, since the core of the particles is assumed to be preserved. Due to the superposition of \(T_\text {g}\), the on-set temperatures offer a more distinct visualization of the particle´s gradual composition change and correlation to the color-loss method (Fig. 4h).
Broadening the scope of CrEEP, we also want to demonstrate its applicability beyond emulsion polymerization. Since silica particles are also synthesized via a seed-mediated growth reaction, the concept of CrEEP can be transferred to the Stöber synthesis [31]. The different reaction mechanism of the controlled extraction Stöber process (CrESP) requires several adaptions to the setup. As the Stöber process is based on a two-step condensation reaction, as mentioned before, its moderate kinetics promote secondary nucleation. We accounted for this by decelerating the precursor feed rate, reducing the precursor concentration in the feed, and elongating the overall reaction time to 120 min. Since the reaction uses an ethanol-water mixture in the presence of ammonia instead of pure water, a silicone tube cannot be utilized any longer. Upon storage, ethanol and ammonia would diffuse through the silicone. Therefore, we switched to a polyethylene (PE) tube. However, this significantly changes the wetting behavior of the suspension during the extraction process. As a consequence, we increased the volumes of fraction and air separation to retain a stable fractionation (cf. Section 2, Fig. 6).
Similar to the previous experiments, we measured SEM to evaluate the size gradient throughout the fractions. SEM images (Fig. 5a) reveal a structural order of the silica particles, which is a first indication of their narrow size distribution. However, we see a few smaller particles due to minor secondary nucleation after a reaction time of approximately 80 min. From the initial seed particles with 384 nm, the particle diameter increases to 500 nm (Fig. 5b). Furthermore, the violin plots indicate a narrow and consistent particle size distribution across all fractions. The undulations in the violins correspond to the pixel size of the SEM micrographs. DLS measurements (Fig. 5c) affirm the linear growth of the fractions. However, the determined particle size is shifted toward larger diameters. We assume that this effect is a combination of the hydration shell and the water/ethanol mixture in the dispersing media. Furthermore, the size-dependent scattering intensity of silica particles might be in a range that cannot be compensated by the instrument. The PDI shows values below 0.05 until a reaction time of 75 min. Subsequently, the PDI increases to a maximum of approximately 0.25 at 105 min. As the SEM images in Fig. 5a have already revealed, secondary nucleation occurs.
Conclusion
We presented a method for the synthesis of nanosized particles with a gradual change in size and composition. With our setup, we were able to precisely and reproducibly create a continuous size gradient over a range of approximately 100 nm. Further, we showed the option to exclude a certain size range from fractionation. Filtration of mixtures of a full-range gradient and an extraction pause gradient led to complex modulations of the diffuse reflectance spectra. Compared to the state-of-the-art semibatch syntheses the CrEEP process allows for the fabrication of particle dispersions with a gradient in each individual bead and within the whole particle dispersion. For example, the time-resolved composition variation translates to a spatially resolved glass transition temperature gradient in each particle’s multilayered shell in parallel to the continuous size discrimination. Going a step further, we transferred the concept of CrEEP to Stöber silica particle synthesis. Yet, the silica reaction relies on a quite different mechanism, the controlled extraction Stöber process (CrESP) yielded a narrowly distributed size gradient. With our gained insights about the control of particle size and composition, we pave the way for a facile and up-scaleable approach to create new gradient- and complex-structured materials.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.
References
Jiménez N, Ballard N, Asua JM (2024) Hard coatings from soft latexes: A review of routes to overcome the film formation dilemma. Macromol Mater Eng 2400026
Schöttle M, Tran T, Feller T, Retsch M (2021) Time-temperature integrating optical sensors based on gradient colloidal crystals. Adv Mater 33:2101948
Sápi A et al (2021) Metallic nanoparticles in heterogeneous catalysis. Catal Lett 151:2153–2175
Joannopoulos JD, Villeneuve PR, Fan S (1997) Photonic crystals: Putting a new twist on light. Nature 386:143–149
García PD, Sapienza R, Blanco A, López C (2007) Photonic glass: A novel random material for light. Adv Mater 19:2597–2602
Engineering and observation of localized plasmon modes (2011) Sonnefraud, Y., Leen Koh, A., McComb, D. W. & Maier, S. A. Nanoplasmonics. Laser Photonics Rev. 6:277–295
Woo HY, Choi Y, Chung H, Lee DW, Paik T (2023) Colloidal inorganic nano- and microparticles for passive daytime radiative cooling. Nano Converg 10:17
Jaramillo-Fernandez J et al (2019) A self-assembled 2D thermofunctional material for radiative cooling. Small 15:1905290
Zhou J et al (2022) Diminishing interfacial turbulence by colloid-polymer electrolyte to stabilize zinc ion flux for deep-cycling Zn metal batteries. Adv Mater 34:2200131
Lu Z et al (2023) Langmuir-Blodgett-mediated formation of antibacterial microneedles for long-term transdermal drug delivery. Adv Mater 35:2303388
Gaumet M, Vargas A, Gurny R, Delie F (2008) Nanoparticles for drug delivery: The need for precision in reporting particle size parameters. Eur J Pharm Biopharm 69:1–9
Wang Y, Gao N, Li X, Ling G, Zhang P (2024) Metal organic framework-based variable-size nanoparticles for tumor microenvironment-responsive drug delivery. Drug Deliv Transl Res 14:1737–1755
Lee SY, Kim S-H, Hwang H, Sim JY, Yang S-M (2014) Controlled pixelation of inverse opaline structures towards reflection-mode displays. Adv Mater 26:2391–2397
Arsenault AC, Puzzo DP, Manners I, Ozin GA (2007) Photonic-crystal full-colour displays. Nat Photonics 1:468–472
Ghimire PP, Jaroniec M (2021) Renaissance of Stöber method for synthesis of colloidal particles: New developments and opportunities. J Colloid Interface Sci 584:838–865
