
Abstract
Unmodified reperfusion therapy for acute myocardial infarction (AMI) is associated with irreversible myocardial injury beyond that sustained during ischemia. Studies in experimental models of ischemia/reperfusion and in humans undergoing reperfusion therapy for AMI have examined potential beneficial effects of nitric oxide (NO) supplemented at the time of reperfusion. Using a rigorous systematic search approach, we have identified and critically evaluated all the relevant experimental and clinical literature to assess whether exogenous NO given at reperfusion can limit infarct size. An inclusive search strategy was undertaken to identify all in vivo experimental animal and clinical human studies published in the period 1990–2014 where NO gas, nitrite, nitrate or NO donors were given to ameliorate reperfusion injury. Articles were screened at title and subsequently at abstract level, followed by objective full text analysis using a critical appraisal tool. In twenty-one animal studies, all NO treatments except nitroglycerin afforded protection against measures of reperfusion injury, including infarct size, creatinine kinase release, neutrophil accumulation and cardiac dysfunction. In three human AMI RCT’s, there was no consistent evidence of infarct limitation associated with NO treatment as an adjunct to reperfusion. Despite experimental evidence that most NO treatments can reduce infarct size when given as adjuncts to reperfusion, the value of these interventions in clinical AMI is unproven. Our study raises issues for the design of further clinical studies and emphasises the need for improved design of animal studies to reflect more accurately the comorbidities and other confounding factors seen in clinical AMI.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Early management of acute myocardial infarction (AMI) focuses on achieving rapid reperfusion of the ischemic risk zone in order to minimise irreversible tissue injury [65]. Although early reperfusion is undoubtedly beneficial after AMI, it can be associated with patterns of reperfusion injury. The deleterious effects of reperfusion on the myocardium occur as a result of the rapid reintroduction of oxygenated blood into the ischemic tissue. There are likely to be multiple underlying mechanisms of reperfusion injury but the most studied aspect is the formation of reactive oxygen species (ROS), in particular superoxide (O2 −) and hydrogen peroxide [49]. These highly reactive species cause oxidative damage to the sarcoplasmic reticulum, mitochondria, cell membrane, nuclear DNA and sarcomeric proteins, leading to calcium overload of the cardiomyocytes [50] and opening of the mitochondrial permeability transition pore (mPTP) [14]. Ultimately, unmodified reperfusion is associated with cardiomyocyte apoptosis and accelerated necrosis of cells already damaged by ischemia. Furthermore, damage to the microvasculature causes a reduction in blood flow leading to the “no-reflow phenomenon” [55].
Nitric oxide (NO) is endogenously produced within myocardium, principally from l-arginine under the influence of nitric oxide synthases (NOS). It can also be produced via NOS-independent mechanisms including the reduction of tissue reservoirs of nitrite (NO2 −) or nitrate (NO3 −) to liberate NO under hypoxic conditions [6], such as occurs in the ischemic myocardium. The production of NO from NO2 − has been shown to reduce myocardial injury [8, 33] and the reduction of NO2 − is thought to be facilitated by molecules including deoxymyoglobin [5] and the enzyme xanthine oxidoreductase [66] among others. NO has a short half-life in vivo and the conversion of NOS derived NO into a variety of storage forms by oxidase enzymes [57] is an important reservoir of NO.
Nitric oxide has been shown in many experimental studies to modulate ischemia/reperfusion injury. Administration of NOS inhibitors has been reported to exacerbate myocardial necrosis [23] supporting the notion that endogenous NO is protective against ischemia/reperfusion injury [18]. In experimental studies, endogenous NO has been shown to contribute in the protective pathways activated in classical and delayed ischemic preconditioning [10] and also hibernation [19]. These potential protective effects of endogenous NO have given rise to a multitude of experimental and clinical studies focusing on the delivery of exogenous NO, in the form of various NO species and NO-donor compounds, to limit ischemia/reperfusion injury [7] with the general hypothesis being that NO ameliorates ischemia/reperfusion injury.
The current study addresses the question of whether NO treatments/namely gaseous NO, NO2 −, NO3 − or organic NO donor compounds, as adjuncts to reperfusion following ischemia, provide consistent cardioprotection against reperfusion injury, when assessed primarily as a reduction in infarct size. We addressed this question by undertaking a systematic qualitative review of experimental and clinical studies that have investigated the effects of NO treatments, when given specifically in a manner that could modify reperfusion injury in (a) in vivo animal models of ischemia/reperfusion or (b) in patients undergoing reperfusion therapy for AMI. We identified articles against predefined, highly selective inclusion criteria and critically analysed relevant articles to evaluate the quality of the studies. Those studies subjected to full text analysis were then synthesised to form the basis of this review.
Methods
Study design
The study design was based on the preferred reporting items for systematic reviews and meta-analyses (PRISMA) 27-point guidance [36] together with review protocols published by the Cochrane Collaboration [1]. A systematic methodological approach was designed in order to reduce reviewer bias when selecting articles for inclusion and to appraise the included articles against predefined inclusion criteria to create an objective synthesis of the current published data.
