
Abstract
Cardiac arrhythmias remain a common challenge and are associated with significant morbidity and mortality. Effective and safe rhythm control strategies are a primary, yet unmet need in everyday clinical practice. Despite significant pharmacological and technological advances, including catheter ablation and device-based therapies, the development of more effective alternatives is of significant interest to increase quality of life and to reduce symptom burden, hospitalizations and mortality. The mechanistic understanding of pathophysiological pathways underlying cardiac arrhythmias has advanced profoundly, opening up novel avenues for mechanism-based therapeutic approaches. Current management of arrhythmias, however, is primarily guided by clinical and demographic characteristics of patient groups as opposed to individual, patient-specific mechanisms and pheno-/genotyping. With this state-of-the-art paper, the Working Group on Cellular Electrophysiology of the German Cardiac Society aims to close the gap between advanced molecular understanding and clinical decision-making in cardiac electrophysiology. The significance of cellular electrophysiological findings for clinical arrhythmia management constitutes the main focus of this document. Clinically relevant knowledge of pathophysiological pathways of arrhythmias and cellular mechanisms of antiarrhythmic interventions are summarized. Furthermore, the specific molecular background for the initiation and perpetuation of atrial and ventricular arrhythmias and mechanism-based strategies for therapeutic interventions are highlighted. Current “hot topics” in atrial fibrillation are critically appraised. Finally, the establishment and support of cellular and translational electrophysiology programs in clinical rhythmology departments is called for to improve basic-science-guided patient management.
Similar content being viewed by others

Introduction
Cardiac arrhythmias remain a clinical challenge and source of morbidity and mortality in developed countries [1]. Research into molecular mechanisms of arrhythmias began more than 40 years ago with the discovery of the cellular mechanisms of delayed afterdepolarization-induced triggered activity and the mechanisms of reentry [2]. Since then, there has been great progress in the understanding of the molecular basis of arrhythmias, fueled by technological advances and new experimental methods ranging from cellular electrophysiology, genetics and genomics to high-resolution microscopy.
In parallel, there have been amazing considerable developments in treatment options with significant pharmacological and technological advances, including catheter ablation and device-based therapies [3,4,5,6,7,8]. However, progress has been largely based on empirical rather than targeted mechanism-based approaches. The rapid progress in device and ablation therapies compared to the relatively poor outcome of pharmacological rhythm control treatment may have reduced the interaction between clinical electrophysiologists—whose primary interest is improving the health and quality of life of patients—and basic electrophysiologists, who are driven by novel scientific discoveries, which may take years to reach the clinical setting [2].
This state-of-the-art paper summarizes the current knowledge about basic cellular electrophysiology and its clinical implications. We will highlight examples where basic cellular electrophysiology has contributed to the development of novel translational therapeutic strategies. Finally, we will emphasize the importance and advantage of a close interaction between clinical and basic electrophysiologists, in order to solve current and future challenges in cardiac arrhythmia management [1].
Fundamental mechanisms of cardiac arrhythmias
Pronounced heterogeneities exist in mechanisms, presentation, treatment and outcome, between different cardiac arrhythmias and between patients with the same type of arrhythmia. Nonetheless, basic research has identified several important fundamental mechanisms of cardiac arrhythmogenesis [9]. In particular, arrhythmias require a vulnerable substrate characterized by structural or electrical abnormalities and an acute initiating event. Both components can be genetic or, more commonly, acquired due to advancing age or concomitant cardiovascular risk factors and can promote abnormal impulse formation (ectopic activity) and/or abnormal impulse conduction (resulting in reentrant activity), which are considered the major fundamental mechanisms of arrhythmia.
