
Abstract
Ascending thoracic aortic aneurysm (ATAA) remains a significant medical concern, with its asymptomatic nature posing diagnostic and monitoring challenges, thereby increasing the risk of aortic wall dissection and rupture. Current management of aortic repair relies on an aortic diameter threshold. However, this approach underestimates the complexity of aortic wall disease due to important knowledge gaps in understanding its underlying pathologic mechanisms.
Since traditional risk factors cannot explain the initiation and progression of ATAA leading to dissection, local vascular factors such as extracellular matrix (ECM) and vascular smooth muscle cells (VSMCs) might harbor targets for early diagnosis and intervention. Derived from diverse embryonic lineages, VSMCs exhibit varied responses to genetic abnormalities that regulate their contractility. The transition of VSMCs into different phenotypes is an adaptive response to stress stimuli such as hemodynamic changes resulting from cardiovascular disease, aging, lifestyle, and genetic predisposition. Upon longer exposure to stress stimuli, VSMC phenotypic switching can instigate pathologic remodeling that contributes to the pathogenesis of ATAA.
This review aims to illuminate the current understanding of cellular and molecular characteristics associated with ATAA and dissection, emphasizing the need for a more nuanced comprehension of the impaired ECM–VSMC network.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
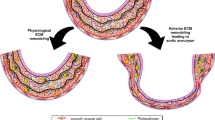
An ascending thoracic aortic aneurysm (ATAA) is a localized dilation in the proximal segment of the aorta. Aortic aneurysms represent weakened areas within the aorta, posing a significant risk of tearing or rupturing and resulting in severe, potentially life-threatening internal bleeding. If left untreated, ATAA can lead to severe complications such as aortic dissection (ATAAD) and rupture, with mortality rates of 50% within 24 h, including 21% mortality in patients who arrived alive in the hospital (Fig. 1) [137]. Known risk factors for ATAA development include advanced age > 65 years, systemic hypertension, and male sex (Fig. 2) [40]. Indeed, ATAA is diagnosed more frequently in men, which also is reflected in 70% of individuals with ATAAD [152]. On the contrary, ATAA severity has been indicated to be worse in women compared to men with faster aneurysm growth [32] and increased in-hospital mortality rates [134]. The estimated incidence of ATAA ranges from 5–10 per 100,000 individuals/year [37, 156], with a currently increasing trend [107]. Compared to abdominal aortic aneurysm (AAA), ATAA exhibits different flow patterns [176], regional variations [185], and developmental origins [193].

Central illustration: Development of Precision Medicine in Thoracic Aortic Diseases gives a summary of the current clinical patient screening management based on diameter threshold (including the risk of aneurysm and rupture) and the proposed screening management with the use of clinical biomarkers as an add-in to imaging modalities to prevent invasive surgical repair and high-risk of mortality

Schematic overview of major clinical risk factors in ATAA. Besides genetic syndromes, connective tissue disease, and bicuspid aortic valve morphology, these include hypertension, smoking, male sex, age, and COPD
Notably, prophylactic treatment options of ATAA are limited to aortic surgery in which a diameter threshold of 5.5 cm and aortic diameter growth rate ≥ 0.3 cm/year are recognized as cut-off values [37]. However, it is recognized that most patients develop ATAAD before reaching these thresholds (Fig. 1) [75, 161, 163].
Therefore, clinical biomarkers, representing early detection of patients at risk of ATAAD are highly anticipated. From an etiological point of view, less than 30% of all ATAA cases are genetically triggered, and thus more than 70% are sporadic or degenerative [35]. It is tempting to speculate that unknown (epi)genetic factors play a key role in initiating and progressing degenerative ATAA.
In patients with bicuspid aortic valves (BAV) or genetic mutations, ATAA is more commonly observed at a younger age [198]. Heritable ATAA has been associated with over 15 genes, including those encoding ECM-proteins such as fibrillin 1 (FBN1), type III collagen (COL3A1), or transforming growth factor-β (TGF-β) receptor proteins such as TGFBR1 and TGFBR2 [204]. It also involves VSMC proteins such as smooth muscle cell actin (ACTA2), myosin heavy chain 11 (MYH11), myosin light chain kinases (MYLK), and protein kinase cGMP-dependent type 1 (PRKG1) [159].
Furthermore, arteries are exposed to wall shear stress (WSS), which is induced by blood flow and exerted at the valvular and vascular endothelial layers [10]. Unequal distribution of WSS at the outer curvature of the ascending aorta has been associated with degenerative ATAA [171]. Blood pressure also exerts a key hemodynamic influence, i.e., wall stress, on the integrity of aortic wall tissue [61].
In the medial layer, vascular smooth muscle cells (VSMC) play an important role in vascular remodeling, exhibiting characteristic plasticity to adapt to changing flow and pressure conditions, e.g., by switching between a contractile and a synthetic phenotype [177]. VSMCs are further supported by the extracellular matrix (ECM) which plays a crucial role in regulating mechanical behavior and resilience, providing elasticity, and imparting arterial wall strength [2]. Once the ECM–VSMC network is disrupted, mechanosensing and mechano-signaling are impaired, leading to VSMC phenotype switching [153]. A compromised structure and function of the ECM leads to mechanical abnormalities and functional changes at the tissue level associated with aortic disease [208]. Progressive loss of arterial wall strength eventually culminates in the development of ATAA and even ATAAD.
There is an urgent need to identify novel biomarkers to screen patients at high-risk for ATAAD. Here, we summarize the current literature on the pathophysiology of ATAA and ATAAD with a focus on ECM–VSMC dysfunction. We highlight gaps in current diagnostic approaches, as well as recommend potential clinical biomarkers that may contribute to advancing our understanding of the development of early-stage ATAA, ultimately to predict and prevent morbidity and mortality associated with ATAAD.
Challenges in clinical management
In clinical practice, the characterization of ATAA is predominantly confined to diameter measurements. The aortic diameter is the principal decision-making criterion for surgical intervention within the Multidisciplinary Aortic Team. According to the latest clinical guidelines (EACTS/STS) for aortic diseases published in 2024, surgical replacement of the aorta is recommended when the aortic diameter is greater than 5.5 cm [37]. In high-risk patients with the presence of connective tissue disorders (e.g., Marfan or Loeys-Dietz), earlier intervention is recommended (≥ 4.5 cm). Patients with low-risk are monitored by imaging every year for timely detection of the surgical threshold (Fig. 1). However, up to 96% of ATAAD occurs in vessels with diameters below the surgical interventional threshold (< 5.5 cm) and 60% of ATAAD occurs in aortas with normal diameters (< 4 cm) [180]. Also, most patients with ascending aortic aneurysms of > 4 cm show little to no further growth during annual follow-up [1]. This is a major unmet clinical challenge for cardiologists and cardiac surgeons in assessing and managing ATAA.
Recently, several new parameters such as aortic elongation and aortic volume have been identified as potentially important predictors of ATAAD [74, 87]. Although these parameters alongside other morphologic characteristics of the vasculature such as the vertebral artery tortuosity show promising results [147], they still represent patients at a later stage of disease development and earlier detection of patients at risk requires a holistic approach implementing multi-scale analysis including vessel wall characterization.
In recent years, imaging modalities such as computed tomography (CT) combined with positron emission tomography (PET), using fluorodeoxyglucose (18F-FDG) or 18F-sodium fluoride (18F-NaF) administration provide geometrical, molecular, and functional information of aortic disease [56]. Several studies showed promising results regarding aortitis [80, 81], aneurysm growth and future clinical events [103], atherosclerosis [179], and detecting malignant tumors of the aorta [199]. Photon Counting-CT is another promising technique, which is still in clinical trial, yet has proven its clinical value regarding improved spatial resolution, and optimized spectral imaging [221]. Furthermore, this method offers precise tissue characterization and improved perfusion imaging while minimizing radiation exposure.
Blood-flow characteristics play a key role in ATAA formation, with effects on endothelial cell (EC) homeostasis and the response of VSMCs. Therefore, a functional assessment of detailed hemodynamic measurements is required to investigate flow characteristics and biomechanical forces. Four-dimensional flow magnetic resonance imaging (4D)-flow MRI, where phase-contrast methods are used to encode blood flow velocities along all dimensions in the aorta [42, 110], has been introduced as a powerful non-invasive technique in cardiovascular imaging for the assessment of local WSS [30]. WSS, which refers to the force per unit area exerted by moving fluid in the vessel, can be estimated as a product of wall shear rate (WSR) and local blood viscosity. Yet, it has not been validated as a clinical screening tool. One of the limitations of 4D flow MRI is insufficient spatial resolution which may underestimate WSS values and affect the accuracy of flow patterns [86]. Broader application of 4D flow MRI has been impeded by long scan times, costs, and data processing and analysis requiring special software. These obstacles hinder both reproducibility and clinical application [131]. Nevertheless, some longitudinal studies showed that 4D flow MRI can be used as a predictive tool to distinguish low and high WSS in ATAA patients with BAV [14, 20, 67, 144] or without BAV [171] compared to healthy volunteers.
Recently, the left ventricular outflow tract angle (LVOT-angle) has been a region of interest in ATAA pathology. Aortas less aligned to the axis of the heart were associated with ATAA [97]. Another study showed a positive correlation between LVOT-aortic angle and WSS on the outer curvature, indicating that increased LVOT-aortic angles (> 58.5º) were linked to elevated levels of WSS [184]. Geometrical biomarkers combined with flow patterns may improve the prediction of ATAA at risk or those who need an earlier intervention.
Pathophysiology
VSMC phenotypic switching in ATAA
In the adult arterial vessel wall, VSMCs are present in both the contractile and synthetic phenotype. Contractile VSMCs are connected via integrins to the ECM and are in a quiescent, non-proliferative state and facilitate contraction and dilation of resistance vessels and microvasculature, thereby regulating blood flow [160]. Contractile phenotype markers include smooth muscle-myosin heavy chain 11 (MYH11), calponin, smooth muscle-22 α (SM22α), and α‐smooth muscle actin (ACTA2) (Table 1). After VSMCs are released from the ECM, integrins trigger intracellular signaling and regulate VSMC phenotypic switching from “contractile” to “synthetic” phenotype. Synthetic VSMCs are characterized by reduced expression of contractile markers (Table 1) [57], and increased production of matrix metalloproteinase (MMP) thereby shifting the balance towards cell migration and ECM remodeling [27]. This transition of VSMCs toward synthetic phenotype can be assessed by integrin detection by flow cytometry, immunocytochemistry, and immunoprecipitation [88, 194].
In larger and so-deemed ‘elastic’ arteries and especially in the aorta, the precise role and relevance of the contractile phenotype and contractile responses to mechanical vessel wall stress and biomechanical stretch are not fully understood. Notably varying sites of the aorta are derived developmentally from different embryonic origins. VSMCs of the aortic root are predominantly derived from the lateral mesoderm. In contrast, VSMCs of the aortic arch are derived from the neural crest, and the descending aorta VSMCs originate from the paraxial mesoderm (Fig. 3) [193]. Notwithstanding there is overlap in the descending aorta wherein spatially distinct domains, have been noted by lineage fate tracing in mice [186]. However, the use of lineage-specific differentiation to VSMCs from human-induced pluripotent stem cells (hiPSCs) has yielded distinct cellular phenotypes suggesting lateral mesoderm malformations correlating to root dilation in Loeys-Dietz and neural crest VSMCs in Marfan associated ATAA [65, 234].

Regional heterogeneity and embryological diversity within human aorta: LM lateral mesoderm (green, located in aortic root), NC neural crest (pink, located in ascending/arch), PM paraxial mesoderm (red, located in descending aorta)
Disruption of homeostasis for vascular repair may result in a key role of VSMCs contributing to vascular pathology. Owens and colleagues showed that the high plasticity of VSMCs facilitates phenotypic switching towards synthetic VSMCs to adapt to environmental stress [160]. Furthermore, it has been shown in a co-culture model of ECs-VSMCs, that altered flow on ECs already induces a synthetic VSMC phenotype [183]. Also, the local inflammatory cascade can induce a phenotypic switch of VSMCs and transform them into synthetic VSMCs with fibroblast-like characteristics [195]. This phenotypic transition often leads to an increase in MMP production [192].
The impact of aberrant wall shear stress on mechanotransduction
Mechanobiology implies that cellular mechanosensing and ECM regulation are critical for maintaining mechanical homeostasis and proper vascular function [80]. Mechanotransduction is the biochemical process through which ECs and VSMCs convert mechanical stimuli through the cytoskeleton, leading to intracellular responses and extracellular changes [166]. In addition, EC integrins play a crucial role in the mechanotransduction of VSMC contractility [151].
Shear stress activates EC integrins by switching them to an active conformation, thereby increasing affinity to ECM proteins [220]. During systole, ECs, and VSMCs experience both longitudinal and circumferential mechanical deformation (‘strain’). The disruption of elastin-VSMC connections plays a critical role in aneurysm formation, not only by damaging the structural integrity of the aortic wall but also by altering cellular processes such as mechanotransduction and cytoskeletal remodeling of VSMCs [241]. ECs regulate these processes by activating mechanosensors, including vascular endothelial growth factor receptor 2 (VEGFR2), vascular endothelial-cadherin (VE-cadherin), and platelet EC adhesion molecule (PECAM-1) [232]. It has been demonstrated that ECs derived from the aortic wall of an aneurysm present decreased levels of VE-cadherin, von Willebrand factor (vWF), and PECAM-1 [134]. These decreased levels disrupt mechanotransduction and induce macrophage infiltration in the media and adventitia through nuclear factor-kB NF-κB activation [190, 200].
Different studies on BAV patients with ATAA have confirmed increased WSS in the greater curvature of the ascending aorta [45, 46]. In this greater curvature region, there was evidence of increased medial degeneration with reduced collagen type I and III and increased VSMC apoptosis [45, 46]. Moreover, it has been demonstrated that the WSS effect on media degeneration and VSMC phenotype change (expressing synthetic marker MYH10) has been shown in ATAA patient samples [102]. High WSS is a frictional force at the EC surface produced by blood flow which induces impaired mechanotransduction leading to vascular remodeling and potentially ATAA formation [192]. The initial sensing event and transduction of the mechano-signaling pathway are as follows: under constant laminar flow, the mechanosome is quiescent and inactive. However, when shear stress changes, the mechanosome consisting of PECAM-1, VEGFR2, and VE-cadherin triggers the activation of NOX2 and eNOS, resulting in the release of ROS and NO [31]. In addition, increased WSS corresponded with changes in the ascending aorta using pre-operative WSS mapping [71]. Here, increased elastin degradation in regions of high WSS, as well as increased TGF-β1 and MMP-1, MMP-2, and MMP-3 were reported [71].
However, the relation between WSS and gene expression, and protein synthesis remains unclear and needs further investigation. Such information is key for fundamental research on shear stress–mechanotransduction mechanisms. Clinically, it could aid in explaining why certain patients with an aortic diameter below the current intervention criteria still develop acute aortic complications.
ECM degradation
The major histopathological features associated with ATAA are abnormalities of cellular and matrix constituents of the media. These include elastin degradation and fragmentation, collagen disorganization, and loss of VSMC number [132]. In addition, mucoid ECM accumulation is a common pathologic finding in TAA and can serve as a marker for ECM degradation [72]. Furthermore, elastic fiber fragmentation has been reported to be greatest in the proximal aneurysmal ascending aorta compared to the middle or distal regions [197]. Furthermore, fibulin-4 (Fbln4), a component of elastic fibers essential for maintaining aortic wall integrity has been implicated in aneurysm formation. Loss of Fbln4 was associated with significantly upregulated levels of thrombospondin-1 (Thbs1), a homotrimeric glycoprotein [227]. Fhbs4 expression is induced by mechanical stretch resulting in disruption of elastin-VSMC connections and decreased mechanosensing. Under physiologic conditions, VSMCs maintain ECM homeostasis by a balanced secretion of MMPs and their inhibitors TIMPs to maintain a load-bearing mechanical state.