Search strategy
Following a pilot study to scope the approximate period and scale of the relevant scientific literature, search terms were agreed by all reviewers (BRJ, GRC, JSB, GFB). Terms were expanded with Boolean operators, as well as adjacency and tree techniques, in order to combine search terms and narrow the specific literature to be included. Due to differences in search functionality between the databases some search terms were adapted or omitted for individual databases. Two reviewers (BRJ, GRC) used the final agreed search terms to search the Cochrane library, Medline, Embase, Web of Science, and the clinical trials databases CT.gov. The strategy was limited exclusively to exogenous sources of NO and their specific effects on myocardial reperfusion injury; as such, terms relating to precursors such as l-arginine and endogenous NO, or pre-ischemic treatment were excluded.
Eligibility criteria
We included all animal and human studies utilising exogenous administration of gaseous NO, organic NO-donors, NO2 −, NO3 − or OONO− during periods relevant to reperfusion injury. Original articles in the serial literature published in English during the period January 1st 1985 to August 15th 2014 were included. Review articles were screened to identify relevant publications once the articles reached the full text level of the screening process. We did not search university dissertation or thesis repositories. Published outputs were included if they met the criteria listed in Table 1.
A critical appraisal tool was developed to allow a comprehensive qualitative critique of the articles at full text level (Table 2).
Article selection and data extraction
Article screening was undertaken in a standardised, non-blinded manner by the two primary reviewers (BRJ, GRC) who independently screened the search results for relevance by reviewing the titles (11,539) and subsequently the abstracts (548) of the identified studies using the eligibility criteria. Following relevance screening, the two reviewers compared results to identify any disagreements or queries and the secondary reviewers (JSB, GFB) gave input until a consensus was reached. All articles deemed relevant (58 animal studies and 35 human studies) underwent full text critical analysis independently by BRJ and GRC who concluded which articles were appropriate for inclusion (Table 3). Each of the articles was then discussed between the primary reviewers and any disagreements were resolved by the secondary reviewers (JSB, GFB). The finalised included and excluded articles were then sampled by the secondary reviewers (JSB, GFB) to confirm the consistency of the data analysis process. Once the sampling process had been completed, the included studies (21 animal and three human) were critiqued.
Meta-analysis
A random effects model was used as it was considered that heterogeneity would be demonstrated due to varying treatments and animal models. Data are reported as mean difference. Authors were contacted to clarify data values if SEM or SD were not published. Statistical heterogeneity was determined using I 2. Sub analysis of grouped studies [by species, NO donor (data not shown)] did not cause significant deviation from the mean difference reported here.
Results
Study inclusion/exclusion
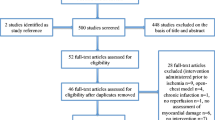
The results of the article selection and data extraction process are summarised in Fig. 1. The database search provided a total of 24,969 citations (from both animal and human studies), and after removing duplicate reports, 11,539 remained of which 10,991 were discarded at title level, leaving 548 articles. Of these, 463 studies were excluded at abstract level since they did not meet the eligibility criteria (Table 1). The remaining 50 animal studies and 35 clinical studies were obtained in full text and an additional eight animal studies were identified from the reference lists (“snowballing”) of the remaining animal studies. A total of 58 animal and 35 human studies were therefore appraised at full text level (see Fig. 1). Following this appraisal, 37 animal and 32 human studies were not deemed to have appropriate methodology or quality for inclusion, leaving 21 animal and three human studies to be included in the two arms of the review (Table 3).

Results of database searches and appraisal at different stages of the review process
Characteristics of experimental animal studies
Table 4 summarises the characteristics and outcomes of 21 in vivo animal studies analysed. Animal studies examined the role of NO treatments in anesthetised animals subjected to ischemia/reperfusion with infarct size assessment as a major endpoint. The species used were cat, dog, mouse, rat, rabbit and pig. In the majority of studies, myocardial ischemia was induced by reversible ligation of the left anterior descending coronary artery (or similar in rodents). A few studies used alternative methods for induction of ischemia including clamping of the aorta or inflation of a balloon in the coronary artery. Duration of ischemia ranged from 30 to 120 min with reperfusion lasting between 2 and 24 h. Injury was assessed by infarct size determination, predominantly by TTC contrast staining.
Six studies administered inhaled NO (iNO), six administered sodium nitrite (NaNO2), five administered novel organic NO donors, two administered ONOO−, one administered sodium nitroprusside (SNP) and one administered nitroglycerin (NTG). Routes of administration of these agents included inhaled, intravenous and intraventricular administration, with timings of administration ranging from time points during ischemia but before reperfusion, to 10 s after the point of reperfusion. The concentration of NO treatments varied according to which agent was used (e.g. iNO 20–80 ppm, NaNO2 2.4 nmol–12.5 mmol kg/h, ONOO− 0.2–20 µM).
Meta-analysis of all experimental studies suggests that infarct size was limited compared to relative controls [mean difference of −17.93 % (95 % confidence interval: −22.05, −13.81)] (Fig. 2) except when NTG was administered. Sensitivity analysis demonstrated that grouping of publications to animal model or specific NOx had little effect on the outcome of the analysis (data not shown). Statistical heterogeneity was high in all sub-group analysis, yet the mean difference in effect size was consistently similar.