Ectopic activity and reentry
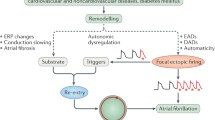
Ectopic activity is the local generation of action potentials (APs) outside the normal activation sequence, which can serve as an initiator of reentry-mediated arrhythmias or maintain the fibrillatory process when occurring repetitively at high frequency. Polymorphic ventricular tachycardia (VT) or ventricular fibrillation often originate from the Purkinje system, which is characterized by distinctive electrophysiological characteristics that promote the generation of ectopic excitations and which can be targeted by catheter ablation [10]. In addition, ectopic excitations may trigger premature ventricular contractions which worsen myocardial function and contribute to increased mortality in patients with structural heart disease [11]. Mechanistically, ectopic activity results from abnormal automaticity or triggered activity resulting from early afterdepolarizations (EADs) or delayed afterdepolarizations (DADs) [12] (Fig. 1). Abnormal automaticity, i.e. spontaneous AP generation from normally quiescent tissue, is often mediated by a reduction in ion currents that stabilize the resting membrane potential (RMP; e.g. the basal inward-rectifier potassium current, IK1) or an increased activity of hyperpolarization-activated pacemaker channels responsible for the funny current, If. On the other hand, EADs typically result from excessive AP duration (APD), providing time for reactivation of L-type calcium channels. Finally, DADs result from calcium-handling abnormalities, whereby spontaneous calcium-release events from the sarcoplasmic reticulum (SR) through so-called ryanodine receptor type-2 (RyR2) channels activate a transient-inward current, mediated largely by the sodium/calcium-exchanger (NCX), which depolarizes RMP and can trigger a new AP. An increased incidence of spontaneous calcium-release events can be due to RyR2 dysfunction or increased SR calcium load. A classical example of the latter is arrhythmic activity induced by cardiac glycoside intoxication, where inhibition of the sodium–potassium-ATPase increases the intracellular sodium concentration. This leads to increased cytosolic calcium by reducing the driving force of calcium efflux via NCX at resting membrane potential and favoring reverse-mode NCX-mediated calcium entry during the AP. The resulting increased SR calcium load not only explains the positive inotropic effect of cardiac glycosides but also favours spontaneous calcium release and DADs [13, 14].

(Replotted with kind permission from [89])
Cellular mechanisms underlying ectopic activity. A Abnormal automaticity is caused by spontaneous diastolic depolarization to a threshold value for activation. B, C Abnormal membrane depolarizations preceding full repolarization (B early afterdepolarizations [EADs]) or after completion of the action potential (C delayed afterdepolarizations [DADs]) may also cause spontaneous activity.
Reentry is considered the primary arrhythmia-maintaining mechanism [15]. Conceptually, reentry describes a stable circulating excitation. Reentry can occur around an anatomically defined inexcitable core when the product of the effective refractory period (ERP) and conduction velocity (the so-called wavelength) is sufficiently small, allowing the tissue to become re-excitable before the reentering impulse arrives. As such, structural remodelling, particularly fibrosis, strongly promotes reentry. Reentry may also occur on purely a functional basis (Fig. 2). The classical “leading-circle” concept describes reentry around a continuously refractory core with a circuit size equal to the wavelength [15]. However, the notion that sodium channel-blocking agents are effective in cardioversion of atrial fibrillation (AF) challenged this concept, since conduction slowing is believed to stabilize reentry acording to the “leading-circle” concept rather than suppressing it. Based on this paradox the “spiral wave” concept has been developed, wherein reentry proceeds around an excitable but continuously unexcited center and maintenance of reentry depends on the balance between the wavefront’s excitatory currents (the ‘source’) and the properties of the unexcited tissue (the ‘sink’) [16,17,18] .
Spiral wave reentry, often visualized as rotors, represents a major pathophysiological concept in AF and rotor ablation has been suggested as a potential ablation strategy of persistent AF [19, 20], which is presently evaluated in various clinical trials (ClinicalTrials.gov numbers: NCT02101541, NCT02274857, NCT01924377, NCT02386345, NCT02497248, NCT02113761).
Slow conduction
Cardiac fibrosis is a hallmark of structural remodelling, promoting slow, heterogeneous conduction and spatial dispersion of repolarization by isolating muscle-bundles. Advances in clinical imaging methods have helped to establish the important role of fibrosis in arrhythmogenesis [12]. In parallel, basic science has identified the mechanisms underlying the proliferation of fibroblasts and their differentiation into myofibroblasts, which is a central response to numerous stress signals activated in many cardiovascular diseases, resulting in excessive collagen production and fibrosis [21]. Experimental models have identified the major underlying signalling pathways and have indicated that targeting these pathways, either using clinically approved drugs (e.g. angiotensin-converting enzyme inhibitors, aldestorone antagonists) or through new targets (e.g. microRNAs such as miR-21 or miR-29) may limit the development of a proarrhythmic substrate [21]. However, there is increasing awareness that such therapy would need to be applied early in the disease process, before extensive fibrosis occurs. Other observations suggest that fibrosis and structural alteration can be partly reversible, if the underlying condition is effectively treated.