The disturbed balance between TIMPs/MMPs
In ATAA, dysfunction of VSMCs causes an imbalance between the production of active MMPs [219], especially MMP-2 and MMP-9 [83, 123], and a decreased expression of TIMPs, mainly TIMP-1[6]. Other proteolytic enzymes have also been found to modulate both ECM and VSMC function in ATAA, such as A-disintegrin metalloproteinase (ADAMTS-1, ADAMTS-4) [172]. Recent data have demonstrated that MMP-1, -2, -3, -9, -12, and -13 play roles in the progression of ATAA [111, 114, 169, 216]. Specifically, MMP-2 and MMP-9 are known to degrade collagen fragments and MMP-2, MMP-9, and MMP-12 elastin fragments [122]. This, in turn, facilitates the disengagement of VSMCs resulting in aortic tissue remodeling [4, 139, 177, 181]. In a mouse model deficient in MMP-2, ANG-II infusion resulted in exacerbated ATAA. The same study unveils the dual role of MMP-2 in both degrading and synthesizing ECM, showcasing its multifaceted role in tissue remodeling [191]. Nevertheless, MMPs and TIMPs are widely distributed throughout physiologic processes in different organs, suggesting that MMP and TIMP blood levels may not represent reliable biomarkers to correlate with aortic aneurysm levels.
In individuals with BAVs, MMP-2 levels are higher compared to those with TAVs. However, MMP-13 levels in TAV samples are significantly higher than in BAVs [84]. MMP-13 is a member of the collagenase subgroup within the MMP family, and previous studies have shown its upregulation in both human AAA [130, 206] and ATAAD [232] tissues. MMP-13 is primarily synthesized by VSMCs present in the aortic wall [130] triggered by JNK, ERK, and p38 kinases of the MAPK family. It not only degrades elements of the aortic collagen network, such as type I and III collagen [34], but also proteins within the elastic fiber networks, such as fibrillin 1[9], fibronectin [196], and decorin. This degradation has a significant impact on the structure of the ECM, potentially contributing to the growth and dissection of the aorta. A separate study provided initial evidence indicating that the collagenase MMP-13 contributes to aneurysm development in mouse models of Marfan syndrome. Pharmaceutical inhibition of MMP-13 in Fbn1 GT8 Marfan mice effectively prevents aortic root dilatation, underling the relevance of MMP-13 as a potential therapeutic target for managing aortic aneurysms [237].
In ATAA, a decrease in the elastin-to-collagen (ELN/COL) ratio is associated with increased aortic stiffness [108]. Typically, the ELN/COL ratio in the media of healthy aorta is some 1.7–1.9, whereas in the media of ATAA aortas, ELN/COL ratios are as low as 0.83–0.81 [223]. Increased collagen expression in the vasculature is most likely a compensatory response to elastin degradation and thus vascular remodeling [19] which results in the thickening of the arterial wall and increased vascular stiffness [78, 236].
Vascular calcification in ATAA
Vascular calcification has been suggested as a potential measure strongly associated with atherosclerosis [190], ATAA, and AAA [13]. It has been reported that ATAA patients develop extensive medial calcification associated with a phenotypic switch of VSMCs into osteoblastic-like- cells therefore creating a pro-calcifying environment [151, 224]. There is emerging evidence indicating that Krüppel-like factor 4 (KLF4), a potent tumor repressor, regulates the transition of VSMCs into osteogenic phenotypes in both murine and humans [5]. Initial calcifications, often referred to as micro-calcifications, typically measure less than 50 μm in size [98] and primarily originate from extracellular vesicles of osteochondrogenic VSMCs [89]. Increased medial micro-calcification was associated with mild and moderate histopathological degeneration (mild/moderate elastin fragmentation) [53]. Instead, patients with severe histopathological degeneration (severe elastin fragmentation), exhibited reduced medial micro-calcification [53]. This mechanism relies on intact elastin fibers for the deposition of micro-calcification.
In the early stage of the disease, micro-calcification can be measured ex vivo by the expression of osteogenic VSMC markers alkaline phosphatase (AP) and osteopontin (OP), or in vivo using 18F-sodium fluoride autoradiography (18F-NaF) [53]. The deposition of micro-calcification in combination with the local fragmentation of elastin fibers is associated with an increased risk of aortic wall rupture, as a result of stiffened regions of the soft ECM in the vessel wall [53, 220].
Matrix Gla-protein (MGP) is an inhibitor of medial micro-calcification that is widely recognized for its importance. It is predominantly secreted by VSMCs and is a vitamin K-dependent protein (VKDP). For MGP to become biologically active, it must undergo post-translational modification via vitamin K-dependent carboxylation by the enzyme gamma-glutamyl carboxylase (GGCX) [21]. Oral anticoagulation or a deficiency in vitamin K can result in inactive MGP. This is indicated by increased levels of dephosphorylated undercarboxylated MGP (dp-ucMGP) in the circulation. It should be noted that dp-ucMGP is a biomarker of vitamin K status and has been related to vascular calcification [91]. In humans, MGP deficiency is known as Keutel Syndrome, a genetic condition characterized by soft tissue calcification [79]. It has been demonstrated that MGP deficiency in humans may exhibit a gradual onset of calcification in both arteries [91] and heart valves [29]. Further, there appears to be a correlation between MGP deficiency and elastin calcification [188]. This data indicates a link between impaired carboxylation of MGP and the development of calcification starting around elastin fibers in the tunica media of patients who underwent percutaneous coronary intervention. It also implies the crucial role of vitamin K in activating MGP to effectively prevent vascular calcification.
Regulatory pathways
Arterial remodeling modulated by TGF-β signaling
Various molecular pathways involved in the synthesis of the ECM exhibit alterations in ATAA. This is evident for example through the upregulation of fibrogenic growth factors like TGF-β, platelet-derived growth factors (PDGF), and connective tissue growth factor (CTGF) [77, 95, 174]. Platelet-derived growth factor-BB (PDGF-BB) and TGF-β serve as pivotal mediators in VSMC phenotypic switching [27, 214, 235]. For example, it has been reported that in Marfan syndrome, an increased expression of TGF-β in VSMCs, results in the activation of SMAD3 and Erk signaling contributing to aneurysm progression [149, 229]. Other factors that alter the aortic wall integrity include angiogenic factors including angiopoietin-1, angiopoietin-2, thrombospondin-1, and fibroblast growth factor-1 [100].
Activation of TGF-β can be triggered by multiple factors, such as thrombospondin [187], and reactive oxygen species (ROS) [51, 120]. Increased expression of TGF-β in VSMCs of patients with Marfan syndrome has been associated with increases in ROS production [93, 229]. In addition, TGF-β activation occurs through the proteolytic degradation of the latent TGF-β complex by MMP-2 and MMP-9. Also, integrin αV can activate TGF-β1 by establishing a close connection between the latent TGF-β complex and MMPs [222, 230]. It thus appears that TGF-β1 induces VSMC senescence via ROS-mediated activation of NF-κB signaling, potentially contributing to aneurysm formation in Marfan patients.
Another pathway involved in TGF-β signaling is the PRKG1 which regulates VSMC relaxation through type I cGMP-dependent protein kinase (PKG-1) [69]. In ATAA, the impaired PRKG1 pathway inhibits Rho-associated protein kinase (ROCK), ensuring the myosin light chain remains in a relaxed state and leading to a reduction in VSMC contractility [202].
Moreover, SMAD3 is a critical transcription factor in the TGF-β signaling pathway, regulating VSMC differentiation and matrix deposition. Heterozygous SMAD3 mutations increase the risk of aortic root aneurysms, which may progress to type A aortic dissections without surgical intervention [62]. Hence, TGF-β through SMAD3 signaling stimulates the proliferation and differentiation of neural crest-derived VSMCs in the ascending aorta [207]. Patients with Loeys–Dietz syndrome (LDS) are characterized by mutations in genes encoding for TGF-β receptors 1 and 2 [214]. In patients with LDS, increased secretion of TGF-β ligands activates TGFBR1/TGFBR2 complexes and enhances TGF-β signaling [60]. In vitro, VSMC explants from patients with heterozygous mutations in TGFBR2 showed decreased expression of VSMC contractile proteins and displayed a synthetic VSMC phenotype [85]. Further, in human ATAAD genetic variants in SMAD4, a secondary messenger of the TGF-β pathway, correlate with VSMC apoptosis, reduced contractile markers, and ECM degradation [49].
Down-regulation of YAP in response to mechanical stress
Involvement of the Hippo pathway in ATAA has been seldom reported, which is surprising due to the pivotal role of mechanobiological processes in aneurysm formation. The Hippo/Yes Associated Protein (YAP) signaling pathway is a highly evolutionary conserved mechanism with a central role in regeneration, proliferation, migration, and cell fate biology [59]. By initiating a cascade of several kinases, YAP, and its WW-domain-containing transcription regulator 1 (WWTR1; also known as and hereafter referred to as TAZ) are controlled in mammalian cells. Diverse upstream biomechanical and mechanobiological cues, such as WSS, vascular stiffness, or hypertrophic responses, regulate the Hippo/YAP signaling pathway which results in a dynamic interaction between vascular cells and their surrounding ECM [73]. YAP was identified as a key transcription factor in a mouse model, driving a pivotal adaptive response mechanism that appeared to be critical for maintaining aortic homeostasis and preventing ATAAD in mice [231]. This study demonstrates that YAP signaling plays a crucial role in the vascular remodeling of aneurysmal specimens, as evidenced by the elevated medial thickness, indicating an adaptive response to increased wall stress.
Mechanical stress-induced YAP down-regulation has been reported in human aortic samples from patients with type A aortic dissection [116]. The induced aortic stress initiated a YAP nuclear translocation which led to the protection of the aorta from medial degeneration and the development of aneurysm and dissection.
The Hippo/YAP pathway alters ECM production or degradation and the growth, death, and migration of VSMC and endothelial cells, which contributes to vascular remodeling in aortic aneurysms. A similar phenomenon was observed in a mouse BAPN-induced Stanford type A aortic dissection model [92].
Via KEGG pathway identification, a series of different target genes and pathways were identified in human tissues linked to aneurysm formation, including the Hippo pathway [3, 23]. In line with the notion that Hippo’s transcriptional activator YAP and extracellular signal-regulated kinase 1/2 (Erk1/2) activities are related, Bertrand et al. showed that impaired mechanotransduction results in a hyperinduction of mechanical stress, subsequently activating YAP and increasing Erk1/2 signaling [17].
In addition, the Hippo pathway is a convergence point of cellular signaling with multiple major pathways, including Wnt/β-catenin, insulin-like growth factor (IGF), Phosphoinositide 3-kinases—RACα serine/threonine-protein kinase (Pi3K-AKT), and mammalian target of rapamycin (mTOR) signaling [138]. The regulation of YAP through these diverse pathways may expand the known mechanisms of vascular remodeling regulated by the Hippo/YAP pathway.
Alteration in notch signaling
The notch signaling pathway in ATAA is not well-defined in patients. However, several in vitro investigations have illustrated the involvement of Jagged–Notch signaling in impaired mechanosensing, resulting in the initiation of phenotype switching and differentiation of VSMCs [121, 148]. This compromised Notch signaling has been observed in human tissues originating from both ATAA and ATAAD [128, 238]. The Notch pathway and Wnt signaling are involved in vascular development and physiology and play a critical role in controlling phenotypic switching of VSMCs [11, 48, 119]. Through interactions with TGF-β [222] and PDGF [94], Notch signaling regulates the migration and differentiation of VSMCs. In addition, Wnt inhibitory factor-1 acts as an inhibitor, suppressing PDGF-BB-induced proliferation of VSMCs [209]. These pathways collectively contribute to the intricate regulation of VSMC behavior. Key mediators in this highly conserved pathway in ATAA are Notch 1, Notch 3, and Jagged 1 (Fig. 4). Notch 1 and Notch 3 [64, 127] regulate the migration and proliferation of VSMCs in vascular injury models, and mutations in these receptors lead to defects in VSMC development [24, 118, 218].

An overview of suggested intra- and intercellular mechanosensitive pathways involved in vascular homeostasis (black arrows) and pathologic condition in ATAA (red arrows). In impaired vascular homeostasis, intracellular pathways leading to nuclear translocation of Hippo pathway effector YAP inducing proliferation. Downregulation of Notch1 and DLL1/4 proteins can have significant effects on cellular function and may impact various physiologic processes. When Notch1 and DLL1/4 proteins are downregulated, it can lead to reduced activation of Notch Intracellular Domain (NICD). In this scenario, the transcriptional repressor Hes1 fails to activate, which is pivotal for VSMC proliferation. Therefore, dysregulation of the Notch signaling pathway could potentially contribute to pathologic processes involved in the development and progression of ATAA
There is supporting evidence indicating that the mechanosensor Piezo1 plays a crucial role in responding to shear stress [28]. This response involves the activation of a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), ultimately leading to the cleavage of Notch1, which then translocates to the nucleus, initiating the transcriptional activation of downstream targets [28]. These cellular processes are crucial in maintaining vascular integrity and may be implicated in the context of ATAA where structural changes occur.
Importantly, Notch is severely affected by biomechanical stimulation, inhibiting VSMC proliferation, while increasing apoptosis [148]. Although Notch is linked to vascular development, there is still no evidence of the precise mechanisms involved during mechanosensitive cell cycle entry and phenotypic switching. Elucidation of mechanisms by which Notch exploits these processes is of critical importance for understanding both normal VSMC development as well as the underlying causes of significant human vascular conditions such as ATAA. So far, the exact mechanism and more specifically the connection and enhancement/inhibition of pathways involved in mechanosensing and response underlying vascular pathology and the interaction of the ECM with vascular cells in the context of ATAA remains elusive.
Identified genes in familial ATAA
In recent years, major progress has been made in unraveling gene mutations as molecular markers for predisposition to ATAA. This has been by the identification of a variety of single nucleotide polymorphisms (SNPs) from genome-wide association studies (GWAS) suggestive of having a role in ATAA pathophysiology [165]. Also, genetic mutations in ATAA pathophysiology including systemic features are frequently classified by clinicians as a syndromic disease with clear connective tissue anomalies [129]. In the absence of these features, gene mutations in ATAA are often described as causative of a monogenetic connective tissue disorder, affecting proteins encoding for VSMC contractile apparatus or ECM of the aortic wall (Table 2) [213].
Genome-wide association studies identifying SNPs in ATAA
In the context of GWAS, various SNPs have been discovered to be associated with either a decreased or elevated risk of ATAA progression. In addition, several novel genes, including CD40 [36], ESR1 [233], COQB1 [109], ENTPD1, PDLIM5 (PDZ and LIM domain 5), ACTN4 (alpha-actinin-4), and GLRX [33] have been identified in the context of ATAA.
The association of ESR1 with CD8 + T-cells has been identified as a positive correlate with ATAA [233]. However, exploring the reduced inflammatory pathogenesis in ATAA requires additional investigation. The mechanistic involvement of COQB1 in the association with ATAA is explained by the negative feedback of rs386542, which elevates COQB1 expression. This increase in COQB1 expression leads to heightened VSMC metabolic activity, ultimately resulting in a decreased risk of ATAA [109].
The increased risk of developing ATAA has been linked to genetic loci, with several genes implicated in predisposing to ATAA, including VKORC1, CTNNA3, FRMD6, MBP, TCF7L2, TGF-B2, and FBN1 [8, 82, 113, 178, 189, 212]. Further, the genetic loci of 9q21, 18q11, 15q21, and 2q35, have been identified as risk regions in ATAA. Interestingly FBN1, the predominate genetic source (including gene loci 15q21) of Marfan syndrome has been identified by multiple studies on separate populations as strongly associated SNPs with ATAA progression and development [82, 113, 212].
GWAS has pinpointed various shared factors in the development of ATAA, whether involving VSMC or ECM roles. However, apart from FBN1, there has been limited subsequent exploration to reinforce earlier discoveries of the ATAA association. Notably, no GWAS has identified markers related to VSMC contractility as implicated in ATAA.