Infarct size in animal models in groups treated with NOx adjuncts compared to control experiments. N. B. NTG treatment for Liu et al. [37] not reported as no separate control group
Characteristics of human clinical studies
The characteristics and outcomes of the three clinical studies which met the criteria for analysis are summarised in Table 5. The earlier studies by Hildebrandt et al. [21] and Morris et al. [37] administered isosorbide dinitrate over 24–48 h whilst in the most recent NIAMI study [59] NaNO2 was administered as a bolus. Reperfusion therapy in the earlier studies was carried out by thrombolysis in contrast to the NIAMI trial in which patients received PPCI 5 min after sodium nitrite. There was no reduction in infarct size in human studies following NOx administration.
Discussion
Experimental animal studies
The key finding of the 21 in vivo animal studies critically reviewed is that, with the exception of NTG, NO treatment prior to or during the early reperfusion period can limit infarct size. However, considerable heterogeneity of effect was observed, related to both treatment (agent, dose, regimen) and species (notably whether collateralised or not).
Our analysis of the combined effects of all animal studies used a random-effects model and was reported as mean difference. Although random-effects models typically provide larger confidence intervals, the assumption made here was that studies were heterogeneous but effects followed some distribution. Indeed the analytical approach here provides an answer to the question “what is the average intervention effect?” The large degree of statistical heterogeneity is likely due to the differences in animal model and NO treatment utilised. However for the purposes of this review, in which we are interested in the overall picture, a summary effect of all interventions provides meaningful insight into targeting NO signalling in I/R.
Sydnonimine nitric oxide donors
Two sydnonimine NO donors, C87-3754 and SIN-1, produced a marked reduction in infarct size compared to both conventional controls and non-NO donating analogues [29, 60] suggesting that protection is afforded by NO, when administered at relatively low doses (1 mg/kg/h IV). However both studies were conducted in cats, a species with a collateralised coronary circulation [34]. Collateralisation does not completely prevent infarction, but may alter processes during early ischemia [15] so modifying infarct size. Conversion of sydnonimines to release NO is sensitive to low pH, conditions found during early reperfusion [54]. Their use in contemporary studies is limited and haemodynamic profile in I/R unreported, however treatment exhibits a reduction in endothelial dysfunction, likely caused by NO quenching of free radical species [60].
Inhaled gaseous NO
iNO significantly reduced infarct size at concentrations ranging from 40 to 80 ppm [16, 32, 38–40, 56] as well as decreasing creatine kinase (CK) concentrations and rate of apoptosis of cardiomyocytes [32] which was seen even when iNO was administered during short periods (e.g. 5 min prior to reperfusion) [39]. However beneficial effects were not seen when iNO was administered immediately before reperfusion. Therefore it is possible bioactive carriers of NO, such as nitrite [12] and S-nitrosylated [62] proteins, provide protective effects rather than molecular NO itself. Indeed the mechanism by which iNO is converted to a more stable nitrogen oxide molecule before entering the blood stream and eliciting extra-pulmonary effects remains to be fully elucidated [41]. The suitability of inhaled NO as an adjunct to reperfusion in the clinic is therefore questionable.
Nitrite
NO2 − was shown to exert a dose dependent infarct-limiting effect, which peaked at 48 nmol when administered intraventricularly, providing significant reduction in infarct size compared to control [8]. However, the control treatment used in this study was NO3 −, which was previously shown to exert a beneficial effect at high doses [27]. A contemporary study by the same group using similar timings of reperfusion showed comparable infarct size for a vehicle control group, suggesting that NO3 − at a concentration of 48 nmol had no cardioprotective effect over control. These results are corroborated by a more recent study by Hendgen-Cotta et al. who further demonstrated that 48 nmol NaNO2 − could limit infarct size in mice [17].
When NO2 − was co-administered with an NO scavenger, cardioprotection was abolished, suggesting the beneficial effects are NOS independent but NO-dependent [8]. However, despite studies showing NO2 − to be beneficial, when administered at the point of reperfusion it exerted no significant effect on infarct size when administered immediately after reperfusion [2] yet LV function after AMI was preserved [64]. This may be due to a difference in timing of administration, or possibly due to differences between rodents and dogs; the latter have a variably collateralised coronary circulation. Another possible interpretation may be the time for the nitrite species to be converted into a cytoprotective nitrogen oxide species if the mechanism of cyoprotection is not mediated by s-nitrosylation (for a comprehensive review of nitrite mediated protection the reader is directed to Rassaf et al. [52]). Acidified NaNO2 and NO in solution have also been demonstrated to limit infarct size in feline models of LAD occlusion [25, 26].
Peroxynitrite
ONOO− is formed when NO reacts with O2 − [35] and shows protective effects when administered at low micromolar concentrations while increasing infarct size at higher concentrations [44]. Maximal physiological concentrations have been previously documented in the order of 2–5 µM [44, 45]. A significant reduction in infarct size was observed when ONOO− was administered via intraventricular infusion. However when infused intravenously no cardioprotection was afforded [45], suggesting ONOO− acts locally rather than systemically. Furthermore, the short half-life and immediate interaction with plasma proteins such as glutathione would suggest that intravenous injection would fail to elicit the same response. Production of S-nitrosothiols from ONOO− to from more stable nitrogen oxide resevoirs is a possible mechanism for affording cytoprotection [43]. The generation of ONOO− during early reperfusion from ROS and NO and further ROS induced ROS release suggest that ONOO− may not be suitable as a therapeutic agent.