Giesche H (1994) Synthesis of monodispersed silica powders II. Controlled growth reaction and continuous production process. J Eur Ceram Soc 14:205–214
Hagemans F, Vlug W, Raffaelli C, van Blaaderen A, Imhof A (2017) Sculpting silica colloids by etching particles with nonuniform compositions. Chem Mater 29:3304–3313
Schork FJ, Ray WH (1987) The dynamics of the continuous emulsion polymerization of methylmethacrylate. J Appl Polym Sci 34:1259–1276
Li JQ, Salovey R (2000) “Continuous” emulsifier-free emulsion polymerization for the synthesis of monodisperse polymeric latex particles. J Polym Sci Part A Polym Chem 38:3181–3187
Dorfs D, Salant A, Popov I, Banin U (2008) ZnSe quantum dots within CdS nanorods: A seeded-growth type-II system. Small 4:1319–1323
Wohlleben W, Bartels FW, Altmann S, Leyrer RJ (2007) Mechano-optical octave-tunable elastic colloidal crystals made from core-shell polymer beads with self-assembly techniques. Langmuir 23:2961–2969
Vogel N, Retsch M, Fustin C-A, del Campo A, Jonas U (2015) Advances in colloidal assembly: The design of structure and hierarchy in two and three dimensions. Chem Rev 115:6265–6311
Roemling LJ, Bleyer G, Goerlitzer ESA, Onishchukov G, Vogel N (2023) Quantitative optical and structural comparison of 3D and (2+1)D colloidal photonic crystals. Langmuir 39:5211–5221
Kubrin R et al (2016) Bottom-up fabrication of multilayer stacks of 3D photonic crystals from titanium dioxide. ACS Appl Mater Interfaces 8:10466–10476
Chen M, Cölfen H, Polarz S (2015) Centrifugal field-induced colloidal assembly: From chaos to order. ACS Nano 9:6944–6950
Chen M, Hagedorn K, Cölfen H, Polarz S (2016) Functional gradient inverse opal carbon monoliths with directional and multinary porosity. Adv Mater 29:1603356
Bahner J, Hug N, Polarz S (2021) Anisotropic magnetism in gradient porous carbon composite aerogels. C 7:22
Bahner J, Klinkenberg N, Frisch M, Brauchle L, Polarz S (2019) Creating directionality in nanoporous carbon materials: Adjustable combinations of structural and chemical gradients. Adv Funct Mater 29:1904058
Ding H et al (2016) Free-standing photonic crystal films with gradient structural colors. ACS Appl Mater Interfaces 8:6796–6801
Schöttle M, Lauster T, Roemling LJ, Vogel N, Retsch M (2022) A continuous gradient colloidal glass. Adv Mater 35:2208745
Stöber W, Fink A, Bohn E (1968) Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci 26:62–69
Nakonechnyi I, Sluydts M, Justo Y, Jasieniak J, Hens Z (2017) Mechanistic insights in seeded growth synthesis of colloidal core/shell quantum dots. Chem Mater 29:4719–4727
Zhu J et al (2019) Spectrally tunable solid state fluorescence and room-temperature phosphorescence of carbon dots synthesized via seeded growth method. Adv Opt Mater 7
González-Rubio G et al (2019) Disconnecting symmetry breaking from seeded growth for the reproducible synthesis of high quality gold nanorods. ACS Nano 13:4424–4435
Jin X-H et al (2018) Long-range exciton transport in conjugated polymer nanofibers prepared by seeded growth. Science 360:897–900
Burgard M et al (2019) Mesostructured nonwovens with penguin downy feather-like morphology – top-down combined with bottom-up. Adv Funct Mater 29:1903166
Thickett SC, Gilbert RG (2007) Emulsion polymerization: State of the art in kinetics and mechanisms. Polymer 48:6965–6991
Han Y et al (2017) Unraveling the growth mechanism of silica particles in the Stöber method. In Situ Seeded Growth Model. Langmuir 33:5879–5890
Goodwin JW, Hearn J, Ho CC, Ottewill RH (1974) Studies on the preparation and characterisation of monodisperse polystyrene laticee: III. Preparation without added surface active agents. Colloid Polym Sci 252:464–471
von Freymann G, Kitaev V, Lotsch BV, Ozin GA (2013) Bottom-up assembly of photonic crystals. Chem Soc Rev 42:2528–2554
Velev OD, Gupta S (2009) Materials fabricated by micro- and nanoparticle assembly - the challenging path from science to engineering. Adv Mater 21:1897–1905
Zhang L et al (2015) Layer-by-layer approach to (2+1)D photonic crystal superlattice with enhanced crystalline integrity. Small 11:4910–4921
Bohren CF, Huffman DR (1998) Absorption and scattering of light by small particles. Wiley
Liu S-S et al (2020) Surface patterning of self-healing P(MMA/nBA) copolymer for dynamic control cell behaviors. Chin J Polym Sci 38:696–703
Penzel E, Rieger J, Schneider HA (1997) The glass transition temperature of random copolymers: 1. Experimental data and the Gordon-Taylor equation. Polymer 38:325–337
Nutz FA, Retsch M (2017) Tailor-made temperature-dependent thermal conductivity via interparticle constriction. Sci Adv 3:eaao5238
Schöttle M, Tran T, Oberhofer H, Retsch M (2023) Machine learning enabled image analysis of time-temperature sensing colloidal arrays. Adv Sci 10:2205512
Kim J, Mok MM, Sandoval RW, Woo DJ, Torkelson JM (2006) Uniquely broad glass transition temperatures of gradient copolymers relative to random and block copolymers containing repulsive comonomers. Macromolecules 39:6152–6160
Guo Y, Zhang J, Xie P, Gao X, Luo Y (2014) Tailor-made compositional gradient copolymer by a many-shot RAFT emulsion polymerization method. Polym Chem 5:3363–3371
Acknowledgements
AB, HvW, and TR thank the Elite Network of Bavaria (ENB). AB and TR appreciate the support of the University of Bayreuth graduate school. Help from Keylab Electron Microscopy in conducting SEM measurements is appreciated. The authors thank Stefan Rettinger for his exceptional support and lab work.
Funding
Open Access funding enabled and organized by Projekt DEAL. MT received funding from the International Research Training Group (IRTG) OPTEXC, project number 464648186 (German Research Foundation).
Author information
Authors and Affiliations
Contributions
AB: conceptualization, investigation, methodology, writing original draft, visualization; MT: conceptualization, investigation, methodology, writing original draft, visualization; HvW: investigation, methodology, writing review/editing; TR: investigation, methodology, writing review/editing; GP: resources, writing review/editing; JS: resources, writing review/editing; MR: conceptualization, resources, funding, writing review/editing, supervision.
Corresponding author
Ethics declarations
Consent for publication
Not applicable
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file 1 (mp4 53608 KB)
Appendix A: Extended Data
Appendix A: Extended Data