Mutations in genes encoding for the contractile apparatus of VSMCs
VSMCs consist of thin filaments such as α-actin (encoded by ACTA2) and thick filament myosin heavy chains (encoded by MYH11), connected by two essential light chains (LC) and regulatory light chains. Contraction of VSMCs is initiated by calcium-calmodulin complex (Ca-CaM) and the force is generated by ATP-dependent cyclic interactions between isoforms of α-actin and myosin heavy chain [143]. The impact of ACTA2 mutations on α-actin function was studied using in vitro assays [124]. Mutant α-actin showed functional defects, such as disrupted force generation and defective contractile VSMC function [68, 124, 146]. Besides ACTA2 mutations, also MYH11 mutations have been shown to disrupt cyclic interaction, which is predisposed in ATAA [172, 235]. Interestingly, increased expression of MYH11 increases the risk of ATAAD by approximately tenfold [105]. Mutations in the genes ACTA2 and MYH11 are the two most common mutations causing familial ATAAD. However, heterozygous loss-of-function mutations in myosin light chain kinase (MYLK) [217] and type I cGMP-dependent protein kinase (PRKG1) [69] have been reported in heritable ATAAD as well. Loss-of-function mutations in MYLK and PRKG1 disrupt kinase binding to calmodulin (CaM) and reduce kinase activity. Taken together, this demonstrates that proper VSMC-contractile function is critical for maintaining the integrity of the thoracic aorta throughout life.
Mutations in genes encoding for ECM proteins
As mentioned above, the contractile apparatus of VSMCs binds microfibrils surrounding elastin fibers through focal adhesions on the cell surface of VSMCs. Fibrillin-1 (encoded FBN1), a large glycoprotein, is the major protein in microfibrils. Heterozygous FBN1 mutations in the gene coding for this protein predispose to ATAAD in patients with Marfan syndrome [113, 182]. Moreover, the use of pluripotent stem cells to model various embryonic origins of VSMCs has revealed an inherent upregulation of FBN1 expression in neural crest VSMCs that drives the incidence of ATAA in Marfan syndrome [65].
The role of FBN1 mutations in ATAA has been thoroughly investigated and confirmed that mutations in FBN1 disrupt the structure and deposition of ECM microfibrils [76, 141]. It has been suspected that the aortic wall of Marfan patients contains low levels of fibrillin-1, which corresponds with findings in undifferentiated VSMCs. [66] These findings have been confirmed in animal studies, where VSMCs from Marfan mice showed VSMC detachment from the ECM causing VSMC phenotypic switching [26] and impaired cytoskeleton and focal adhesion organization [213]. This is supportive of findings from the use of iPSC-VSMCs derived from various lineages, wherein increased expression of FBN1 in neural crest VSMCs was found. In addition, it was reported that neural crest VSMCs of Marfan harboring mutations to FBN1 resulted in increased apoptosis compared to VSMC that had been FBN1 CRISPR corrected as well as to wild-type neural crest VSMCs [65]. It is well known that these signaling pathways are involved in the proliferation, apoptosis, inflammation, and phenotypic switching of VSMCs. Besides FBN1, there are other genes known to affect the ECM with pathogenic outcomes, such as COL5A1/2, COL1A1, and COL1A2 in Ehlers–Danlos syndrome [38]. Furthermore, lysyl oxidase (LOX) mutations encoding for an enzyme aiding the cross-linking of collagen and elastin in the aortic wall may lead to ATAAD [112].
The role of aging
Aging is the biggest risk factor for impaired cardiovascular health, with cardiovascular disease being the cause of death in 40% of individuals over 65 years old. The remodeling of the human thoracic aorta correlates with aging [115, 140] and ATAA related to aging is often labeled as a degenerative disease. As a main component of the vessel wall, elastin fulfills a key role in the remodeling process. Elastin content progressively decreases with a half-life of some 75 years in humans [203]. Several studies reported that with progressing age, alterations in the quantity and quality of elastin and collagen cause a decrease in total arterial compliance [208, 225]. Aging is associated with the destruction of interlaminar fibrillar elastic structures as well as a decreased amount of medial VSMCs [208]. This results in a reduction or loss of elastic and vasoactive function of the vascular system [58]. Loss of aortic elasticity is not only related to damage of elastin but also changes in collagen content. As individuals age, collagen type I remains consistently predominant, while the quantity of collagen type III declines gradually from the heart to the distal portion of the aorta [133].
Senescence-associated β-galactosidase (SA-β-Gal) activity is used as a tool for in vivo assessment of aging. The increase of SA-β-Gal is a result of increased lysosomal content, the expression of cyclin-dependent kinase inhibitors, the presence of DNA damage, or the presence of critically short telomere length [44, 63]. Telomere length provides a potential marker for an individual’s biologic age. Several studies suggested that telomerase plays a protective role in AAA [47] and ATAA [7]. Telomeres shortening, reduced telomerase function, and cellular senescence of VSMCs play a crucial role in the development of ATAAs. Significantly higher expressions of stress-induced senescence markers p16(INK4a) and p19(ARF) in telomerase-deficient mice were shown compared to wild-type mice [18]. Further, telomere shortening in human blood leukocytes reveals its use as a potential biomarker for ATAA [12].
Other important macromolecules contributing to the pathogenesis of ATAAD, are glycosaminoglycans (GAGs) and proteoglycans (PG), fundamental contributors to the structure and function of the aortic wall. There is contradictory data regarding changes in GAGs upon aging, most studies reported an increase in GAGs, often followed by a decrease upon further in an aging aorta [16, 154]. In ATAAD, multiple structural disruptions are reported as a result of localized GAG accumulation leading to increased interlamellar pressure [175]. Mitochondria also play an important role in aging. Tyrrell et al. demonstrated that with aging mitochondrial dysfunction may activate innate immune pathways including the TLR9, inflammasome, and STING pathways [210]. Recently, it was demonstrated in mice that the mitochondrial function of VSMCs is controlled by the ECM and drives the development of aortic aneurysms in Marfan syndrome [157].
Alteration in connective fibers within the aorta impairs the elastic recoil and reduces adhesive strength between the aortic wall layers. This may impair the functionality of aortic cells and subsequently lead to ATAA formation or aortic rupture in case pressure-induced wall stresses exceed this strength.
Sex differences
Sex differences play a significant role in the development, management, and clinical outcomes of aorta pathology. Biologically, women are protected against ATAA due to premenopausal levels of estrogen and therefore often present with ATAA at an older age than men [150]. Although ATAA is less prevalent in women, a recent epidemiologic study demonstrates that women have a 40% increased risk of mortality [152] and a threefold increased risk of ATAAD or rupture compared to men [43]. Although heritable ATAA growth rates were similar, ATAA growth rates were over three-fold higher in women than in men with degenerative ATAA [32]. These differences in sex etiology can be explained by anatomic differences such as aorta size and proportional dilation between genders. Despite the correction of aneurysm size to body size, acute aortic syndromes occur at smaller aneurysm sizes in women than in men with worse ATAA-related outcomes [55]. Thus, a smaller diameter of the aorta can progress more rapidly in women requiring close monitoring. In vitro and animal studies have indicated that estrogen can reduce collagen deposition and increase elastin in the aortic wall, potentially contributing to the prevention of TAA development [150, 168]. However, during and after menopause women exhibit a greater aortic stiffening and impairment of elastic properties, which correlates with declining levels of estrogen [215]. This may explain why women have a more progressed state of aortic disease and need to undergo surgery for ATAA at an older age [15].
Sporadic and genetic biomarkers in ATAA(D)
Sporadic or nonfamilial biomarkers
Matrix metalloproteinases
MMPs have emerged as valuable circulating markers for ATAA pathology. Studies have highlighted the significance of circulating MMP-1 and MMP-2 [162], MMP-3 [200], and MMP-9 [117] as potential indicators in assessing and monitoring ATAA. Specifically, circulating MMP-3 and insulin-like growth factor binding protein 2 (IGFBP-2) have been associated with aortic diameter in patients with ATAA [200]. Following the acute phase of aortic dissection, there is an increase in circulating MMP-9 levels, with plasma MMP-9 expression reaching its maximum approximately 2 weeks after the onset of symptoms [205]. In addition, increased expression of MMP-1, TIMP-1, and MMP-12 was positively associated with systolic WSS and TAWSS observed in the proximal ascending aorta (Table 3) [162], further underlining the importance of MMPs in assessing and monitoring aortic diseases. In the context of AAA, it is noteworthy that the administration of a pan-MMP inhibitor resulted in a slight exacerbation of aneurysm severity, in terms of aneurysm growth [136]. This suggests that the mechanism underlying MMPs and aneurysm formation/progression is complex and that a targeted approach may be required to effectively modulate MMPs in the context of AAA.
α-1-Antitrypsin protein
α1-Antitrypsin (A1AT) is a circulating serine proteinase inhibitor crucial for maintaining connective tissue integrity. A deficiency in A1AT is characterized by decreased levels, potentially leading to arterial wall degradation due to insufficient protection against the proteolytic effects of elastase and collagenase. Notably, heightened levels of MMP-9 have been identified in the vessel walls of aortic aneurysms, and these levels correlate with aortic diameter [126]. Researchers indicate that A1AT may inhibit MMP-9 activity by deactivating elastase and restraining gelatinase B within neutrophils [90]. The first controlled study investigating the relationship between A1AT deficiency and ascending aortic diameter has recently been published [39]. In this study, serum A1AT levels in the aneurysmal group were approximately 9.5 times lower than those in the nonaneurysmal group [39]. The link between reduced A1AT levels and aortic aneurysm provides additional support for its significance in evaluating the risk of ATAAD (Table 3).
Proteoglycans
Plasma levels of aggrecan (ACAN), a multimodular proteoglycan (PG) protein, were significantly enhanced in plasma samples of ATAAD patients compared to samples from healthy patients [104]. Also, increased levels of PG) and glycosaminoglycan (GAG) were detected in the serum of ATAAD patients (Table 3) [170].
Desmosine (DES)and isodesmosine (IDES)
As the aorta contains elastin, novel biomarkers for thoracic aortopathies are potentially the breakdown products of elastin: desmosine (DES) and isodesmosine (IDES), which are released in plasma, urine, or sputum [125]. Desmosine plays a pivotal role in cross-linking tropoelastin, offering valuable insights into disease mechanisms [54]. Researchers have investigated the use of desmosine as a biomarker to assess the extent of elastin degradation in the aorta, helping in the diagnosis and monitoring of aortic aneurysm progression [52, 145]. Elevated levels of DES in blood or urine samples may indicate increased elastin turnover, suggesting ongoing damage to the aortic wall. DES and IDES have been previously associated with AAA size, risk of rupture [50, 145, 211], and as a prognostic marker in acute myocardial infarction [5].
Interestingly, when combined with MRI, DES may enable the direct visualization of biologic processes at precise anatomic sites. This has been demonstrated in a Marfan mouse model [155]. Currently, there is a lack of available data on DES/IDES in ATAA. The exploration of plasma DES concentrations in studies to predict dissection or rupture in thoracic aortopathy holds significant value (Table 3).
Microcalcification
An association has been identified between reduced levels of alpha-2-HS-glycoprotein (AHSG), also known as Fetuin-A, in human blood plasma, as determined through mass spectrometry-based proteomic analysis, and an increased risk of ATAA formation [99]. AHSG binds to calciprotein particles (CPPs), forming essential complexes for regulating mineral metabolism. This interaction is essential for stabilizing and facilitating the clearance of calcium and phosphate from the circulation [106].
Plasma AHSG concentrations can differentiate between patients with ATAA and healthy controls [99]. AHSG deficiency is associated with inflammation and links vascular calcification to mortality in patients on dialysis [101]. This suggests that it might be a promising bloodborne biomarker for early ATAA diagnosis. It is worth noting that during the vascular calcification process, VSMCs may undergo phenotypic changes from a synthetic state to a chondrogenic state, which may be accompanied by the release of EVs in the bloodstream [96]. Small extracellular vesicle-derived miR-574-5p was significantly up-regulated in the serum of patients with ATAA compared to the control, and this up-regulation was higher in patients with large aneurysms (> 49 mm) [22].
Furthermore, circulating dp-ucMGP has been associated with elastin degradation, although it has not been studied in the context of ATAA [164, 188]. Findings indicate that dp-ucMGP serves as a potential biomarker for identifying individuals at risk of developing arterial and valvular calcification, suggesting its potential utility in the clinical assessment of diseases before their clinical manifestation. Although the association between dp-ucMGP and ATAAD remains unclear, it's worth noting that vitamin K deficiency, as indicated by dp-ucMGP, correlates with circulating plasma DES and IDES levels in both CVD and COPD [166]. This unexplored avenue presents an opportunity for further research to elucidate the potential implications of circulating dp-ucMGP in the context of ATAA, bridging the gap between vitamin K deficiency, vitamin K antagonist use, and elastin degradation. In addition, the conjugation of circulating dp-ucMGP with 18F-NaF PET presents the potential to develop a non-invasive imaging tool capable of precisely quantifying and colocalizing active micro-calcification within the arterial wall. This innovative approach holds promise for advancing our understanding of micro-calcification dynamics and its role in vascular health (Table 3).
Inflammation
Although the role of inflammation in ATAA is currently insufficient, several markers suggest activation of the innate immune system and the subsequent development of a low-grade chronic inflammatory reaction, which may lead to the evolution of ATAA. In this cross-sectional study, the blood of 158 patients with ATAAD was analyzed, and several circulating blood biomarkers were associated with the maximal thoracic aortic diameter estimated by CT-angiography or transthoracic echocardiography. The biomarkers that were found to be significantly associated with aortic size were primarily inflammatory markers IL-6 and GDF-15 [135]. Another study associated elevated levels of TLT-2 expressed in cells of the immune system [200] and IL-11[226] with ATAAD. Yang et al., demonstrated an increase in C18-ceramide in ATAAD, suggesting its role in aortic inflammation via association with NLRP3 in the NLR family [228]. Moreover, a novel comparison of a targeted proteomic approach has shown that patients with ATAA have increased serum levels of several inflammatory markers, such as IL-8, intracellular adhesion molecule-1 (ICAM1), C–C motif chemokine ligand 5 (CCL5), and human beta-defensin 1 (HBD1) (Table 3) [41].
Genetic biomarkers
Over the last 2 decades, there has been an emergence of newly discovered causative genes and syndromes associated with subtle or even non-existent external phenotypes. Genetic heterogeneity of hereditary ATAAD has been established by the ClinGen Aortopathy Working Group [173]. The genes were selected based on the published data and genes tested on clinical aortopathy gene panels that are currently available. Out of the 53 genes subjected to testing, the following 11 genes were conclusively identified as having a definite association with heritable ATAAD, and are clinically actionable listed in highly penetrant risk category (A1): ACTA2, COL3A1, FBN1, MYH11, SMAD3, TGF-B2, TGFBR1, TGTBR2, MYLK, LOX, PRKG1 [173]. These genes were identified over three years ago, and their association with ATAAD has been well-documented [25, 70, 142]. These genes play a role in encoding proteins associated with contraction and adhesion of VSMCs to ECM. In additiony, they contribute to TGF-β signaling pathways and VSMC metabolism. Recently, novel genes have been discovered. Tomida et al. [201] unveiled a previously overlooked mechanism connecting familial thoracic aortic aneurysm and dissection to impaired calcium ion uptake by MYH11, suggesting that elevating cytosolic Ca2 + levels could potentially prevent ATAAD [201]. Two additional studies have reported evidence suggesting that specific genetic variation at the rs2118181 locus within the FBN1 gene may be associated with an increased risk of developing ATAAD (Table 3) [82, 113].
Future outlook
Significant progress has been made over the last decades in understanding the pathophysiology of ATAA. However, important gaps remain in the early detection of acute aortic pathologies such as ATAAD. The autopsy reports indicate that up to 25% of patients with ATAAD die before diagnosis [158], and these cases often involve younger individuals [167]. Relying solely on aortic diameter for risk assessment is inadequate to distinguish between different pathologic processes with varying risks of acute complications.