Other nitric oxide donor compounds
Several studies have suggested that novel NO donors may have advantages, such as increased potency and reduced tolerance compared to traditional NO donors [3, 31]. However whether this is of relevance to the setting of ischemia/reperfusion is unclear, as generally agents are not administered over long periods of time. Nevertheless all studies using other donors showed a significant reduction in infarct size [29–31, 47, 60]. There were however discrepancies in the results with respect to neutrophil accumulation and activation: this was seen in all the other NO donor studies, except the work by Siegfried et al. [60], and the animal model used (feline or canine) is a potentially confounding factor. Lefer et al. [29, 30] diverted coronary collateral blood flow away from the ischaemic area by inserting an open cannula through the arteriotomy distal to the occluded LAD and therefore suggested that the protective effect occurred independently of collateral blood flow. However other studies that utilised feline myocardial models failed to measure collateral flow and so it is difficult to conclude whether this would have contributed to infarct limitation at reperfusion. It may therefore be more appropriate to consider these agents with respect to a more representative animal model, such as pig in the future.
Traditional nitric oxide donating compounds
In two studies, NTG did not reduce infarct size when administered at reperfusion [32, 53] which may be due to tolerance induced through continuous infusion or due to a relative reduction in its bioavailability [32]. There is sustained contradiction as to precisely how NTG causes vasodilation via NO signalling i.e. cGMP or nitrosylation. At clinical plasma concentrations evidence suggests that free NO is not released [46], but possibly a mechanism by which NTG nitrosylates other proteins which may lead to its vasoactive actions, a similar mechanism to that proposed for NTG tolerance following chronic administration [61]. Interestingly, NTG could afford late preconditioning in conscious rabbits, an observation that was sustained in NO tolerant rabbits [22].
Downstream targets
These data support the overriding thesis that NOx is a successful candidate for targeting the injurious effects of ischaemia reperfusion injury in animal models. Evidence that suggests that endogenous production and maintenance of cofactors of NOS are compromised during injury, and the consequential reduction in NO bioavailability further supports this rationale. Addition of both l-arganine and tetrahydrobiopterin just prior to reperfusion in both rats and swine limit infarct size [63]. Increased NO availability and the subsequent reduction in superoxide production provides favourable conditions. Arginase inhibition has similarly been shown to limit infarct size by increased NO production [13].
Modification of the electron transport chain by S-nitrosation has also been well documented as a means of cytoprotection, ultimately inhibiting mitochondrial transition pore opening and reducing cyctochrome-c release [17, 58]. The reduction in pH and hypoxic environment during ischaemia favours nitrite reduction providing an environment suitable for NO2 − to afford infarct limitation by targeting complex I. Furthermore, NO has been shown to regulate the respiratory complexes and improve myocardial oxygen consumption [4]. Cyclophilin D can be S-nitrosylated at Cys203 which results in a reduction in mPTP opening in mouse fibroblasts, which is critical in reducing cell death [42].
Human clinical trials
Three high quality clinical studies which met the criteria for inclusion were identified. The primary endpoint in all three studies was infarct size; there was no evidence of infarct size reduction in patients treated with NO compounds immediately prior to reperfusion. There was a considerable period of time between the earliest study in 1992 and the most recent study in 2014. Measurement of infarct size in each of the studies was performed in a different way. Enzyme release into plasma was used in the earlier studies to measure CK-MB or HBDH [21, 37] whilst cardiac magnetic ressonance (CMR) was used in the 2014 NIAMI trial [59]. Unlike the experimental setting where infarct size measurement is reliably measured by post mortem histological staining and direct imaging techniques, there is as yet no consistent, gold standard technique for assessing infarct size relative to risk zone size in the clinical setting [20].
A reperfusion protocol formed part of the inclusion criteria in this review. However both Hildebrandt et al. [21] and Morris et al. [37] performed subgroup analysis on patients in which thrombolysis was ineffective or reperfusion was limited. Hildebrandt et al. [21] reported in this sub group of patients that isosorbide dinitrate did afford some infarct limitation. Morris et al. [37] however, suggest that in their sub-group analysis of patients with incomplete reperfusion, judged by ST segment resolution, isosorbide dinitrate had no effect on infarct size. They further reported that patients with an intermediate ST elevation benefited from isosorbide dinitrate in contrast to patients with large ST elevation in which isosorbide dinitrate was deleterious. Siddiqi et al. [59] reported that infarct size in their patients was relatively large compared to placebo treated patients in a remote conditioning study from 2010, yet there was no relationship between patients with smaller or larger infarcts, varying risk areas or chest pain duration.