Images of the currently used setups for CrEEP and CrESP. Below, the corresponding particle fractions in the tubes are displayed

Particle size characterization via SEM images of the CrEEP standard full size gradient. The peaks in the violin plots correlate with the pixel size of the SEM micrographs

Measured µ-vis spectra of every fifth fraction of the standard full size gradient. a The spectra are corrected for the baseline, with respect to their stopband minimum, and normalized to the value at 700nm. b Extracted stop-band positions from a as a function of the reaction time

Measured µ-vis spectra of every fifth fraction of the extraction pause gradient and theoretical calculation of particle number and volume count. a The spectra are corrected for the baseline, with respect to their stop band minimum, and normalized to the value at 700 nm. b Extracted stop-band positions from a as a function of reaction time. c Calculated number and d volume count from Gaussian distributions of particle sizes. The gradients are calculated with superimposed single particle sizes with a bin size of 1nm. For volume count, the size distributions are multiplied with the cubic particle size \(r^3\) and normalized to the area. It is apparent, that larger particles dominate the volume count

Particle size evaluation of the extraction pause gradient via a DLS and b SEM. The peaks in the violin plots correlate with the pixel size of the SEM micrographs

Theoretical and experimental characterization of the monomer composition in the reservoir and the particles. a Calculated monomer content in mol% in the reservoir for synthesis. b Calculated monomer content in mol% added to the reaction flask. The plateau correlates with the dead volume of 1 mL in the peristaltic tube. c Chemical structure of the (PMMA-r-nBA) copolymer with highlighted methyl and ethyl groups for d NMR spectroscopy characterization. The colored regions correspond to the highlighted functional groups in the chemical structure

Particle size evaluation of the glass transition temperature gradient. a Measured µ-vis spectra of every fifth fraction of the glass transition temperature gradient. The spectra are baseline corrected, with respect to their stop-band minimum, and normalized to the value at 700 nm. b Extracted stop-band positions from a as a function of the reaction time. c SEM images depicting beam damage for different samples of the \(T_\text {g}\) gradient. d SEM and e DLS size evaluation of the \(T_\text {g}\) gradient. The peaks in the violin plots correlate with the pixel size of the SEM micrographs

Thermal characterization of the \(T_\text {g}\) gradient via DSC measurements. The curves are normalized and plotted with an offset
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Berger, A., Theis, M., von Wedel, H. et al. A versatile method for facile and reliable synthesis of colloidal particles with a size and composition gradient. Colloid Polym Sci (2024). https://doi.org/10.1007/s00396-024-05282-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00396-024-05282-7