Most of the recent literature on ATAA focused on identifying circulating biomarkers to improve diagnosis. While these biomarkers show promising results, their isolated use may lack specificity in indicating disease progression due to their involvement in multiple processes throughout the body. Understanding the patient-specific cellular and molecular mechanisms and integrating complementary diagnostic tools by combining circulating biomarkers, with advanced imaging tools, such as molecular imaging probes could enable direct visualization of biologic processes at specific anatomic locations.
Further investigation emphasizes the need for more personalized strategies to improve risk assessment such as integrating imaging data with genotypes and circulating biomarkers to identify patients at high-risk and guide surgical decision-making.
Abbreviations
- AAA:
-
Abdominal aortic aneurysm
- ACTA2:
-
Actin Alpha 2, Smooth Muscle
- ACA:
-
Aggrecan
- ADmax:
-
Absolute max diameter
- AHA:
-
American Heart Association
- AHSG:
-
Alpha-2-HS-glycoprotein
- A1AT:
-
α1-Antitrypsin
- ATAA:
-
Ascending Thoracic Aortic Aneurysm
- ATAAD:
-
Acute type A aortic dissection
- BAV:
-
Bicuspid aortic valve
- CADESIL:
-
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- COL3A1:
-
Collagen type III
- dp-ucMGP:
-
Dephosphorylated undercarboxylated Matrix Gla protein
- DES:
-
Desmosine
- ECM:
-
Extracellular matrix
- ECs:
-
Endothelial cells
- ELN/COL:
-
Elastin-to-collagen
- FBN1:
-
Fibrillin 1
- 18F-FDG:
-
18F-fluorodeoxyglucose
- 18F-NaF:
-
18F–sodium fluoride
- GAGs:
-
Glycosaminoglycans
- hiPSCs:
-
Human-induced pluripotent stem cells
- IDES:
-
Isodesmosine
- KLF4:
-
Krüppel like factor 4
- LDS:
-
Loeys–Dietz Syndrome
- LVOT-angle:
-
Left ventricular outflow tract angle
- MMP:
-
Matrix metalloproteinases
- MYH11:
-
Myosin heavy chain 11
- MYLK:
-
Myosin light chain kinases
- MGP:
-
Matrix Gla-protein
- NICD:
-
Notch intracellular domain
- PECAM-1:
-
Platelet EC adhesion molecule
- PG:
-
Proteoglycans
- PRKG1:
-
Protein kinase cGMP-dependent type 1
- RANTES:
-
Regulated on activation, normal T-cell expression, and secreted
- ROS:
-
Reactive oxygen species
- SA-β-Gal:
-
Senescence-associated β-galactosidase
- TGF-β:
-
Transforming growth factor-β
- TAV:
-
Tricuspid aortic valve
- VE-cadherin:
-
Vascular endothelial-cadherin
- VEGFR2:
-
Vascular endothelial growth factor receptor 2
- VKOR:
-
Vitamin K epoxide reductase
- VSMC:
-
Vascular smooth muscle cell
- vWF:
-
Willebrand factor
- WSR:
-
Wall shear rate
- WSS:
-
Wall shear stress
References
Adriaans BP, Ramaekers M, Heuts S, Crijns H, Bekkers S, Westenberg JJM, Lamb HJ, Wildberger JE, Schalla S (2021) Determining the optimal interval for imaging surveillance of ascending aortic aneurysms. Neth Heart J 29:623–631. https://doi.org/10.1007/s12471-021-01564-9
Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL, Lau CL, Owens GK (2009) Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg 138:1392–1399. https://doi.org/10.1016/j.jtcvs.2009.07.075
Albinsson S, Della Corte A, Alajbegovic A, Krawczyk KK, Bancone C, Galderisi U, Cipollaro M, De Feo M, Forte A (2017) Patients with bicuspid and tricuspid aortic valve exhibit distinct regional microrna signatures in mildly dilated ascending aorta. Heart Vessels 32:750–767. https://doi.org/10.1007/s00380-016-0942-7
Alexander MR, Owens GK (2012) Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol 74:13–40. https://doi.org/10.1146/annurev-physiol-012110-142315
Ali K, Israr MZ, Ng LL, Mordi I, Lang CC, Kuzmanova E, Huang JT, Choy AM (2022) Plasma desmosine for prediction of outcomes after acute myocardial infarction. Front Cardiovasc Med 9:992388. https://doi.org/10.3389/fcvm.2022.992388
Allaire E, Forough R, Clowes M, Starcher B, Clowes AW (1998) Local overexpression of TIMP-1 prevents aortic aneurysm degeneration and rupture in a rat model. J Clin Invest 102:1413–1420. https://doi.org/10.1172/jci2909
Aschacher T, Salameh O, Enzmann F, Messner B, Bergmann M (2017) Telomere biology and thoracic aortic aneurysm. Int J Mol Sci. https://doi.org/10.3390/ijms19010003
Ashvetiya T, Fan SX, Chen YJ, Williams CH, O’Connell JR, Perry JA, Hong CC (2021) Identification of novel genetic susceptibility loci for thoracic and abdominal aortic aneurysms via genome-wide association study using the UK Biobank Cohort. PLoS ONE 16:e0247287. https://doi.org/10.1371/journal.pone.0247287
Ashworth JL, Murphy G, Rock MJ, Sherratt MJ, Shapiro SD, Shuttleworth CA, Kielty CM (1999) Fibrillin degradation by matrix metalloproteinases: implications for connective tissue remodelling. Biochem J 340(Pt 1):171–181
Bäck M, Gasser TC, Michel JB, Caligiuri G (2013) Biomechanical factors in the biology of aortic wall and aortic valve diseases. Cardiovasc Res 99:232–241. https://doi.org/10.1093/cvr/cvt040
Baeten JT, Lilly B (2017) Notch signaling in vascular smooth muscle cells. Adv Pharmacol 78:351–382. https://doi.org/10.1016/bs.apha.2016.07.002
Balistreri CR, Pisano C, Merlo D, Fattouch K, Caruso M, Incalcaterra E, Colonna-Romano G, Candore G (2012) Is the mean blood leukocyte telomere length a predictor for sporadic thoracic aortic aneurysm? Data from a preliminary study. Rejuvenation Res 15:170–173. https://doi.org/10.1089/rej.2011.1273
Ballegaard CR, Pham MHC, Sigvardsen PE, Kühl JT, Sørgaard M, Taudorf M, Fuchs A, Nordestgaard BG, Køber LV, Kofoed KF (2022) Aortic enlargement and coronary artery calcification in a general population cohort. Eur Heart J Cardiovasc Imaging 23:855–862. https://doi.org/10.1093/ehjci/jeab122
Barker AJ, Markl M, Bürk J, Lorenz R, Bock J, Bauer S, Schulz-Menger J, von Knobelsdorff-Brenkenhoff F (2012) Bicuspid aortic valve is associated with altered wall shear stress in the ascending aorta. Circ Cardiovasc Imaging 5:457–466. https://doi.org/10.1161/circimaging.112.973370
Beller CJ, Farag M, Wannaku S, Seppelt P, Arif R, Ruhparwar A, Karck M, Weymann A, Kallenbach K (2015) Gender-specific differences in outcome of ascending aortic aneurysm surgery. PLoS ONE 10:e0124461. https://doi.org/10.1371/journal.pone.0124461
Bertelsen S (1968) Chemical studies on the arterial wall in relation to atherosclerosis. Ann N Y Acad Sci 149:643–654. https://doi.org/10.1111/j.1749-6632.1968.tb53824.x
Bertrand AT, Ziaei S, Ehret C, Duchemin H, Mamchaoui K, Bigot A, Mayer M, Quijano-Roy S, Desguerre I, Lainé J, Ben Yaou R, Bonne G, Coirault C (2014) Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J Cell Sci 127:2873–2884. https://doi.org/10.1242/jcs.144907
Bhayadia R, Schmidt BM, Melk A, Hömme M (2016) Senescence-induced oxidative stress causes endothelial dysfunction. J Gerontol A Biol Sci Med Sci 71:161–169. https://doi.org/10.1093/gerona/glv008
Bishop JE, Lindahl G (1999) Regulation of cardiovascular collagen synthesis by mechanical load. Cardiovasc Res 42:27–44. https://doi.org/10.1016/s0008-6363(99)00021-8
Bissell MM, Hess AT, Biasiolli L, Glaze SJ, Loudon M, Pitcher A, Davis A, Prendergast B, Markl M, Barker AJ, Neubauer S, Myerson SG (2013) Aortic dilation in bicuspid aortic valve disease: flow pattern is a major contributor and differs with valve fusion type. Circ Cardiovasc Imaging 6:499–507. https://doi.org/10.1161/circimaging.113.000528
Bjørklund G, Svanberg E, Dadar M, Card DJ, Chirumbolo S, Harrington DJ, Aaseth J (2020) The role of matrix gla protein (MGP) in vascular calcification. Curr Med Chem 27:1647–1660. https://doi.org/10.2174/0929867325666180716104159
Boileau A, Lino Cardenas CL, Courtois A, Zhang L, Rodosthenous RS, Das S, Sakalihasan N, Michel JB, Lindsay ME, Devaux Y (2019) MiR-574–5p: a Circulating marker of thoracic aortic aneurysm. Int J Mol Sci. https://doi.org/10.3390/ijms20163924
Borghini A, Foffa I, Pulignani S, Vecoli C, Ait-Ali L, Andreassi MG (2017) miRNome profiling in bicuspid aortic valve-associated aortopathy by next-generation sequencing. Int J Mol Sci. https://doi.org/10.3390/ijms18112498
Boucher J, Gridley T, Liaw L (2012) Molecular pathways of notch signaling in vascular smooth muscle cells. Front Physiol 3:81. https://doi.org/10.3389/fphys.2012.00081
Brownstein AJ, Ziganshin BA, Kuivaniemi H, Body SC, Bale AE, Elefteriades JA (2017) Genes associated with thoracic aortic aneurysm and dissection: an update and clinical implications. Aorta (Stamford) 5:11–20. https://doi.org/10.12945/j.aorta.2017.17.003
Bunton TE, Biery NJ, Myers L, Gayraud B, Ramirez F, Dietz HC (2001) Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res 88:37–43. https://doi.org/10.1161/01.res.88.1.37
Cao G, Xuan X, Hu J, Zhang R, Jin H, Dong H (2022) How vascular smooth muscle cell phenotype switching contributes to vascular disease. Cell Commun Signal 20:180. https://doi.org/10.1186/s12964-022-00993-2
Caolo V, Debant M, Endesh N, Futers TS, Lichtenstein L, Bartoli F, Parsonage G, Jones EA, Beech DJ (2020) Shear stress activates ADAM10 sheddase to regulate Notch1 via the Piezo1 force sensor in endothelial cells. Elife. https://doi.org/10.7554/eLife.50684
Capoulade R, Côté N, Mathieu P, Chan KL, Clavel MA, Dumesnil JG, Teo KK, Tam JW, Fournier D, Després JP, Pibarot P (2014) Circulating levels of matrix gla protein and progression of aortic stenosis: a substudy of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Can J Cardiol 30:1088–1095. https://doi.org/10.1016/j.cjca.2014.03.025
Catapano F, Pambianchi G, Cundari G, Rebelo J, Cilia F, Carbone I, Catalano C, Francone M, Galea N (2020) 4D flow imaging of the thoracic aorta: is there an added clinical value? Cardiovasc Diagn Ther 10:1068–1089. https://doi.org/10.21037/cdt-20-452
Chatterjee S, Fisher AB (2014) Mechanotransduction in the endothelium: role of membrane proteins and reactive oxygen species in sensing, transduction, and transmission of the signal with altered blood flow. Antioxid Redox Signal 20:899–913. https://doi.org/10.1089/ars.2013.5624
Cheung K, Boodhwani M, Chan KL, Beauchesne L, Dick A, Coutinho T (2017) Thoracic aortic aneurysm growth: role of sex and aneurysm etiology. J Am Heart Assoc. https://doi.org/10.1161/JAHA.116.003792
Chou EL, Chaffin M, Simonson B, Pirruccello JP, Akkad AD, Nekoui M, Lino Cardenas CL, Bedi KC Jr, Nash C, Juric D, Stone JR, Isselbacher EM, Margulies KB, Klattenhoff C, Ellinor PT, Lindsay ME (2022) Aortic cellular diversity and quantitative genome-wide association study trait prioritization through single-nuclear rna sequencing of the aneurysmal human aorta. Arterioscler Thromb Vasc Biol 42:1355–1374. https://doi.org/10.1161/atvbaha.122.317953
Chung L, Dinakarpandian D, Yoshida N, Lauer-Fields JL, Fields GB, Visse R, Nagase H (2004) Collagenase unwinds triple-helical collagen prior to peptide bond hydrolysis. Embo j 23:3020–3030. https://doi.org/10.1038/sj.emboj.7600318
Coady MA, Rizzo JA, Goldstein LJ, Elefteriades JA (1999) Natural history, pathogenesis, and etiology of thoracic aortic aneurysms and dissections. Cardiol Clin 17(615–635):vii. https://doi.org/10.1016/s0733-8651(05)70105-3
Cui X, Xuan T, Chen S, Guo X (2022) Causal associations between CD40/CD40L and aortic diseases: a mendelian randomization study. Front Genet 13:998525. https://doi.org/10.3389/fgene.2022.998525
Czerny M, Grabenwöger M, Berger T, Aboyans V, Della Corte A, Chen EP, Desai ND, Dumfarth J, Elefteriades JA, Etz CD, Kim KM, Kreibich M, Lescan M, Di Marco L, Martens A, Mestres CA, Milojevic M, Nienaber CA, Piffaretti G, Preventza O, Quintana E, Rylski B, Schlett CL, Schoenhoff F, Trimarchi S, Tsagakis K (2024) EACTS/STS Guidelines for diagnosing and treating acute and chronic syndromes of the aortic organ. Eur J Cardiothorac Surg. https://doi.org/10.1093/ejcts/ezad426
D’Hondt S, Van Damme T, Malfait F (2018) Vascular phenotypes in nonvascular subtypes of the ehlers-danlos syndrome: a systematic review. Genet Med 20:562–573. https://doi.org/10.1038/gim.2017.138
Dako F, Zhao H, Mulvenna A, Gupta YS, Simpson S, Kueppers F (2021) Relationship between α(1)-antitrypsin deficiency and ascending aortic distention. Mayo Clin Proc Innov Qual Outcomes 5:590–595. https://doi.org/10.1016/j.mayocpiqo.2021.03.004
Danyi P, Elefteriades JA, Jovin IS (2012) Medical therapy of thoracic aortic aneurysms. Trends Cardiovasc Med 22:180–184. https://doi.org/10.1016/j.tcm.2012.07.017
Daskalopoulou A, Giotaki SG, Toli K, Minia A, Pliaka V, Alexopoulos LG, Deftereos G, Iliodromitis K, Dimitroulis D, Siasos G, Verikokos C, Iliopoulos D (2023) Targeted proteomic analysis of patients with ascending thoracic aortic aneurysm. Biomedicines. https://doi.org/10.3390/biomedicines11051273
Davies PF (1995) Flow-mediated endothelial mechanotransduction. Physiol Rev 75:519–560. https://doi.org/10.1152/physrev.1995.75.3.519
Davies RR, Goldstein LJ, Coady MA, Tittle SL, Rizzo JA, Kopf GS, Elefteriades JA (2002) Yearly rupture or dissection rates for thoracic aortic aneurysms: simple prediction based on size. Ann Thorac Surg. https://doi.org/10.1016/s0003-4975(01)03236-2
de Magalhães JP, Passos JF (2018) Stress, cell senescence and organismal ageing. Mech Ageing Dev 170:2–9. https://doi.org/10.1016/j.mad.2017.07.001