Reentry-promoting slow conduction is also mediated by gap-junction remodelling, resulting in impaired electrical cell-to-cell coupling. Preclinical studies have shown that the expression of connexin-43, the predominant constituent of ventricular gap-junction channels, is reduced under pathological conditions (e.g. post-myocardial infarction), contributing to slowed conduction, and that gene transfer of connexin-43 reduces the inducibility of ventricular tachycardia in pigs, providing an important proof-of-concept for the antiarrhythmic effects of targeting the mechanisms underlying slow conduction [22].
Alternans
Cardiac alternans is evident on the surface ECG as beat-to-beat alternations in the ST segment or the T-wave. At the cellular level, APD alternans underlies T-wave alternans (TWA) in the surface ECG. When APD alternans become spatially discordant, a small reduction of cycle length may result in unidirectional block followed by reentry and initiation of ventricular fibrillation, thus providing a mechanistic link between TWA and sudden cardiac death (SCD) [23]. Basic science has revealed that cardiac alternans is often caused by perturbed cellular calcium signalling [24]. In support of this notion, normalization of SR calcium handling in animal models of heart failure (HF) has been shown to reduce the incidence of cardiac alternans and ventricular arrhythmias [25]. Thus, normalization of RyR2 function and SR calcium handling may be promising targets to prevent development of alternans and SCD.
Inherited channelopathies
Inherited channelopathies are prime examples of cardiac diseases in which basic science has provided important mechanistic understanding of pathophysiology. This has led to (1) the development of specific diagnostic tests such as the ajmaline challenge in Brugada syndrome [26] and QTc evaluation during stress test in long and short QT syndromes (LQTS, SQTS), and importantly (2) the continued exploration and development of mechanism-based therapeutic approaches such as late sodium channel block in LQT3 [27] and the use of flecainide in catecholaminergic polymorphic ventricular tachycardia (CPVT) [28] which have already partially entered the clinical guidelines [29].
Long and short QT syndrome
Both, pathological prolongation (LQTS) and acceleration (SQTS) of cardiac repolarization can predispose patients to atrial and ventricular tachycardia (AT/VT) and SCD [29]. Since the link of mutations in KCNQ1 and KCNH2/HERG potassium channel and SCN5A sodium channel genes to inherited LQTS was established in the 1990s [30, 31], mutations in 15 genes have been linked to LQTS and mutations in 6 genes to SQTS [29]. An increased understanding of the biophysical consequences of these mutations (complete loss-of-function vs. change in voltage-dependence of the respective currents) and the exact location of the mutations within the channels (pore-region vs. C- and N-terminus) may have clinical relevance for risk prediction [32, 33].
Characterization of the biophysical properties of the mutated channels and their response to sympathetic activity has helped to identify the importance of EADs and sympathetic stimulation as proarrhythmic triggers, providing the scientific basis for beta-blocker therapy (or left cardiac sympathetic denervation) as a mainstay of antiarrhythmic therapy in LQTS [29].
The subsequent generation of genetic (knock-out/knock-in) and transgenic (mouse and rabbit) models of LQTS has provided important insights into the major role of spatial and temporal APD heterogeneities as an arrhythmogenic substrate [34]. Mechanisms underlying genotype-specific differences in arrhythmia initiation [sustained sympathetic activity (sports), LQT1; sudden sympathetic activation (startle), LQT2] [35] contributing to clinically observed genotype differences in the efficacy of beta-blocker therapy [36] have been identified. The detrimental role of bradycardia and short-long-short sequences as triggers for VTs has been observed in patients and confirmed in (animal) models [27, 37]. In addition, the potential of genotype-specific therapeutic approaches has been explored: in genetic murine LQT3 models, with pathologically enhanced late INa, sodium-channel blockers mexiletine, flecainide and the more selective GS967 exert a mechanism-directed, genotype-specific antiarrhythmic effect [38,39,40]. Similarly, recent clinical data demonstrate that LQT3 patients benefit from QT-shortening and anti-arrhythmic effects of sodium channel blockers mexiletine, flecainide, ranolazine and eleclazine [41,42,43,44,45,46]. The 2015 ESC guidelines for the prevention of SCD thus mention sodium-channel blockers as a potential add-on therapy in patients with LQT3 [47]. Moreover, proarrhythmic effects of estradiol due to increased APD heterogeneities and EADs have been revealed in transgenic LQT2 rabbit models, contrasting with anti-arrhythmic, protective effects of progesterone due to shortening of cardiac refractoriness and reduced EAD formation [48]. These data suggest progesterone-based therapies might constitute novel antiarrhythmic approaches in female LQTS patients.