All clinical studies were conducted double-blind. In all studies patient populations were heterogeneous, with similar mean ages and sex distribution. In each of these studies, infarct size, time to reperfusion, age, and the presence of comorbidities was variable. This is a criticism of translational science generally, which may in part explain the disparity between clinical and animal studies. The animal studies included in this review reported data from healthy juvenile animals with no comorbidities and highly regulated infarct size and location. The clinical trials reviewed here, like most others, have a study cohort of patients with numerous comorbidities and, as the current clinical studies report, varying degrees of infarct size, location and indeed reperfusion success. A recent phase 2 trial which was published after our literature screening and analysis, in which 82 patients were randomized to sodium nitrite or placebo just prior to PPCI reflected the outcomes of the human trials included in this review. No reduction in infarct size was observed, measured by CK and troponin and subsequent CMR. However a reduction in major adverse cardiac events was reported [28].
The complexity and number of comorbidities that present alongside AMI create significant challenges when translating therapies to the clinic. Disruption to cytoprotecitve signalling as a consequence of metabolic disturbances and other pathological processes have commanded much interest due to unsuccessful translation of cardioprotection strategies. Indeed, of particular interest to NOx signalling is the downstream sGC associated kinase PKG. The protection afforded by NO donor SNAP was abolished in a hyperlipidaemic rat model, via possible oxidative dimerization of PKG in rats fed a cholesterol rich diet [11]. Similarly, diabetes has been shown to impair pharmacological postconditioning in an in vivo rabbit model. Isoflurane induced infarct size limitation was abrogated in hyperglycaemic rabbits, which was associated with impaired Akt/eNOS signalling [51].
Concomitant pharmacotherapy with pharmacological agents such as antihypertensives, anti-anginal drugs, lipid-lowering drugs, anti-platelet aspirin, and drugs used for the treatment of diabetes among others, modify the signalling cascades that are of interest to limit the injurious effects of AMI and may also confound clinical studies. For example, statins have been extensively studied in both animal models and in humans. Although there is a large body of evidence that suggests that many statins positively modify NO signalling via eNOS induction (comprehensively reviewed in [48]), pravastatin demonstrated opposing effects on myocardial NO levels [24]. Many of these therapies may provide protection against irreversible injury and so additional intervention will only induce small incremental limitation of infarct size [9]. At high micromolar and millimolar concentrations NO can promote cellular injury, a situation that is possible in patients being treated with polypharmacy. Therefore it is essential to define the optimum compound, formulation and dose to minimise toxicity of these compounds when administered in clinical AMI. Timing and administration route are also crucial considerations which may be possible when mechanisms are further understood.
Conclusion
All NO donor agents except NTG exhibit the potential to limit infarct size when given as adjuncts to reperfusion in various in vivo animal models of ischemia/reperfusion. Despite this there is no definitive conclusion to the exact mechanism(s) by which beneficial effects are obtained. The evidence reported in this review emphasises a disparity between preclinical animal studies and the human trials. It is clear that the preclinical models included for review here, do not reflect the complexities and heterogeneity of the human cohort. The lack of standardised infarct size measurement relative to risk zone, marked variation in time to reperfusion/intervention and variation in the ischemic territory all present challenges to assessment of adjunct therapies. Further well designed pre-clinical models which better reflect the complexities of the human setting and subsequent high quality RCTs are needed.
References
Higgins JPT, Green S (eds) (2011) Cochrane handbook for systematic reviews of interventions Version 5.1.0 [updated March 2011]. In: The Cochrane collaboration. Available via http://www.cochranehandbook.org
Baker JE, Su J, Fu X, Hsu A, Gross GJ, Tweddell JS, Hogg N (2007) Nitrite confers protection against myocardial infarction: role of xanthine oxidoreductase, NADPH oxidase and K(ATP) channels. J Mol Cell Cardiol 43:437–444. doi:10.1016/j.yjmcc.2007.07.057