Della Corte A, De Santo LS, Montagnani S, Quarto C, Romano G, Amarelli C, Scardone M, De Feo M, Cotrufo M, Caianiello G (2006) Spatial patterns of matrix protein expression in dilated ascending aorta with aortic regurgitation: congenital bicuspid valve versus marfan’s syndrome. J Heart Valve Dis. https://doi.org/10.1074/jbc.M602749200
Della Corte A, Quarto C, Bancone C, Castaldo C, Di Meglio F, Nurzynska D, De Santo LS, De Feo M, Scardone M, Montagnani S, Cotrufo M (2008) Spatiotemporal patterns of smooth muscle cell changes in ascending aortic dilatation with bicuspid and tricuspid aortic valve stenosis: focus on cell-matrix signaling. J Thorac Cardiovasc Surg 135(8–18):18.e11–12. https://doi.org/10.1016/j.jtcvs.2007.09.009
Dimitroulis D, Katsargyris A, Klonaris C, Avgerinos ED, Fragou-Plemenou M, Kouraklis G, Liapis CD (2011) Telomerase expression on aortic wall endothelial cells is attenuated in abdominal aortic aneurysms compared to healthy nonaneurysmal aortas. J Vasc Surg 54:1778–1783. https://doi.org/10.1016/j.jvs.2011.06.079
Doi H, Iso T, Sato H, Yamazaki M, Matsui H, Tanaka T, Manabe I, Arai M, Nagai R, Kurabayashi M (2006) Jagged1-selective notch signaling induces smooth muscle differentiation via a RBP-Jkappa-dependent pathway. J Biol Chem. https://doi.org/10.1074/jbc.M602749200
Duan XY, Guo DC, Regalado ES, Shen H, Coselli JS, Estrera AL, Safi HJ, Bamshad MJ, Nickerson DA, LeMaire SA, De Backer J, Milewicz DM (2019) SMAD4 rare variants in individuals and families with thoracic aortic aneurysms and dissections. Eur J Hum Genet 27:1054–1060. https://doi.org/10.1038/s41431-019-0357-x
Farrell K, Simmers P, Mahajan G, Boytard L, Camardo A, Joshi J, Ramamurthi A, Pinet F, Kothapalli CR (2019) Alterations in phenotype and gene expression of adult human aneurysmal smooth muscle cells by exogenous nitric oxide. Exp Cell Res 384:111589. https://doi.org/10.1016/j.yexcr.2019.111589
Fiorillo C, Becatti M, Attanasio M, Lucarini L, Nassi N, Evangelisti L, Porciani MC, Nassi P, Gensini GF, Abbate R, Pepe G (2010) Evidence for oxidative stress in plasma of patients with Marfan syndrome. Int J Cardiol 145:544–546. https://doi.org/10.1016/j.ijcard.2010.04.077
Fletcher A, Syed MBJ, Iskander Z, Debono S, Dweck MR, Huang J, Chin C, Newby DE, Choy AM (2021) Plasma desmosine as a biomarker in acute aortic syndrome. Eur Heart J. https://doi.org/10.1093/eurheartj/ehab724.2011
Fletcher AJ, Nash J, Syed MBJ, Macaskill MG, Tavares AAS, Walker N, Salcudean H, Leipsic JA, Lim KHH, Madine J, Wallace W, Field M, Newby DE, Bouchareb R, Seidman MA, Akhtar R, Sellers SL (2022) Microcalcification and thoracic aortopathy: a window into disease severity. Arterioscler Thromb Vasc Biol 42:1048–1059. https://doi.org/10.1161/atvbaha.122.317111
Fletcher AJ, Syed MBJ, Aitman TJ, Newby DE, Walker NL (2020) Inherited thoracic aortic disease: new insights and translational targets. Circulation 141:1570–1587. https://doi.org/10.1161/circulationaha.119.043756
Forbes TL, Harris JR, Lawlor DK, Derose G (2010) Evaluation of sex differences in relative dilatation of thoracic aortic aneurysms. Eur J Vasc Endovasc Surg 39:555–558. https://doi.org/10.1016/j.ejvs.2010.02.011
Forsythe RO, Dweck MR, McBride OMB, Vesey AT, Semple SI, Shah ASV, Adamson PD, Wallace WA, Kaczynski J, Ho W, van Beek EJR, Gray CD, Fletcher A, Lucatelli C, Marin A, Burns P, Tambyraja A, Chalmers RTA, Weir G, Mitchard N, Tavares A, Robson JMJ, Newby DE (2018) (18)F-Sodium fluoride uptake in abdominal aortic aneurysms: the SoFIA(3) study. J Am Coll Cardiol 71:513–523. https://doi.org/10.1016/j.jacc.2017.11.053
Forte A, Della Corte A, Grossi M, Bancone C, Maiello C, Galderisi U, Cipollaro M (2013) Differential expression of proteins related to smooth muscle cells and myofibroblasts in human thoracic aortic aneurysm. Histol Histopathol 28:795–803. https://doi.org/10.14670/hh-28.795
Fritze O, Romero B, Schleicher M, Jacob MP, Oh DY, Starcher B, Schenke-Layland K, Bujan J, Stock UA (2012) Age-related changes in the elastic tissue of the human aorta. J Vasc Res 49:77–86. https://doi.org/10.1159/000331278
Fu V, Plouffe SW, Guan KL (2017) The Hippo pathway in organ development, homeostasis, and regeneration. Curr Opin Cell Biol 49:99–107. https://doi.org/10.1016/j.ceb.2017.12.012
Fujiwara T, Takeda N, Ishii S, Morita H, Komuro I (2019) Unique mechanism by which tgfbr1 variants cause 2 distinct system diseases - loeys-dietz syndrome and multiple self-healing squamous epithelioma. Circ Rep 1:487–492. https://doi.org/10.1253/circrep.CR-19-0098
Ganizada BH, Reesink KD, Parikh S, Ramaekers M, Akbulut AC, Saraber P, Debeij GP, Mumc-Taa Student T, Jaminon AM, Natour E, Lorusso R, Wildberger JE, Mees B, Schurink GW, Jacobs MJ, Cleutjens J, Krapels I, Gombert A, Maessen JG, Accord R, Delhaas T, Schalla S, Schurgers LJ, Bidar E (2023) The maastricht acquisition platform for studying mechanisms of cell-matrix crosstalk (MAPEX): an interdisciplinary and systems approach towards understanding thoracic aortic disease. Biomedicines. https://doi.org/10.3390/biomedicines11082095
Gong J, Zhou D, Jiang L, Qiu P, Milewicz DM, Chen YE, Yang B (2020) In vitro lineage-specific differentiation of vascular smooth muscle cells in response to SMAD3 Deficiency: implications for SMAD3-related thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol 40:1651–1663. https://doi.org/10.1161/atvbaha.120.313033
González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D (2021) A guide to assessing cellular senescence in vitro and in vivo. Febs j 288:56–80. https://doi.org/10.1111/febs.15570
Granata A, Bernard WG, Zhao N, McCafferty J, Lilly B, Sinha S (2015) Temporal and embryonic lineage-dependent regulation of human vascular SMC development by NOTCH3. Stem Cells Dev 24:846–856. https://doi.org/10.1089/scd.2014.0520
Granata A, Serrano F, Bernard WG, McNamara M, Low L, Sastry P, Sinha S (2017) An iPSC-derived vascular model of marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet 49:97–109. https://doi.org/10.1038/ng.3723
Grewal N, Gittenberger-de Groot AC (2018) Pathogenesis of aortic wall complications in marfan syndrome. Cardiovasc Pathol 33:62–69. https://doi.org/10.1016/j.carpath.2018.01.005
Guala A, Dux-Santoy L, Teixido-Tura G, Ruiz-Muñoz A, Galian-Gay L, Servato ML, Valente F, Gutiérrez L, González-Alujas T, Johnson KM, Wieben O, Casas-Masnou G, Sao Avilés A, Fernandez-Galera R, Ferreira-Gonzalez I, Evangelista A, Rodríguez-Palomares JF (2022) Wall shear stress predicts aortic dilation in patients with bicuspid aortic valve. JACC Cardiovasc Imaging. https://doi.org/10.1016/j.jcmg.2021.09.023
Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, Kim DH, Pannu H, Willing MC, Sparks E, Pyeritz RE, Singh MN, Dalman RL, Grotta JC, Marian AJ, Boerwinkle EA, Frazier LQ, LeMaire SA, Coselli JS, Estrera AL, Safi HJ, Veeraraghavan S, Muzny DM, Wheeler DA, Willerson JT, Yu RK, Shete SS, Scherer SE, Raman CS, Buja LM, Milewicz DM (2009) Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet 84:617–627. https://doi.org/10.1016/j.ajhg.2009.04.007
Guo DC, Regalado E, Casteel DE, Santos-Cortez RL, Gong L, Kim JJ, Dyack S, Horne SG, Chang G, Jondeau G, Boileau C, Coselli JS, Li Z, Leal SM, Shendure J, Rieder MJ, Bamshad MJ, Nickerson DA, Kim C, Milewicz DM (2013) Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet 93:398–404. https://doi.org/10.1016/j.ajhg.2013.06.019
Guo DC, Regalado ES, Minn C, Tran-Fadulu V, Coney J, Cao J, Wang M, Yu RK, Estrera AL, Safi HJ, Shete SS, Milewicz DM (2011) Familial thoracic aortic aneurysms and dissections: identification of a novel locus for stable aneurysms with a low risk for progression to aortic dissection. Circ Cardiovasc Genet 4:36–42. https://doi.org/10.1161/circgenetics.110.958066
Guzzardi DG, Barker AJ, van Ooij P, Malaisrie SC, Puthumana JJ, Belke DD, Mewhort HE, Svystonyuk DA, Kang S, Verma S, Collins J, Carr J, Bonow RO, Markl M, Thomas JD, McCarthy PM, Fedak PW (2015) Valve-related hemodynamics mediate human bicuspid aortopathy: insights from wall shear stress mapping. J Am Coll Cardiol 66:892–900. https://doi.org/10.1016/j.jacc.2015.06.1310
Halushka MK, Angelini A, Bartoloni G, Basso C, Batoroeva L, Bruneval P, Buja LM, Butany J, d’Amati G, Fallon JT, Gallagher PJ, Gittenberger-de Groot AC, Gouveia RH, Kholova I, Kelly KL, Leone O, Litovsky SH, Maleszewski JJ, Miller DV, Mitchell RN, Preston SD, Pucci A, Radio SJ, Rodriguez ER, Sheppard MN, Stone JR, Suvarna SK, Tan CD, Thiene G, Veinot JP, van der Wal AC (2016) Consensus statement on surgical pathology of the aorta from the Society for Cardiovascular Pathology and the Association For European Cardiovascular Pathology: II. noninflammatory degenerative diseases–nomenclature and diagnostic criteria. Cardiovasc Pathol 25:247–257. https://doi.org/10.1016/j.carpath.2016.03.002
He J, Bao Q, Yan M, Liang J, Zhu Y, Wang C, Ai D (2018) The role of Hippo/yes-associated protein signalling in vascular remodelling associated with cardiovascular disease. Br J Pharmacol 175:1354–1361. https://doi.org/10.1111/bph.13806
Heuts S, Adriaans BP, Gerretsen S, Natour E, Vos R, Cheriex EC, Crijns H, Wildberger JE, Maessen JG, Schalla S, Sardari Nia P (2018) Aortic elongation part II: the risk of acute type A aortic dissection. Heart 104:1778–1782. https://doi.org/10.1136/heartjnl-2017-312867
Heuts S, Adriaans BP, Rylski B, Mihl C, Bekkers S, Olsthoorn JR, Natour E, Bouman H, Berezowski M, Kosiorowska K, Crijns H, Maessen JG, Wildberger J, Schalla S, Sardari Nia P (2020) Evaluating the diagnostic accuracy of maximal aortic diameter, length and volume for prediction of aortic dissection. Heart 106:892–897. https://doi.org/10.1136/heartjnl-2019-316251
Hollister DW, Godfrey M, Sakai LY, Pyeritz RE (1990) Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan syndrome. N Engl J Med 323:152–159. https://doi.org/10.1056/nejm199007193230303
Humphrey JD, Dufresne ER, Schwartz MA (2014) Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol 15:802–812. https://doi.org/10.1038/nrm3896
Humphrey JD, Tellides G (2019) Central artery stiffness and thoracic aortopathy. Am J Physiol Heart Circ Physiol 316:H169-h182. https://doi.org/10.1152/ajpheart.00205.2018
Hur DJ, Raymond GV, Kahler SG, Riegert-Johnson DL, Cohen BA, Boyadjiev SA (2005) A novel MGP mutation in a consanguineous family: review of the clinical and molecular characteristics of Keutel syndrome. Am J Med Genet A 135:36–40. https://doi.org/10.1002/ajmg.a.30680
Husmann L, Huellner MW, Eberhard N, Ledergerber B, Kaelin MB, Anagnostopoulos A, Kudura K, Burger IA, Mestres CA, Rancic Z, Hasse B (2021) PET/CT in therapy control of infective native aortic aneurysms. Sci Rep 11:5065. https://doi.org/10.1038/s41598-021-84658-z
Husmann L, Huellner MW, Ledergerber B, Eberhard N, Kaelin MB, Anagnostopoulos A, Kudura K, Burger IA, Mestres CA, Rancic Z, Hasse B (2020) Diagnostic accuracy of PET/CT and contrast enhanced ct in patients with suspected infected aortic aneurysms. Eur J Vasc Endovasc Surg 59:972–981. https://doi.org/10.1016/j.ejvs.2020.01.032
Iakoubova OA, Tong CH, Rowland CM, Luke MM, Garcia VE, Catanese JJ, Moomiaie RM, Sotonyi P, Ascady G, Nikas D, Dedelias P, Tranquilli M, Elefteriades JA (2014) Genetic variants in FBN-1 and risk for thoracic aortic aneurysm and dissection. PLoS ONE 9:e91437. https://doi.org/10.1371/journal.pone.0091437
Ikonomidis JS, Gibson WC, Butler JE, McClister DM, Sweterlitsch SE, Thompson RP, Mukherjee R, Spinale FG (2004) Effects of deletion of the tissue inhibitor of matrix metalloproteinases-1 gene on the progression of murine thoracic aortic aneurysms. Circulation. https://doi.org/10.1161/01.Cir.0000138384.68947.20
Ikonomidis JS, Jones JA, Barbour JR, Stroud RE, Clark LL, Kaplan BS, Zeeshan A, Bavaria JE, Gorman JH 3rd, Spinale FG, Gorman RC (2007) Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with bicuspid or tricuspid aortic valves. J Thorac Cardiovasc Surg 133:1028–1036. https://doi.org/10.1016/j.jtcvs.2006.10.083
Inamoto S, Kwartler CS, Lafont AL, Liang YY, Fadulu VT, Duraisamy S, Willing M, Estrera A, Safi H, Hannibal MC, Carey J, Wiktorowicz J, Tan FK, Feng XH, Pannu H, Milewicz DM (2010) TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res 88:520–529. https://doi.org/10.1093/cvr/cvq230
Isoda H, Fukuyama A (2022) Quality control for 4D flow MR imaging. Magn Reson Med Sci 21:278–292. https://doi.org/10.2463/mrms.rev.2021-0165
Isselbacher EM, Preventza O, Hamilton Black J, Augoustides JG, Beck AW, Bolen MA, Braverman AC, Bray BE, Brown-Zimmerman MM, Chen EP, Collins TJ, DeAnda A, Fanola CL, Girardi LN, Hicks CW, Hui DS, Schuyler Jones W, Kalahasti V, Kim KM, Milewicz DM, Oderich GS, Ogbechie L, Promes SB, Gyang Ross E, Schermerhorn ML, Singleton Times S, Tseng EE, Wang GJ, Woo YJ (2022) 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. https://doi.org/10.1161/cir.0000000000001106
Jain M, Chauhan AK (2022) Role of integrins in modulating smooth muscle cell plasticity and vascular remodeling: from expression to therapeutic implications. Cells. https://doi.org/10.3390/cells11040646
Jaminon A, Reesink K, Kroon A, Schurgers L (2019) The role of vascular smooth muscle cells in arterial remodeling: focus on calcification-related processes. Int J Mol Sci. https://doi.org/10.3390/ijms20225694