Recent clinical and basic science data suggest subclinical mechanical dysfunction in the “electrical” disease LQTS due to close electro-mechanical coupling [49] and a correlation between the extent of mechanical dysfunction and arrhythmic risk, strongly suggesting that assessment of regional mechanical dysfunction may be helpful to improve future clinical risk stratification [50].
In SQTS, mechanisms of arrhythmogenesis are less well-understood than in LQTS and therefore effective antiarrhythmic treatment options are sparse. The recent development of genetic/transgenic animal models of SQTS (zebrafish, rabbit) have the potential to close this gap and help to develop mechanism-based antiarrhythmic therapeutic approaches [51].
Brugada syndrome
Brugada syndrome (BrS) is characterized by typical precordial ST-segment elevations on the surface ECG and an increased risk for SCD caused by ventricular arrhythmia [52]. The identification of SCN5A as an important disease gene [53] in BrS suggested that the syndrome was a monogenetic disorder, similar to LQTS. “Loss-of-function” mutations in other genes, implicated in cardiac sodium (GPD1L, SCN1B, SCN3B) or calcium (CACNA1C, CACNB2) channel expression, were also associated with BrS, as well. “Gain-of-function” mutations in KCND3, by contrast, demonstrated an involvement of increased transient-outward potassium current Ito, in the pathogenesis of BrS [54, 55]. Interestingly, recent data suggested SCN10A and its gene product Nav1.8, primarily expressed in the nervous system and only to a minor extent in the heart, as an important disease gene in BrS [56, 57], although another study did not confirm this view [58]. However, only ~ 30% of BrS cases may be explained by the 22 disease-associated genes known so far, pointing to a multifactorial origin of the disorder. This view was fueled by recent findings that hearts of BrS patients often show subtle structural changes, in particular in the right ventricular outflow tract (RVOT), suggesting a distinct cardiomyopathy [59]. This is pathophysiologically important as such regions-either caused by channelopathy or even more complex mechanisms-may give rise to areas of low voltage and electrical zig-zag conduction that constitute a substrate for microreentry as the arrhythmogenic basis of the syndrome. Observations in BrS patients undergoing electrophysiological study and reports of successful radiofrequency ablation of critical areas in the RVOT [60] further substantiated this pathomechanistic idea, building the backbone of the depolarization hypothesis. Accordingly, localized slow conduction in the RVOT constitutes the origin of typical ECG changes in right precordial leads.
The repolarization hypothesis, by contrast, postulates an imbalance of ion currents during phase 1 of the right ventricular epicardial AP, mediated by transmural differences in the outward potassium current Ito and genetic or drug-induced reduction of INa, underlying the ECG pattern and the induction of arrhythmias in BrS [61].
Regardless which of the two hypotheses more precisely reflects the underlying mechanisms, it is an important finding of basic electrophysiological research that a mismatch of depolarizing current (in particular INa) and Ito, as well as distinct phases of bradycardia, are major drivers of arrhythmogenicity in BrS. Therefore, modulation of the INa/Ito relation has an important role in current clinical diagnostic risk stratification and therapeutic approaches in BrS, including (1) the application of sodium-channel blockers as a diagnostic test for inducibility of Brugada ECG (ajmaline or flecainide challenge) [26], (2) the use of quinidine to decrease arrhythmic burden in BrS patients by inhibiting Ito [62], (3) the use of isoproterenol or other β-adrenergic stimuli in arrhythmogenic storm to increase depolarizing current (in particular L-type calcium current; ICa,L) and avoid bradycardia [62], (4) avoidance of drugs that decrease INa or increase Ito (see http://www.brugadadrugs.org) and (5) avoidance of fever to prevent temperature-induced INa/Ito mismatch [63]. However, currently, the only way to safely prevent SCD is the implantation of an implantable cardioverter defibrillator (ICD), indicating an urgent need for basic and translational research to provide better mechanism-based diagnostic and therapeutic strategies.