Bohn H, Brendel J, Martorana PA, Schonafinger K (1995) Cardiovascular actions of the furoxan CAS 1609, a novel nitric oxide donor. Br J Pharmacol 114:1605–1612. doi:10.1111/j.1476-5381.1995.tb14946.x
Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cocheme HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RAJ, Krieg T, Brookes PS, Murphy MP (2013) Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med 19:753–759. doi:10.1038/nm.3212
Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO 3rd, Gladwin MT (2003) Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med 9:1498–1505. doi:10.1038/nm954
Curtis E, Hsu LL, Noguchi AC, Geary L, Shiva S (2012) Oxygen regulates tissue nitrite metabolism. Antioxid Redox Signal 17:951–961. doi:10.1089/ars.2011.4242
Dezfulian C, Raat N, Shiva S, Gladwin MT (2007) Role of the anion nitrite in ischemia-reperfusion cytoprotection and therapeutics. Cardiovasc Res 75:327–338. doi:10.1016/j.cardiores.2007.05.001
Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, Gladwin MT, Lefer DJ (2005) Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest 115:1232–1240. doi:10.1172/JCI22493
Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R (2014) Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev 66:1142–1174. doi:10.1124/pr.113.008300
Ferdinandy P, Schulz R (2003) Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br J Pharmacol 138:532–543. doi:10.1038/sj.bjp.0705080
Giricz Z, Görbe A, Pipis J, Burley DS, Ferdinandy P, Baxter GF (2009) Hyperlipidaemia induced by a high-cholesterol diet leads to the deterioration of guanosine-3′,5′-cyclic monophosphate/protein kinase G-dependent cardioprotection in rats. Br J Pharmacol 158:1495–1502. doi:10.1111/j.1476-5381.2009.00424.x
Gladwin MT, Schechter AN (2004) NO contest: nitrite versus S-nitroso-hemoglobin. Circ Res 94:851–855. doi:10.1161/01.RES.0000126697.64381.37
Gonon AT, Jung C, Katz A, Westerblad H, Shemyakin A, Sjoquist PO, Lundberg JO, Pernow J (2012) Local arginase inhibition during early reperfusion mediates cardioprotection via increased nitric oxide production. PLoS One 7:e42038. doi:10.1371/journal.pone.0042038
Halestrap AP, Kerr PM, Javadov S, Woodfield KY (1998) Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim Biophys Acta 1366:79–94
Harken AH, Simson MB, Haselgrove J, Wetstein L, Harden WR 3rd, Barlow CH (1981) Early ischemia after complete coronary ligation in the rabbit, dog, pig, and monkey. Am J Physiol 241:H202–H210
Hataishi R, Rodrigues AC, Neilan TG, Morgan JG, Buys E, Shiva S, Tambouret R, Jassal DS, Raher MJ, Furutani E, Ichinose F, Gladwin MT, Rosenzweig A, Zapol WM, Picard MH, Bloch KD, Scherrer-Crosbie M (2006) Inhaled nitric oxide decreases infarction size and improves left ventricular function in a murine model of myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 291:H379–H384. doi:10.1152/ajpheart.01172.2005
Hendgen-Cotta UB, Merx MW, Shiva S, Schmitz J, Becher S, Klare JP, Steinhoff HJ, Goedecke A, Schrader J, Gladwin MT, Kelm M, Rassaf T (2008) Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc Natl Acad Sci USA 105:10256–10261. doi:10.1073/pnas.0801336105
Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116:674–699. doi:10.1161/circresaha.116.305348
Heusch G, Post H, Michel MC, Kelm M, Schulz R (2000) Endogenous nitric oxide and myocardial adaptation to ischemia. Circ Res 87:146–152. doi:10.1161/01.res.87.2.146
Heusch P, Nensa F, Heusch G (2015) Is MRI really the gold standard for the quantification of salvage from myocardial infarction? Circ Res 117:222–224. doi:10.1161/circresaha.117.306929
Hildebrandt P, Torp-Pedersen C, Joen T, Iversen E, Jensen G, Jeppesen D, Melchior T, Schytten HJ, Ringsdal V, Jensen J et al (1992) Reduced infarct size in nonreperfused myocardial infarction by combined infusion of isosorbide dinitrate and streptokinase. Am Heart J 124:1139–1144
Hill M, Takano H, Tang X-L, Kodani E, Shirk G, Bolli R (2001) Nitroglycerin induces late preconditioning against myocardial infarction in conscious rabbits despite development of nitrate tolerance. Circulation 104:694–699. doi:10.1161/hc3201.092218
Hoshida S, Yamashita N, Igarashi J, Nishida M, Hori M, Kamada T, Kuzuya T, Tada M (1995) Nitric oxide synthase protects the heart against ischemia-reperfusion injury in rabbits. J Pharmacol Exp Ther 274:413–418
Ikeda U, Shimpo M, Ikeda M, Minota S, Shimada K (2001) Lipophilic statins augment inducible nitric oxide synthase expression in cytokine-stimulated cardiac myocytes. J Cardiovasc Pharmacol 38:69–77
Johnson G III, Tsao P, Lefer AM (1990) Synergism between superoxide-dismutase and sodium-nitrite in cardioprotection following ischemia and reperfusion. Am Heart J 119:530–537. doi:10.1016/S0002-8703(05)80275-3
Johnson G III, Tsao PS, Lefer AM (1991) Cardioprotective effects of authentic nitric oxide in myocardial ischemia with reperfusion. Crit Care Med 19:244–252