Janciauskiene S, Wrenger S, Immenschuh S, Olejnicka B, Greulich T, Welte T, Chorostowska-Wynimko J (2018) The multifaceted effects of alpha1-antitrypsin on neutrophil functions. Front Pharmacol 9:341. https://doi.org/10.3389/fphar.2018.00341
Jespersen T, Møllehave LT, Thuesen BH, Skaaby T, Rossing P, Toft U, Jørgensen NR, Corfixen BL, Jakobsen J, Frimodt-Møller M, Linneberg A (2020) Uncarboxylated matrix Gla-protein: a biomarker of vitamin K status and cardiovascular risk. Clin Biochem 83:49–56. https://doi.org/10.1016/j.clinbiochem.2020.05.005
Jiang WJ, Ren WH, Liu XJ, Liu Y, Wu FJ, Sun LZ, Lan F, Du J, Zhang HJ (2016) Disruption of mechanical stress in extracellular matrix is related to Stanford type A aortic dissection through down-regulation of Yes-associated protein. Aging (Albany NY) 8:1923–1939. https://doi.org/10.18632/aging.101033
Jiménez-Altayó F, Meirelles T, Crosas-Molist E, Sorolla MA, Del Blanco DG, López-Luque J, Mas-Stachurska A, Siegert AM, Bonorino F, Barberà L, García C, Condom E, Sitges M, Rodríguez-Pascual F, Laurindo F, Schröder K, Ros J, Fabregat I, Egea G (2018) Redox stress in Marfan syndrome: dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic Biol Med 118:44–58. https://doi.org/10.1016/j.freeradbiomed.2018.02.023
Jin S, Hansson EM, Tikka S, Lanner F, Sahlgren C, Farnebo F, Baumann M, Kalimo H, Lendahl U (2008) Notch signaling regulates platelet-derived growth factor receptor-beta expression in vascular smooth muscle cells. Circ Res 102:1483–1491. https://doi.org/10.1161/circresaha.107.167965
Jones JA, Spinale FG, Ikonomidis JS (2009) Transforming growth factor-beta signaling in thoracic aortic aneurysm development: a paradox in pathogenesis. J Vasc Res 46:119–137. https://doi.org/10.1159/000151766
Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RT, Alvarez-Hernandez D, Shroff R, Yin X, Muller K, Skepper JN, Mayr M, Reutelingsperger CP, Chester A, Bertazzo S, Schurgers LJ, Shanahan CM (2015) Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res 116:1312–1323. https://doi.org/10.1161/circresaha.116.305012
Kauhanen SP, Liimatainen T, Kariniemi E, Korhonen M, Parkkonen J, Vienonen J, Vanninen R, Hedman M (2020) A smaller heart-aorta-angle associates with ascending aortic dilatation and increases wall shear stress. Eur Radiol 30:5149–5157. https://doi.org/10.1007/s00330-020-06852-3
Kawtharany L, Bessueille L, Issa H, Hamade E, Zibara K, Magne D (2022) Inflammation and microcalcification: a never-ending vicious cycle in atherosclerosis? J Vasc Res 59:137–150. https://doi.org/10.1159/000521161
Kazamia R, Keravnou A, Moushi A, Sokratous K, Michailidou K, Yiangou K, Soteriou M, Xenophontos S, Cariolou MA, Bashiardes E (2023) Tissue and plasma proteomic profiling indicates AHSG as a potential biomarker for ascending thoracic aortic aneurysms. BMC Cardiovasc Disord 23:138. https://doi.org/10.1186/s12872-023-03154-6
Kessler K, Borges LF, Ho-Tin-Noé B, Jondeau G, Michel JB, Vranckx R (2014) Angiogenesis and remodelling in human thoracic aortic aneurysms. Cardiovasc Res 104:147–159. https://doi.org/10.1093/cvr/cvu196
Ketteler M, Bongartz P, Westenfeld R, Wildberger JE, Mahnken AH, Böhm R, Metzger T, Wanner C, Jahnen-Dechent W, Floege J (2003) Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet 361:827–833. https://doi.org/10.1016/s0140-6736(03)12710-9
Kiema M, Sarin JK, Kauhanen SP, Torniainen J, Matikka H, Luoto ES, Jaakkola P, Saari P, Liimatainen T, Vanninen R, Ylä-Herttuala S, Hedman M, Laakkonen JP (2022) Wall shear stress predicts media degeneration and biomechanical changes in thoracic aorta. Front Physiol 13:934941. https://doi.org/10.3389/fphys.2022.934941
Kim J, Song HC (2018) Role of PET/CT in the evaluation of aortic disease. Chonnam Med J 54:143–152. https://doi.org/10.4068/cmj.2018.54.3.143
König KC, Lahm H, Dreßen M, Doppler SA, Eichhorn S, Beck N, Kraehschuetz K, Doll S, Holdenrieder S, Kastrati A, Lange R, Krane M (2021) Aggrecan: a new biomarker for acute type A aortic dissection. Sci Rep 11:10371. https://doi.org/10.1038/s41598-021-89653-y
Kuang SQ, Guo DC, Prakash SK, McDonald ML, Johnson RJ, Wang M, Regalado ES, Russell L, Cao JM, Kwartler C, Fraivillig K, Coselli JS, Safi HJ, Estrera AL, Leal SM, LeMaire SA, Belmont JW, Milewicz DM (2011) Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet. https://doi.org/10.1371/journal.pgen.1002118
Kutikhin AG, Feenstra L, Kostyunin AE, Yuzhalin AE, Hillebrands JL, Krenning G (2021) Calciprotein particles: balancing mineral homeostasis and vascular pathology. Arterioscler Thromb Vasc Biol 41:1607–1624. https://doi.org/10.1161/atvbaha.120.315697
Kuzmik GA, Sang AX, Elefteriades JA (2012) Natural history of thoracic aortic aneurysms. J Vasc Surg 56:565–571. https://doi.org/10.1016/j.jvs.2012.04.053
Lacolley P, Regnault V, Laurent S (2020) Mechanisms of arterial stiffening: from mechanotransduction to epigenetics. Arterioscler Thromb Vasc Biol 40:1055–1062. https://doi.org/10.1161/atvbaha.119.313129
Landis BJ, Lai D, Guo DC, Corvera JS, Idrees MT, Stadler HW, Cuevas C, Needler GU, Vujakovich CE, Milewicz DM, Hinton RB, Ware SM (2022) Identification of a common polymorphism in COQ8B acting as a modifier of thoracic aortic aneurysm severity. HGG Adv. https://doi.org/10.1016/j.xhgg.2021.100057
Langille BL (1996) Arterial remodeling: relation to hemodynamics. Can J Physiol Pharmacol 74:834–841
Lee J, Shen M, Parajuli N, Oudit GY, McMurtry MS, Kassiri Z (2014) Gender-dependent aortic remodelling in patients with bicuspid aortic valve-associated thoracic aortic aneurysm. J Mol Med (Berl) 92:939–949. https://doi.org/10.1007/s00109-014-1178-6
Lee VS, Halabi CM, Hoffman EP, Carmichael N, Leshchiner I, Lian CG, Bierhals AJ, Vuzman D, Mecham RP, Frank NY, Stitziel NO (2016) Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc Natl Acad Sci U S A 113:8759–8764. https://doi.org/10.1073/pnas.1601442113
LeMaire SA, McDonald ML, Guo DC, Russell L, Miller CC 3rd, Johnson RJ, Bekheirnia MR, Franco LM, Nguyen M, Pyeritz RE, Bavaria JE, Devereux R, Maslen C, Holmes KW, Eagle K, Body SC, Seidman C, Seidman JG, Isselbacher EM, Bray M, Coselli JS, Estrera AL, Safi HJ, Belmont JW, Leal SM, Milewicz DM (2011) Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet 43:996–1000. https://doi.org/10.1038/ng.934
Lesauskaite V, Tanganelli P, Sassi C, Neri E, Diciolla F, Ivanoviene L, Epistolato MC, Lalinga AV, Alessandrini C, Spina D (2001) Smooth muscle cells of the media in the dilatative pathology of ascending thoracic aorta: morphology, immunoreactivity for osteopontin, matrix metalloproteinases, and their inhibitors. Hum Pathol 32:1003–1011. https://doi.org/10.1053/hupa.2001.27107
Levi N, Papismadov N, Solomonov I, Sagi I, Krizhanovsky V (2020) The ECM path of senescence in aging: components and modifiers. Febs j 287:2636–2646. https://doi.org/10.1111/febs.15282
Li H, Jiang W, Ren W, Guo D, Guo J, Wang X, Liu Y, Lan F, Du J, Zhang H (2016) Downregulation of the yes-associated protein is associated with extracellular matrix disorders in ascending aortic aneurysms. Stem Cells Int 2016:6786184. https://doi.org/10.1155/2016/6786184
Li T, Jiang B, Li X, Sun HY, Li XT, Jing JJ, Yang J (2018) Serum matrix metalloproteinase-9 is a valuable biomarker for identification of abdominal and thoracic aortic aneurysm: a case-control study. BMC Cardiovasc Disord 18:202. https://doi.org/10.1186/s12872-018-0931-0
Li Y, Takeshita K, Liu PY, Satoh M, Oyama N, Mukai Y, Chin MT, Krebs L, Kotlikoff MI, Radtke F, Gridley T, Liao JK (2009) Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation 119:2686–2692. https://doi.org/10.1161/circulationaha.108.790485
Lindner V, Booth C, Prudovsky I, Small D, Maciag T, Liaw L (2001) Members of the Jagged/Notch gene families are expressed in injured arteries and regulate cell phenotype via alterations in cell matrix and cell-cell interaction. Am J Pathol 159:875–883. https://doi.org/10.1016/s0002-9440(10)61763-4
Liu RM, Desai LP (2015) Reciprocal regulation of TGF-β and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol 6:565–577. https://doi.org/10.1016/j.redox.2015.09.009
Loerakker S, Stassen O, Ter Huurne FM, Boareto M, Bouten CVC, Sahlgren CM (2018) Mechanosensitivity of jagged-notch signaling can induce a switch-type behavior in vascular homeostasis. Proc Natl Acad Sci U S A 115:E3682-e3691. https://doi.org/10.1073/pnas.1715277115
Lu H, Aikawa M (2015) Many faces of matrix metalloproteinases in aortic aneurysms. Arterioscler Thromb Vasc Biol 35:752–754. https://doi.org/10.1161/atvbaha.115.305401
Lu H, Du W, Ren L, Hamblin MH, Becker RC, Chen YE, Fan Y (2021) Vascular smooth muscle cells in aortic aneurysm: from genetics to mechanisms. J Am Heart Assoc 10:e023601. https://doi.org/10.1161/jaha.121.023601
Lu H, Fagnant PM, Bookwalter CS, Joel P, Trybus KM (2015) Vascular disease-causing mutation R258C in ACTA2 disrupts actin dynamics and interaction with myosin. Proc Natl Acad Sci U S A 112:E4168-4177. https://doi.org/10.1073/pnas.1507587112
Ma S, Lieberman S, Turino GM, Lin YY (2003) The detection and quantitation of free desmosine and isodesmosine in human urine and their peptide-bound forms in sputum. Proc Natl Acad Sci U S A 100:12941–12943. https://doi.org/10.1073/pnas.2235344100
Maguire EM, Pearce SWA, Xiao R, Oo AY, Xiao Q (2019) Matrix metalloproteinase in abdominal aortic aneurysm and aortic dissection. Pharmaceuticals (Basel). https://doi.org/10.3390/ph12030118
Majesky MW (2007) Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol 27:1248–1258. https://doi.org/10.1161/atvbaha.107.141069
Malashicheva A, Kostina A, Kostareva A, Irtyuga O, Gordeev M, Uspensky V (2020) Notch signaling in the pathogenesis of thoracic aortic aneurysms: A bridge between embryonic and adult states. Biochim Biophys Acta Mol Basis Dis 1866:165631. https://doi.org/10.1016/j.bbadis.2019.165631
Malecki C, Hambly BD, Jeremy RW, Robertson EN (2020) The Role of inflammation and myeloperoxidase-related oxidative stress in the pathogenesis of genetically triggered thoracic aortic aneurysms. Int J Mol Sci. https://doi.org/10.3390/ijms21207678
Mao D, Lee JK, VanVickle SJ, Thompson RW (1999) Expression of collagenase-3 (MMP-13) in human abdominal aortic aneurysms and vascular smooth muscle cells in culture. Biochem Biophys Res Commun 261:904–910. https://doi.org/10.1006/bbrc.1999.1142
Markl M, Hope MD (2022) 4D flow imaging-state of the art. Ann Cardiothorac Surg 11:468–469. https://doi.org/10.21037/acs-2021-bav-15
Matthias Bechtel JF, Noack F, Sayk F, Erasmi AW, Bartels C, Sievers HH (2003) Histopathological grading of ascending aortic aneurysm: comparison of patients with bicuspid versus tricuspid aortic valve. J Heart Valve Dis 12:54–59; discussion 59–61
Maurel E, Shuttleworth CA, Bouissou H (1987) Interstitial collagens and ageing in human aorta. Virchows Arch A Pathol Anat Histopathol 410:383–390. https://doi.org/10.1007/bf00712757
McClure RS, Brogly SB, Lajkosz K, Payne D, Hall SF, Johnson AP (2018) Epidemiology and management of thoracic aortic dissections and thoracic aortic aneurysms in Ontario, Canada: a population-based study. J Thorac Cardiovasc Surg 155:2254-2264.e2254. https://doi.org/10.1016/j.jtcvs.2017.11.105
Meccanici F, Thijssen CGE, Dekker S, Bons LR, Gökalp AL, de Rijke YB, Takkenberg JJM, Mokhles MM, Bekkers JA, Boersma E, Bouwens E, van der Bosch AE, van Kimmenade RRL, Roos-Hesselink JW (2023) Circulating biomarkers associated with aortic diameter in male and female patients with thoracic aortic disease: a cross-sectional study. Open Heart. https://doi.org/10.1136/openhrt-2023-002317
Meijer CA, Stijnen T, Wasser MN, Hamming JF, van Bockel JH, Lindeman JH (2013) Doxycycline for stabilization of abdominal aortic aneurysms: a randomized trial. Ann Intern Med 159:815–823. https://doi.org/10.7326/0003-4819-159-12-201312170-00007
Melvinsdottir IH, Lund SH, Agnarsson BA, Sigvaldason K, Gudbjartsson T, Geirsson A (2016) The incidence and mortality of acute thoracic aortic dissection: results from a whole nation study. Eur J Cardiothorac Surg 50:1111–1117. https://doi.org/10.1093/ejcts/ezw235
Mia MM, Singh MK (2019) The hippo signaling pathway in cardiac development and diseases. Front Cell Dev Biol 7:211. https://doi.org/10.3389/fcell.2019.00211
Michel JB, Jondeau G, Milewicz DM (2018) From genetics to response to injury: vascular smooth muscle cells in aneurysms and dissections of the ascending aorta. Cardiovasc Res 114:578–589. https://doi.org/10.1093/cvr/cvy006
Mikael LR, Paiva AMG, Gomes MM, Sousa ALL, Jardim P, Vitorino PVO, Euzébio MB, Sousa WM, Barroso WKS (2017) Vascular aging and arterial stiffness. Arq Bras Cardiol 109:253–258. https://doi.org/10.5935/abc.20170091
Milewicz DM, Pyeritz RE, Crawford ES, Byers PH (1992) Marfan syndrome: defective synthesis, secretion, and extracellular matrix formation of fibrillin by cultured dermal fibroblasts. J Clin Invest 89:79–86. https://doi.org/10.1172/jci115589
Milewicz DM, Regalado ES, Shendure J, Nickerson DA, Guo DC (2014) Successes and challenges of using whole exome sequencing to identify novel genes underlying an inherited predisposition for thoracic aortic aneurysms and acute aortic dissections. Trends Cardiovasc Med 24:53–60. https://doi.org/10.1016/j.tcm.2013.06.004