Catecholaminergic polymorphic ventricular tachycardia
CPVT is a highly lethal inherited arrhythmogenic disorder characterized by episodes of polymorphic VT and SCD in the setting of exercise or emotional stress. In recent years significant progress has been made to understand the underlying mechanisms, which has positively impacted the therapy of CPVT patients [64]. The molecular basis of CPVT has mostly been attributed to dysfunction of the RyR2 channel, promoting calcium-handling abnormalities, DADs and triggered activity, particularly during increased sympathetic tone when SR calcium load increases and RyR2 channels become hyperphosphorylated, further promoting their dysfunction [65]. In ≈ 65% of CPVT patients, this RyR2 dysfunction is due to mutations in the RYR2 gene itself (CPVT1), but mutations in proteins regulating RyR2 function as part of the macromolecular complex that controls SR calcium release (e.g. calsequestrin-2 or calmodulin), which can indirectly promote RyR2 dysfunction, have also been described [64, 65].
Recent data from animal models of CPVT suggest that the primary defects in calcium handling may also cause sinoatrial node (SAN) dysfunction, fibrosis, atrial arrhythmias, exaggerated left ventricular (LV) hypertrophy and HF [66]. These findings may explain why some affected patients exhibit bradycardia and atrial arrhythmias, in addition to CPVT. Moreover, they provide a direct mechanistic link between a primary calcium-handling disease and structural remodelling in the heart. This might have more widespread implications for other diseases.
The identification of the underlying arrhythmogenic mechanisms has played an important role in the treatment of CPVT patients, which involves inhibition of sympathetic activity (beta-blockers or left cardiac sympathetic denervation) and the use of flecainide. The role of flecainide in CPVT represents a prime example of the interaction between basic scientists and clinicians. It was initially established in mice and applied as a proof of concept in two CPVT patients [28], but has since then become a class IIa guideline recommendation [29]. However, the exact antiarrhythmic mechanism of flecainide remains a topic of debate and may involve a combination of sodium channel block (preventing triggered activity), inhibition of RyR2 (reducing the likelihood of spontaneous SR calcium-release events), or indirect effects on intracellular calcium handling due to changes in intracellular sodium [67, 68]. Based on the underlying mechanisms of CPVT, RyR2 dysfunction is considered a promising therapeutic target. Several RyR2-stabilizing compounds, including K201/JTV-519, S107 and carvedilol-analogues, have indeed shown promise in preclinical studies [69]. Of particular clinical interest are the effects of dantrolene, an RyR-stabilizing drug approved for the treatment of malignant hyperthermia, which also has antiarrhythmic effects in CPVT patients [70, 71].
Electrical conduction defects
Cardiac conduction defect (CCD) also comprises primary genetic forms, although most cases arise from age-related fibrotic degeneration of the conduction system. The yield of genetic testing in CCD is ~ 30% [72]. Isolated CCD can be caused by mutations in SCN5A (progressive familial heart block 1a) [73] or TRPM4 (progressive familial heart block 1b) [74]. In association with cardiomyopathy, CCD was linked to LMNA [75] and PRKAG2 mutations [76]. Patients with such mutations are also at risk of ventricular arrhythmias rendering further decision-making regarding ICD implementation important. Furthermore, mutations in NKX2-5 [77] and TBX5 [78] cause CCD in association with congenital heart disease. Thus, evidence for genetic forms of CCD can significantly improve stratification of patients, as it constitutes a prognostic indicator for the course of disease with or without a syndromic co-morbidity.
Hereditary sinus node disease
Loss or dysfunction of SAN cells results in sinus node disease (SND), comprising sinus bradycardia, SAN block or arrest and bradycardia-tachycardia syndrome (BTS) [79]. In the majority of cases SND is idiopathic and occurs age-dependently either by cellular dysfunction of SAN cells or degeneration of the formerly intact SAN.