Johnson G III, Tsao PS, Mulloy D, Lefer AM (1990) Cardioprotective effects of acidified sodium nitrite in myocardial ischemia with reperfusion. J Pharmacol Exp Ther 252:35–41
Jones DA, Pellaton C, Velmurugan S, Rathod KS, Andiapen M, Antoniou S, van Eijl S, Webb AJ, Westwood MA, Parmar MK, Mathur A, Ahluwalia A (2015) Randomized phase 2 trial of intracoronary nitrite during acute myocardial infarction. Circ Res 116:437–447. doi:10.1161/CIRCRESAHA.116.305082
Lefer AM, Siegfried MR, Ma XL (1993) Protection of ischemia-reperfusion injury by sydnonimine NO donors via inhibition of neutrophil-endothelium interaction. J Cardiovasc Pharmacol 22(Suppl 7):S27–S33
Lefer DJ, Nakanishi K, Johnston WE, Vinten-Johansen J (1993) Antineutrophil and myocardial protecting actions of a novel nitric oxide donor after acute myocardial ischemia and reperfusion of dogs. Circulation 88:2337–2350
Lefer DJ, Nakanishi K, Vinten-Johansen J (1993) Endothelial and myocardial cell protection by a cysteine-containing nitric oxide donor after myocardial ischemia and reperfusion. J Cardiovasc Pharmacol 22(Suppl 7):S34–S43
Liu X, Huang Y, Pokreisz P, Vermeersch P, Marsboom G, Swinnen M, Verbeken E, Santos J, Pellens M, Gillijns H, Van de Werf F, Bloch KD, Janssens S (2007) Nitric oxide inhalation improves microvascular flow and decreases infarction size after myocardial ischemia and reperfusion. J Am Coll Cardiol 50:808–817. doi:10.1016/j.jacc.2007.04.069
Martin C, Schulz R, Post H, Boengler K, Kelm M, Kleinbongard P, Gres P, Skyschally A, Konietzka I, Heusch G (2007) Microdialysis-based analysis of interstitial NO in situ: NO synthase-independent NO formation during myocardial ischemia. Cardiovasc Res 74:46–55. doi:10.1016/j.cardiores.2006.12.020
Maxwell MP, Hearse DJ, Yellon DM (1987) Species variation in the coronary collateral circulation during regional myocardial ischaemia: a critical determinant of the rate of evolution and extent of myocardial infarction. Cardiovasc Res 21:737–746
Miles AM, Bohle DS, Glassbrenner PA, Hansert B, Wink DA, Grisham MB (1996) Modulation of superoxide-dependent oxidation and hydroxylation reactions by nitric oxide. J Biol Chem 271:40–47
Moher D, Liberati A, Tetzlaff J, Altman DG, The PG (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 6:e1000097. doi:10.1371/journal.pmed.1000097
Morris JL, Zaman AG, Smyllie JH, Cowan JC (1995) Nitrates in myocardial infarction: influence on infarct size, reperfusion, and ventricular remodelling. Br Heart J 73:310–319
Nagasaka Y, Buys ES, Spagnolli E, Steinbicker AU, Hayton SR, Rauwerdink KM, Brouckaert P, Zapol WM, Bloch KD (2011) Soluble guanylate cyclase-α1 is required for the cardioprotective effects of inhaled nitric oxide. Am J Physiol Heart Circ Physiol. doi:10.1152/ajpheart.00948.2010
Nagasaka Y, Fernandez BO, Garcia-Saura MF, Petersen B, Ichinose F, Bloch KD, Feelisch M, Zapol WM (2008) Brief periods of nitric oxide inhalation protect against myocardial ischemia-reperfusion injury. Anesthesiology 109:675–682. doi:10.1097/ALN.0b013e318186316e
Neye N, Enigk F, Shiva S, Habazettl H, Plesnila N, Kuppe H, Gladwin MT, Kuebler WM (2012) Inhalation of NO during myocardial ischemia reduces infarct size and improves cardiac function. Intensive Care Med 38:1381–1391. doi:10.1007/s00134-012-2605-1
Ng ESM, Jourd’heuil D, McCord JM, Hernandez D, Yasui M, Knight D, Kubes P (2004) Enhanced S-nitroso-albumin formation from inhaled NO during ischemia/reperfusion. Circ Res 94:559–565. doi:10.1161/01.res.0000117771.63140.d6
Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E (2011) Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem 286:40184–40192. doi:10.1074/jbc.M111.243469
Nossuli TO, Hayward R, Jensen D, Scalia R, Lefer AM (1998) Mechanisms of cardioprotection by peroxynitrite in myocardial ischemia and reperfusion injury. Am J Physiol 275:H509–H519
Nossuli TO, Hayward R, Scalia R, Lefer AM (1998) Cellular mechanisms of cardioprotection by peroxynitrite in myocardial ischemia and reperfusion injury. Faseb J 12:A1113–A1113
Nossuli TO, Hayward R, Scalia R, Lefer AM (1997) Peroxynitrite reduces myocardial infarct size and preserves coronary endothelium after ischemia and reperfusion in cats. Circulation 96:2317–2324. doi:10.1161/01.cir.96.7.2317
Núñez C, Víctor VM, Tur R, Alvarez-Barrientos A, Moncada S, Esplugues JV, D’Ocón P (2005) Discrepancies between nitroglycerin and NO-releasing drugs on mitochondrial oxygen consumption, vasoactivity, and the release of NO. Circ Res 97:1063–1069. doi:10.1161/01.res.0000190588.84680.34
Pabla R, Buda AJ, Flynn DM, Salzberg DB, Lefer DJ (1995) Intracoronary nitric-oxide improves postischemic coronary blood-flow and myocardial contractile function. Am J Physiol Heart Circ Physiol 269:H1113–H1121