Milewicz DM, Trybus KM, Guo DC, Sweeney HL, Regalado E, Kamm K, Stull JT (2017) Altered smooth muscle cell force generation as a driver of thoracic aortic aneurysms and dissections. Arterioscler Thromb Vasc Biol 37:26–34. https://doi.org/10.1161/atvbaha.116.303229
Minderhoud SCS, Roos-Hesselink JW, Chelu RG, Bons LR, van den Hoven AT, Korteland SA, van den Bosch AE, Budde RPJ, Wentzel JJ, Hirsch A (2022) Wall shear stress angle is associated with aortic growth in bicuspid aortic valve patients. Eur Heart J Cardiovasc Imaging. https://doi.org/10.1093/ehjci/jeab290
Mordi IR, Forsythe RO, Gellatly C, Iskandar Z, McBride OM, Saratzis A, Chalmers R, Chin C, Bown MJ, Newby DE, Lang CC, Huang JTJ, Choy AM (2019) Plasma desmosine and abdominal aortic aneurysm disease. J Am Heart Assoc 8:e013743. https://doi.org/10.1161/jaha.119.013743
Morisaki H, Akutsu K, Ogino H, Kondo N, Yamanaka I, Tsutsumi Y, Yoshimuta T, Okajima T, Matsuda H, Minatoya K, Sasaki H, Tanaka H, Ishibashi-Ueda H, Morisaki T (2009) Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Hum Mutat 30:1406–1411. https://doi.org/10.1002/humu.21081
Morris SA, Orbach DB, Geva T, Singh MN, Gauvreau K, Lacro RV (2011) Increased vertebral artery tortuosity index is associated with adverse outcomes in children and young adults with connective tissue disorders. Circulation 124:388–396. https://doi.org/10.1161/circulationaha.110.990549
Morrow D, Sweeney C, Birney YA, Cummins PM, Walls D, Redmond EM, Cahill PA (2005) Cyclic strain inhibits Notch receptor signaling in vascular smooth muscle cells in vitro. Circ Res 96:567–575. https://doi.org/10.1161/01.Res.0000159182.98874.43
Nataatmadja M, West J, Prabowo S, West M (2013) Angiotensin II receptor antagonism reduces transforming growth factor beta and smad signaling in thoracic aortic aneurysm. Ochsner J 13:42–48
Natoli AK, Medley TL, Ahimastos AA, Drew BG, Thearle DJ, Dilley RJ, Kingwell BA (2005) Sex steroids modulate human aortic smooth muscle cell matrix protein deposition and matrix metalloproteinase expression. Hypertension 46:1129–1134. https://doi.org/10.1161/01.Hyp.0000187016.06549.96
New SE, Aikawa E (2011) Cardiovascular calcification: an inflammatory disease. Circ J 75:1305–1313. https://doi.org/10.1253/circj.cj-11-0395
Nienaber CA, Fattori R, Mehta RH, Richartz BM, Evangelista A, Petzsch M, Cooper JV, Januzzi JL, Ince H, Sechtem U, Bossone E, Fang J, Smith DE, Isselbacher EM, Pape LA, Eagle KA (2004) Gender-related differences in acute aortic dissection. Circulation 109:3014–3021. https://doi.org/10.1161/01.Cir.0000130644.78677.2c
Nolasco P, Fernandes CG, Ribeiro-Silva JC, Oliveira PVS, Sacrini M, de Brito IV, De Bessa TC, Pereira LV, Tanaka LY, Alencar A, Laurindo FRM (2020) Impaired vascular smooth muscle cell force-generating capacity and phenotypic deregulation in marfan syndrome mice. Biochim Biophys Acta Mol Basis Dis 1866:165587. https://doi.org/10.1016/j.bbadis.2019.165587
Okada R, Hazato N, Nishijo T, Sugiua M (1973) Acid-mucopolysaccharides and sclerosis of the human aorta. Jpn Circ J 37:253–259. https://doi.org/10.1253/jcj.37.253
Okamura H, Pisani LJ, Dalal AR, Emrich F, Dake BA, Arakawa M, Onthank DC, Cesati RR, Robinson SP, Milanesi M, Kotek G, Smit H, Connolly AJ, Adachi H, McConnell MV, Fischbein MP (2014) Assessment of elastin deficit in a Marfan mouse aneurysm model using an elastin-specific magnetic resonance imaging contrast agent. Circ Cardiovasc Imaging 7:690–696. https://doi.org/10.1161/circimaging.114.001658
Oladokun D, Patterson BO, Sobocinski J, Karthikesalingam A, Loftus I, Thompson MM, Holt PJ (2016) Systematic review of the growth rates and influencing factors in thoracic aortic aneurysms. Eur J Vasc Endovasc Surg 51:674–681. https://doi.org/10.1016/j.ejvs.2016.01.017
Oller J, Gabandé-Rodríguez E, Ruiz-Rodríguez MJ, Desdín-Micó G, Aranda JF, Rodrigues-Diez R, Ballesteros-Martínez C, Blanco EM, Roldan-Montero R, Acuña P, Forteza Gil A, Martín-López CE, Nistal JF, Lino Cardenas CL, Lindsay ME, Martín-Ventura JL, Briones AM, Redondo JM, Mittelbrunn M (2021) Extracellular tuning of mitochondrial respiration leads to aortic aneurysm. Circulation 143:2091–2109. https://doi.org/10.1161/circulationaha.120.051171
Olsson C, Thelin S, Ståhle E, Ekbom A, Granath F (2006) Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation 114:2611–2618. https://doi.org/10.1161/circulationaha.106.630400
Ostberg NP, Zafar MA, Ziganshin BA, Elefteriades JA (2020) The genetics of thoracic aortic aneurysms and dissection: a clinical perspective. Biomolecules. https://doi.org/10.3390/biom10020182
Owens GK, Kumar MS, Wamhoff BR (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84:767–801. https://doi.org/10.1152/physrev.00041.2003
Pape LA, Tsai TT, Isselbacher EM, Oh JK, O’Gara PT, Evangelista A, Fattori R, Meinhardt G, Trimarchi S, Bossone E, Suzuki T, Cooper JV, Froehlich JB, Nienaber CA, Eagle KA (2007) Aortic diameter >or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation 116:1120–1127. https://doi.org/10.1161/circulationaha.107.702720
Pasta S, Agnese V, Gallo A, Cosentino F, Di Giuseppe M, Gentile G, Raffa GM, Maalouf JF, Michelena HI, Bellavia D, Conaldi PG, Pilato M (2020) Shear stress and aortic strain associations with biomarkers of ascending thoracic aortic aneurysm. Ann Thorac Surg 110:1595–1604. https://doi.org/10.1016/j.athoracsur.2020.03.017
Perez ZG, Zafar MA, Velasco JJ, Sonsino A, Ellauzi H, John C, Kalyanasundaram A, Ziganshin BA, Elefteriades JA (2023) Aortic size at the time of type a and type b dissections. Ann Thorac Surg. https://doi.org/10.1016/j.athoracsur.2023.03.037
Petsophonsakul P, Furmanik M, Forsythe R, Dweck M, Schurink GW, Natour E, Reutelingsperger C, Jacobs M, Mees B, Schurgers L (2019) Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arterioscler Thromb Vasc Biol 39:1351–1368. https://doi.org/10.1161/atvbaha.119.312787
Pirruccello JP, Khurshid S, Lin H, Lu-Chen W, Zamirpour S, Kany S, Raghavan A, Koyama S, Vasan RS, Benjamin EJ, Lindsay ME, Ellinor PT (2023) AORTA Gene: polygenic prediction improves detection of thoracic aortic aneurysm. MedRxiv. https://doi.org/10.1101/2023.08.23.23294513
Piscaer I, Wouters EFM, Vermeer C, Janssens W, Franssen FME, Janssen R (2017) Vitamin K deficiency: the linking pin between COPD and cardiovascular diseases? Respir Res 18:189. https://doi.org/10.1186/s12931-017-0673-z
Prakash SK, Haden-Pinneri K, Milewicz DM (2011) Susceptibility to acute thoracic aortic dissections in patients dying outside the hospital: an autopsy study. Am Heart J 162:474–479. https://doi.org/10.1016/j.ahj.2011.06.020
Qi X, Wang F, Chun C, Saldarriaga L, Jiang Z, Pruitt EY, Arnaoutakis GJ, Upchurch GR Jr, Jiang Z (2020) A validated mouse model capable of recapitulating the protective effects of female sex hormones on ascending aortic aneurysms and dissections (AADs). Physiol Rep 8:e14631. https://doi.org/10.14814/phy2.14631
Rabkin SW (2017) The role matrix metalloproteinases in the production of aortic aneurysm. Prog Mol Biol Transl Sci 147:239–265. https://doi.org/10.1016/bs.pmbts.2017.02.002
Rai P, Robinson L, Davies HA, Akhtar R, Field M, Madine J (2022) Is there enough evidence to support the role of glycosaminoglycans and proteoglycans in thoracic aortic aneurysm and dissection?-a systematic review. Int J Mol Sci. https://doi.org/10.3390/ijms23169200
Ramaekers M, Adriaans BP, Juffermans JF, van Assen HC, Bekkers S, Scholte A, Kenjeres S, Lamb HJ, Wildberger JE, Westenberg JJM, Schalla S (2021) Characterization of ascending aortic flow in patients with degenerative aneurysms: a 4D flow magnetic resonance study. Invest Radiol 56:494–500. https://doi.org/10.1097/RLI.0000000000000768
Renard M, Callewaert B, Baetens M, Campens L, MacDermot K, Fryns JP, Bonduelle M, Dietz HC, Gaspar IM, Cavaco D, Stattin EL, Schrander-Stumpel C, Coucke P, Loeys B, De Paepe A, De Backer J (2013) Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFβ signaling in FTAAD. Int J Cardiol 165:314–321. https://doi.org/10.1016/j.ijcard.2011.08.079
Renard M, Francis C, Ghosh R, Scott AF, Witmer PD, Adès LC, Andelfinger GU, Arnaud P, Boileau C, Callewaert BL, Guo D, Hanna N, Lindsay ME, Morisaki H, Morisaki T, Pachter N, Robert L, Van Laer L, Dietz HC, Loeys BL, Milewicz DM, De Backer J (2018) Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J Am Coll Cardiol 72:605–615. https://doi.org/10.1016/j.jacc.2018.04.089
Risinger GM Jr, Updike DL, Bullen EC, Tomasek JJ, Howard EW (2010) TGF-beta suppresses the upregulation of MMP-2 by vascular smooth muscle cells in response to PDGF-BB. Am J Physiol Cell Physiol 298:C191-201. https://doi.org/10.1152/ajpcell.00417.2008
Roccabianca S, Ateshian GA, Humphrey JD (2014) Biomechanical roles of medial pooling of glycosaminoglycans in thoracic aortic dissection. Biomech Model Mechanobiol 13:13–25. https://doi.org/10.1007/s10237-013-0482-3
Rodríguez-Palomares JF, Dux-Santoy L, Guala A, Kale R, Maldonado G, Teixidó-Turà G, Galian L, Huguet M, Valente F, Gutiérrez L, González-Alujas T, Johnson KM, Wieben O, García-Dorado D, Evangelista A (2018) Aortic flow patterns and wall shear stress maps by 4D-flow cardiovascular magnetic resonance in the assessment of aortic dilatation in bicuspid aortic valve disease. J Cardiovasc Magn Reson 20:28. https://doi.org/10.1186/s12968-018-0451-1
Rombouts KB, van Merrienboer TAR, Ket JCF, Bogunovic N, van der Velden J, Yeung KK (2022) The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur J Clin Invest 52:e13697. https://doi.org/10.1111/eci.13697
Roychowdhury T, Lu H, Hornsby WE, Crone B, Wang GT, Guo DC, Sendamarai AK, Devineni P, Lin M, Zhou W, Graham SE, Wolford BN, Surakka I, Wang Z, Chang L, Zhang J, Mathis M, Brummett CM, Melendez TL, Shea MJ, Kim KM, Deeb GM, Patel HJ, Eliason J, Eagle KA, Yang B, Ganesh SK, Brumpton B, Åsvold BO, Skogholt AH, Hveem K, Pyarajan S, Klarin D, Tsao PS, Damrauer SM, Leal SM, Milewicz DM, Chen YE, Garcia-Barrio MT, Willer CJ (2021) Regulatory variants in TCF7L2 are associated with thoracic aortic aneurysm. Am J Hum Genet 108:1578–1589. https://doi.org/10.1016/j.ajhg.2021.06.016
Rudd JH, Warburton EA, Fryer TD, Jones HA, Clark JC, Antoun N, Johnström P, Davenport AP, Kirkpatrick PJ, Arch BN, Pickard JD, Weissberg PL (2002) Imaging atherosclerotic plaque inflammation with [18F]-fluorodeoxyglucose positron emission tomography. Circulation 105:2708–2711. https://doi.org/10.1161/01.cir.0000020548.60110.76
Rylski B, Branchetti E, Bavaria JE, Vallabhajosyula P, Szeto WY, Milewski RK, Desai ND (2014) Modeling of predissection aortic size in acute type A dissection: More than 90% fail to meet the guidelines for elective ascending replacement. J Thorac Cardiovasc Surg 148:944-948.e941. https://doi.org/10.1016/j.jtcvs.2014.05.050
Rzucidlo EM, Martin KA, Powell RJ (2007) Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. https://doi.org/10.1016/j.jvs.2007.03.001
Sakai LY, Keene DR, Renard M, De Backer J (2016) FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene 591:279–291. https://doi.org/10.1016/j.gene.2016.07.033
Sakamoto N, Ohashi T, Sato M (2006) Effect of fluid shear stress on migration of vascular smooth muscle cells in cocultured model. Ann Biomed Eng 34:408–415. https://doi.org/10.1007/s10439-005-9043-y
Salmasi MY, Pirola S, Mahuttanatan S, Fisichella SM, Sengupta S, Jarral OA, Oo A, O’Regan D, Xu XY, Athanasiou T (2023) Geometry and flow in ascending aortic aneurysms are influenced by left ventricular outflow tract orientation: Detecting increased wall shear stress on the outer curve of proximal aortic aneurysms. J Thorac Cardiovasc Surg 166:11-21.e11. https://doi.org/10.1016/j.jtcvs.2021.06.014
Salmasi MY, Sasidharan S, Frattolin J, Edgar L, Stock U, Athanasiou T, Moore J Jr (2022) Regional variation in biomechanical properties of ascending thoracic aortic aneurysms. Eur J Cardiothorac Surg. https://doi.org/10.1093/ejcts/ezac392
Sawada H, Rateri DL, Moorleghen JJ, Majesky MW, Daugherty A (2017) Smooth muscle cells derived from second heart field and cardiac neural crest reside in spatially distinct domains in the media of the ascending aorta-brief report. Arterioscler Thromb Vasc Biol 37:1722–1726. https://doi.org/10.1161/atvbaha.117.309599
Schultz-Cherry S, Murphy-Ullrich JE (1993) Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J Cell Biol 122:923–932. https://doi.org/10.1083/jcb.122.4.923
Schurgers LJ, Teunissen KJ, Knapen MH, Kwaijtaal M, van Diest R, Appels A, Reutelingsperger CP, Cleutjens JP, Vermeer C (2005) Novel conformation-specific antibodies against matrix gamma-carboxyglutamic acid (Gla) protein: undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler Thromb Vasc Biol 25:1629–1633. https://doi.org/10.1161/01.Atv.0000173313.46222.43
Scola L, Di Maggio FM, Vaccarino L, Bova M, Forte GI, Pisano C, Candore G, Colonna-Romano G, Lio D, Ruvolo G, Balistreri CR (2014) Role of TGF-β pathway polymorphisms in sporadic thoracic aortic aneurysm: rs900 TGF-β2 is a marker of differential gender susceptibility. Mediators Inflamm 2014:165758. https://doi.org/10.1155/2014/165758
Shaw LJ, Raggi P, Berman DS, Callister TQ (2006) Coronary artery calcium as a measure of biologic age. Atherosclerosis 188:112–119. https://doi.org/10.1016/j.atherosclerosis.2005.10.010
Shen M, Lee J, Basu R, Sakamuri SS, Wang X, Fan D, Kassiri Z (2015) Divergent roles of matrix metalloproteinase 2 in pathogenesis of thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol 35:888–898. https://doi.org/10.1161/atvbaha.114.305115
Silver AE, Vita JA (2006) Shear-stress-mediated arterial remodeling in atherosclerosis: too much of a good thing? Circulation 113:2787–2789. https://doi.org/10.1161/circulationaha.106.634378
Sinha S, Iyer D, Granata A (2014) Embryonic origins of human vascular smooth muscle cells: implications for in vitro modeling and clinical application. Cell Mol Life Sci 71:2271–2288. https://doi.org/10.1007/s00018-013-1554-3
Skinner MP, Raines EW, Ross R (1994) Dynamic expression of alpha 1 beta 1 and alpha 2 beta 1 integrin receptors by human vascular smooth muscle cells. Alpha 2 beta 1 integrin is required for chemotaxis across type I collagen-coated membranes. Am J Pathol 145:1070–1081
Sorokin V, Vickneson K, Kofidis T, Woo CC, Lin XY, Foo R, Shanahan CM (2020) Role of vascular smooth muscle cell plasticity and interactions in vessel wall inflammation. Front Immunol 11:599415. https://doi.org/10.3389/fimmu.2020.599415
Stepien KL, Bajdak-Rusinek K, Fus-Kujawa A, Kuczmik W, Gawron K (2022) Role of extracellular matrix and inflammation in abdominal aortic aneurysm. Int J Mol Sci. https://doi.org/10.3390/ijms231911078
Surman TL, Abrahams JM, Manavis J, Finnie J, O’Rourke D, Reynolds KJ, Edwards J, Worthington MG, Beltrame J (2021) Histological regional analysis of the aortic root and thoracic ascending aorta: a complete analysis of aneurysms from root to arch. J Cardiothorac Surg 16:255. https://doi.org/10.1186/s13019-021-01641-5
Tadros TM, Klein MD, Shapira OM (2009) Ascending aortic dilatation associated with bicuspid aortic valve: pathophysiology, molecular biology, and clinical implications. Circulation 119:880–890. https://doi.org/10.1161/circulationaha.108.795401
Takahashi T, Watanabe N, Wakasa M, Kajinami K, Tonami H (2016) 18F-FDG PET/CT for detecting sarcoma of the aorta in a patient with takayasu arteritis. Nucl Med Mol Imaging 50:171–172. https://doi.org/10.1007/s13139-015-0347-z
Thijssen CGE, Dekker S, Bons LR, Geenen LW, Gökalp AL, Takkenberg JJM, Mokhles MM, Bekkers JA, Boersma E, Bouwens E, van Kimmenade RRJ, Roos-Hesselink JW (2023) Novel biomarkers associated with thoracic aortic disease. Int J Cardiol 378:115–122. https://doi.org/10.1016/j.ijcard.2023.02.006
Tomida S, Ishima T, Sawaki D, Imai Y, Nagai R, Aizawa K (2023) Multi-omics of familial thoracic aortic aneurysm and dissection: calcium transport impairment predisposes aortas to dissection. Int J Mol Sci. https://doi.org/10.3390/ijms242015213
Toral M, de la Fuente-Alonso A, Campanero MR, Redondo JM (2022) The NO signalling pathway in aortic aneurysm and dissection. Br J Pharmacol 179:1287–1303. https://doi.org/10.1111/bph.15694
Toyama BH, Hetzer MW (2013) Protein homeostasis: live long, won’t prosper. Nat Rev Mol Cell Biol 14:55–61. https://doi.org/10.1038/nrm3496
Tran-Fadulu V, Pannu H, Kim DH, Vick GW 3rd, Lonsford CM, Lafont AL, Boccalandro C, Smart S, Peterson KL, Hain JZ, Willing MC, Coselli JS, LeMaire SA, Ahn C, Byers PH, Milewicz DM (2009) Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J Med Genet 46:607–613. https://doi.org/10.1136/jmg.2008.062844
Trimarchi S, Sangiorgi G, Sang X, Rampoldi V, Suzuki T, Eagle KA, Elefteriades JA (2010) In search of blood tests for thoracic aortic diseases. Ann Thorac Surg 90:1735–1742. https://doi.org/10.1016/j.athoracsur.2010.04.111
Tromp G, Gatalica Z, Skunca M, Berguer R, Siegel T, Kline RA, Kuivaniemi H (2004) Elevated expression of matrix metalloproteinase-13 in abdominal aortic aneurysms. Ann Vasc Surg 18:414–420. https://doi.org/10.1007/s10016-004-0050-5
Tsai S, Hollenbeck ST, Ryer EJ, Edlin R, Yamanouchi D, Kundi R, Wang C, Liu B, Kent KC (2009) TGF-beta through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. Am J Physiol Heart Circ Physiol 297:H540-549. https://doi.org/10.1152/ajpheart.91478.2007
Tsamis A, Krawiec JT, Vorp DA (2013) Elastin and collagen fibre microstructure of the human aorta in ageing and disease: a review. J R Soc Interface 10:20121004. https://doi.org/10.1098/rsif.2012.1004
Tsaousi A, Williams H, Lyon CA, Taylor V, Swain A, Johnson JL, George SJ (2011) Wnt4/β-catenin signaling induces VSMC proliferation and is associated with intimal thickening. Circ Res 108:427–436. https://doi.org/10.1161/circresaha.110.233999
Tyrrell DJ, Chen J, Li BY, Wood SC, Rosebury-Smith W, Remmer HA, Jiang L, Zhang M, Salmon M, Ailawadi G, Yang B, Goldstein DR (2022) Aging alters the aortic proteome in health and thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol 42:1060–1076. https://doi.org/10.1161/atvbaha.122.317643
Umeda H, Aikawa M, Libby P (2011) Liberation of desmosine and isodesmosine as amino acids from insoluble elastin by elastolytic proteases. Biochem Biophys Res Commun 411:281–286. https://doi.org/10.1016/j.bbrc.2011.06.124
van 't Hof FN, Ruigrok YM, Lee CH, Ripke S, Anderson G, de Andrade M, Baas AF, Blankensteijn JD, Böttinger EP, Bown MJ, Broderick J, Bijlenga P, Carrell DS, Crawford DC, Crosslin DR, Ebeling C, Eriksson JG, Fornage M, Foroud T, von Und Zu Fraunberg M, Friedrich CM, Gaál EI, Gottesman O, Guo DC, Harrison SC, Hernesniemi J, Hofman A, Inoue I, Jääskeläinen JE, Jones GT, Kiemeney LA, Kivisaari R, Ko N, Koskinen S, Kubo M, Kullo IJ, Kuivaniemi H, Kurki MI, Laakso A, Lai D, Leal SM, Lehto H, LeMaire SA, Low SK, Malinowski J, McCarty CA, Milewicz DM, Mosley TH, Nakamura Y, Nakaoka H, Niemelä M, Pacheco J, Peissig PL, Pera J, Rasmussen-Torvik L, Ritchie MD, Rivadeneira F, van Rij AM, Santos-Cortez RL, Saratzis A, Slowik A, Takahashi A, Tromp G, Uitterlinden AG, Verma SS, Vermeulen SH, Wang GT, Han B, Rinkel GJ, de Bakker PI. (2016). Shared Genetic Risk Factors of Intracranial, Abdominal, and Thoracic Aneurysms. J Am Heart Assoc. doi:https://doi.org/10.1161/jaha.115.002603
van de Luijtgaarden KM, Heijsman D, Maugeri A, Weiss MM, Verhagen HJ, A IJ, Brüggenwirth HT, Majoor-Krakauer D, (2015) First genetic analysis of aneurysm genes in familial and sporadic abdominal aortic aneurysm. Hum Genet 134:881–893. https://doi.org/10.1007/s00439-015-1567-0
Van Laer L, Dietz H, Loeys B (2014) Loeys-dietz syndrome. Adv Exp Med Biol 802:95–105. https://doi.org/10.1007/978-94-007-7893-1_7
Waddell TK, Dart AM, Gatzka CD, Cameron JD, Kingwell BA (2001) Women exhibit a greater age-related increase in proximal aortic stiffness than men. J Hypertens 19:2205–2212. https://doi.org/10.1097/00004872-200112000-00014
Wang C, Chang Q, Sun X, Qian X, Liu P, Pei H, Guo X, Liu W (2015) Angiotensin II induces an increase in matrix metalloproteinase 2 expression in aortic smooth muscle cells of ascending thoracic aortic aneurysms through JNK, ERK1/2, and p38 MAPK activation. J Cardiovasc Pharmacol 66:285–293. https://doi.org/10.1097/fjc.0000000000000276
Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He WQ, Zhu MS, Offermanns S, Gilchrist D, Elefteriades J, Stull JT, Milewicz DM (2010) Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet 87:701–707. https://doi.org/10.1016/j.ajhg.2010.10.006
Wang W, Campos AH, Prince CZ, Mou Y, Pollman MJ (2002) Coordinate Notch3-hairy-related transcription factor pathway regulation in response to arterial injury. Mediator role of platelet-derived growth factor and ERK. J Biol Chem 277:23165–23171. https://doi.org/10.1074/jbc.M201409200
Wang X, Khalil RA (2018) Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv Pharmacol 81:241–330. https://doi.org/10.1016/bs.apha.2017.08.002
Wanga S, Hibender S, Ridwan Y, van Roomen C, Vos M, van der Made I, van Vliet N, Franken R, van Riel LA, Groenink M, Zwinderman AH, Mulder BJ, de Vries CJ, Essers J, de Waard V (2017) Aortic microcalcification is associated with elastin fragmentation in marfan syndrome. J Pathol 243:294–306. https://doi.org/10.1002/path.4949
Willemink MJ, Persson M, Pourmorteza A, Pelc NJ, Fleischmann D (2018) Photon-counting CT: technical principles and clinical prospects. Radiology 289:293–312. https://doi.org/10.1148/radiol.2018172656
Wipff PJ, Hinz B (2008) Integrins and the activation of latent transforming growth factor beta1 - an intimate relationship. Eur J Cell Biol 87:601–615. https://doi.org/10.1016/j.ejcb.2008.01.012
Wittig C, Szulcek R (2021) Extracellular matrix protein ratios in the human heart and vessels: how to distinguish pathological from physiological changes? Front Physiol 12:708656. https://doi.org/10.3389/fphys.2021.708656
Wu M, Rementer C, Giachelli CM (2013) Vascular calcification: an update on mechanisms and challenges in treatment. Calcif Tissue Int 93:365–373. https://doi.org/10.1007/s00223-013-9712-z
Xu X, Wang B, Ren C, Hu J, Greenberg DA, Chen T, Xie L, Jin K (2017) Age-related Impairment of Vascular Structure and Functions. Aging Dis 8:590–610. https://doi.org/10.14336/ad.2017.0430
Xu Y, Ye J, Wang M, Wang Y, Ji Q, Huang Y, Zeng T, Wang Z, Ye D, Jiang H, Liu J, Lin Y, Wan J (2018) Increased interleukin-11 levels in thoracic aorta and plasma from patients with acute thoracic aortic dissection. Clin Chim Acta 481:193–199. https://doi.org/10.1016/j.cca.2018.03.014
Yamashiro Y, Thang BQ, Shin SJ, Lino CA, Nakamura T, Kim J, Sugiyama K, Tokunaga C, Sakamoto H, Osaka M, Davis EC, Wagenseil JE, Hiramatsu Y, Yanagisawa H (2018) Role of thrombospondin-1 in mechanotransduction and development of thoracic aortic aneurysm in mouse and humans. Circ Res 123:660–672. https://doi.org/10.1161/circresaha.118.313105
Yang H, Yang F, Luo M, Chen Q, Liu X, Zhang Y, Zhu G, Chen W, Li T, Shu C, Zhou Z (2022) Metabolomic profile reveals that ceramide metabolic disturbance plays an important role in thoracic aortic dissection. Front Cardiovasc Med 9:826861. https://doi.org/10.3389/fcvm.2022.826861
You W, Hong Y, He H, Huang X, Tao W, Liang X, Zhang Y, Li X (2019) TGF-β mediates aortic smooth muscle cell senescence in Marfan syndrome. Aging (Albany NY) 11:3574–3584. https://doi.org/10.18632/aging.101998
Yu Q, Stamenkovic I (2000) Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev 14:163–176
Zhang C, Li Y, Chakraborty A, Li Y, Rebello KR, Ren P, Luo W, Zhang L, Lu HS, Cassis LA, Coselli JS, Daugherty A, LeMaire SA, Shen YH (2023) Aortic stress activates an adaptive program in thoracic aortic smooth muscle cells that maintains aortic strength and protects against aneurysm and dissection in mice. Arterioscler Thromb Vasc Biol 43:234–252. https://doi.org/10.1161/atvbaha.122.318135
Zhang X, Wu D, Choi JC, Minard CG, Hou X, Coselli JS, Shen YH, LeMaire SA (2014) Matrix metalloproteinase levels in chronic thoracic aortic dissection. J Surg Res 189:348–358. https://doi.org/10.1016/j.jss.2014.03.027
Zhang Y, Li Y, Miao S, Dai X, Chen L, Ma L (2023) Low expression of ESR1 correlates with ascending aortic dilation and acute type a aortic dissection. Gene 851:147001. https://doi.org/10.1016/j.gene.2022.147001
Zhou D, Feng H, Yang Y, Huang T, Qiu P, Zhang C, Olsen TR, Zhang J, Chen YE, Mizrak D, Yang B (2021) hiPSC Modeling of lineage-specific smooth muscle cell defects caused by TGFBR1(A230T) variant, and its therapeutic implications for loeys-dietz syndrome. Circulation 144:1145–1159. https://doi.org/10.1161/circulationaha.121.054744
Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X (2006) Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet 38:343–349. https://doi.org/10.1038/ng1721
Zieman SJ, Melenovsky V, Kass DA (2005) Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol 25:932–943. https://doi.org/10.1161/01.Atv.0000160548.78317.29
Zimmermann LMA, Furlan AG, Mehrkens D, Geißen S, Zuk AV, Pryymachuk G, Pykarek N, Beers Tv, Sonntag-Bensch D, Marzi J, Schenke Layland K, Brinckmann J, Zigrino P, Grandoch M, Baldus S, Sengle G. (2022). Targeting of MMP-13 prevents aortic aneurysm formation in Marfan mice. doi: https://doi.org/10.1101/2022.11.30.518511
Zou S, Ren P, Nguyen M, Coselli JS, Shen YH, LeMaire SA (2012) Notch signaling in descending thoracic aortic aneurysm and dissection. PLoS ONE 7:e52833. https://doi.org/10.1371/journal.pone.0052833
Funding
This review is financially supported by ZonMW, grant LSHM21078-SGF “CELLSYSTEMICS” (Koen Reesink, Elham Bidar, Leon Schurgers).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
L.J.S. has received consultancy fees from Immuno Diagnostic Systems and grants from Gnosis by Lesaffre, Boehringer Ingelheim, and Bayer and is a shareholder of Coagulation Profile. All other authors have nothing to declare.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ganizada, B.H., J. A. Veltrop, R., Akbulut, A.C. et al. Unveiling cellular and molecular aspects of ascending thoracic aortic aneurysms and dissections. Basic Res Cardiol 119, 371–395 (2024). https://doi.org/10.1007/s00395-024-01053-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-024-01053-1