Pechánová O, Varga ZV, Cebová M, Giricz Z, Pacher P, Ferdinandy P (2015) Cardiac NO signalling in the metabolic syndrome. Br J Pharmacol 172:1415–1433. doi:10.1111/bph.12960
Penna C, Mancardi D, Rastaldo R, Pagliaro P (2009) Cardioprotection: a radical view. Free radicals in pre and postconditioning. Biochimica et Biophysica Acta Bioenerg 1787:781–793. doi:10.1016/j.bbabio.2009.02.008
Piper HM, Abdallah Y, Schafer C (2004) The first minutes of reperfusion: a window of opportunity for cardioprotection. Cardiovasc Res 61:365–371. doi:10.1016/j.cardiores.2003.12.012
Raphael J, Gozal Y, Navot N, Zuo Z (2010) Hyperglycemia inhibits anesthetic-induced postconditioning in the rabbit heart via modulation of phosphatidylinositol-3-kinase/Akt and endothelial nitric oxide synthase signaling. J Cardiovasc Pharmacol 55:348–357. doi:10.1097/FJC.0b013e3181d26583
Rassaf T, Ferdinandy P, Schulz R (2014) Nitrite in organ protection. Br J Pharmacol 171:1–11. doi:10.1111/Bph.12291
Salloum FN, Takenoshita Y, Ockaili RA, Daoud VP, Chou E, Yoshida KI, Kukreja RC (2007) Sildenafil and vardenafil but not nitroglycerin limit myocardial infarction through opening of mitochondrial K-ATP channels when administered at reperfusion following ischemia in rabbits. J Mol Cell Cardiol 42:453–458. doi:10.1016/j.yjmcc.2006.10.015
Schönafinger K (1999) Heterocyclic NO prodrugs. Il Farmaco 54:316–320. doi:10.1016/S0014-827X(99)00031-2
Schwartz BG, Kloner RA (2012) Coronary no reflow. J Mol Cell Cardiol 52:873–882. doi:10.1016/j.yjmcc.2011.06.009
Shinbo T, Kokubo K, Sato Y, Hagiri S, Hataishi R, Hirose M, Kobayashi H (2013) Breathing nitric oxide plus hydrogen gas reduces ischemia-reperfusion injury and nitrotyrosine production in murine heart. Am J Physiol Heart Circ Physiol 305:H542–H550. doi:10.1152/ajpheart.00844.2012
Shiva S, Sack MN, Duranski M, Ringwood LA, Wang X, Raghavachari N, Macarthur P, Burwell L, Brookes PS, Lefer DJ, Gladwin MT (2006) Nitrite mediates both classical and long term preconditioning from ischemia/reperfusion injury at the mitochondrial level. Free Radic Biol Med 41:S62–S62. doi:10.1084/jem.20070198
Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, Wang X, MacArthur PH, Shoja A, Raghavachari N, Calvert JW, Brookes PS, Lefer DJ, Gladwin MT (2007) Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med 204:2089–2102. doi:10.1084/jem.20070198
Siddiqi N, Neil C, Bruce M, MacLennan G, Cotton S, Papadopoulou S, Feelisch M, Bunce N, Lim PO, Hildick-Smith D, Horowitz J, Madhani M, Boon N, Dawson D, Kaski JC, Frenneaux M, investigators N (2014) Intravenous sodium nitrite in acute ST-elevation myocardial infarction: a randomized controlled trial (NIAMI). Eur Heart J 35:1255–1262. doi:10.1093/eurheartj/ehu096
Siegfried MR, Erhardt J, Rider T, Ma XL, Lefer AM (1992) Cardioprotection and attenuation of endothelial dysfunction by organic nitric oxide donors in myocardial ischemia-reperfusion. J Pharmacol Exp Ther 260:668–675
Stamler JS (2008) Nitroglycerin-mediated S-nitrosylation of proteins: a field comes full cycle. Circ Res 103:557–559. doi:10.1161/circresaha.108.184341
Stamler JS, Loscalzo J (1992) Capillary zone electrophoretic detection of biological thiols and their S-nitrosated derivatives. Anal Chem 64:779–785
Tratsiakovich Y, Gonon AT, Kiss A, Yang J, Bohm F, Tornvall P, Settergren M, Channon KM, Sjoquist PO, Pernow J (2013) Myocardial protection by co-administration of l-arginine and tetrahydrobiopterin during ischemia and reperfusion. Int J Cardiol 169:83–88. doi:10.1016/j.ijcard.2013.08.075
Tripathi Y, Hegde BM, Raghuveer CV (1997) Effect of superoxide dismutase and acidified sodium nitrite on infarct size following ischemia and reperfusion in dogs. Indian J Physiol Pharmacol 41:248–256
Van de Werf F, Ardissino D, Betriu A, Cokkinos DV, Falk E, Fox KAA, Julian D, Lengyel M, Neumann F-J, Ruzyllo W, Thygesen C, Underwood SR, Vahanian A, Verheugt FWA, Wijns W (2003) Management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J 24:28–66. doi:10.1016/s0195-668x(02)00618-8
Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A (2004) Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc Natl Acad Sci USA 101:13683–13688. doi:10.1073/pnas.0402927101
Acknowledgments
The authors gratefully acknowledge the advice on study design and analysis provided by Dr Helen Morgan in Cardiff University’s Specialist Unit for Review Evidence (SURE).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bice, J.S., Jones, B.R., Chamberlain, G.R. et al. Nitric oxide treatments as adjuncts to reperfusion in acute myocardial infarction: a systematic review of experimental and clinical studies. Basic Res Cardiol 111, 23 (2016). https://doi.org/10.1007/s00395-016-0540-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-016-0540-y