Loss-of-function mutations in SCN5A are an established pathomechanism (sick sinus syndrome 1). Electrophysiological studies and computational modelling established that mutated channels cause either abnormally slow pacemaking or produce SAN exit block [79]. Furthermore, mutations in HCN4 underlying a significant proportion of the pacemaker current If in the SAN, cause hereditary SND (sick sinus syndrome 2) and BTS, respectively [80,81,82].
HCN4 gain-of-function mutations are associated with inappropriate sinus tachycardia. Functional investigations showed that HCN4-R524Q mutant channels, heterozygously carried by affected family members, have an increased cAMP sensitivity resulting in an augmented funny-current at baseline leading to a faster resting heart rate [83]. Thus, inappropriate sinus tachycardia in part has a genetic basis, which raises the possibility of inheritable traits.
Importantly, investigation of If and its underlying ion channels, built the groundwork for ivabradine, the first clinically available If blocker targeting impulse formation in the SAN. This pharmacological mechanism is successfully used to treat patients with chronic stable angina and HF [84].
Drug-induced, reversible “channelopathies”
Various drugs can phenocopy ECG features and arrhythmias of genetic channelopathies by interacting with different cardiac ion channels, causing for example acquired LQTS and BrS-particularly in patients harbouring single-nucleotide polymorphism variants or even silent disease-causing mutations [85]. Most drugs causing acquired LQTS block HERG-encoded IKr [86]. However, drugs may also cause acquired LQTS by blocking other currents such as IKs (e.g. isoflurane) or IK1 (e.g. midazolam) [87]. Drug-induced BrS is often caused by sodium-channel-blocking class 1 drugs [85]. Overall, acquired drug-induced “channelopathies” and arrhythmias are much more prevalent than rare genetic forms and exploring their mechanisms has important pre-clinical and clinical implications to prevent adverse drug effects.
Safety pharmacology
Every new pharmacological agent under development undergoes extensive cardiac safety testing to exclude any proarrhythmic liability [86]. Until recently, cardiac safety assays predominantly involved high-throughput screening of HERG blockade early on and a so-called ‘thorough QT study’ assessing repolarization prolongation in humans later on in drug development. However, extensive basic science studies have established that HERG screening alone has limited sensitivity and specificity to identify proarrhythmic compounds, which has prompted a more integrative approach to assess the cardiac safety of new compounds. The Comprehensive In vitro Proarrhythmia Assay (CIPA) initiative [88] advocates the use of (1) screening of ion-channel-blocking effects beyond HERG alone, (2) in silico integration of these findings to assess overall effects on ventricular repolarization and (3) use of integrated biological systems such as induced pluripotent stem cell-derived cardiomyocytes (IPSC-CM).
Ventricular arrhythmia in subjects with structural heart disease
Ventricular arrhythmias remain a major contributor to increased mortality in patients with ischaemic or nonischaemic cardiomyopathy [47]. The underlying pathophysiology, risk markers and current treatment options are outlined below. Outflow tract tachycardia and idiopathic LV tachycardia are not discussed due to lack of significant basic science data.
Ischaemic cardiomyopathy (ICM)
Ischaemia results in release of reactive oxygen species, increase in intracellular sodium due to sodium/hydrogen exchange and inhibition of the sodium/potassium-ATPase triggered directly by hypoxia or secondary to ATP depletion. Within hours after ischaemia onset, associated cytokine- and chemoattractant-mediated endothelial dysfunction, apoptosis, autophagy, platelet aggregation and micromebolization and neutrophil accumulation as well as macrophage and T-cell-mediated cell-damage occur [90, 91].
While acute ischaemia/reperfusion injury primarily leads to ventricular fibrillation, myocardial infarction-related scars and especially the border zone between infarcted and vital myocardium are an important substrate for VT occurrence in ICM: infarct-related changes in excitability and conduction velocity as arrhythmogenic substrates lead to perpetual reentrant VT (see also Fig. 2).

(Adapted with kind permission from [9])
Current concepts of functional reentry. A The leading circle concept is based on reentering excitation travelling around a functional refractory and, therefore, unexcitable core. The circuit size is determined by the wavelength, i.e. the distance travelled by the cardiac impulse in one effective refractory period (ERP), given by the product of conduction velocity (CV) and ERP. B In the spiral wave concept reentry occurs around an excitable but continuously unexcited core and maintenance depends on balance of current-source/tissue-excitability (favoring propagation) and current-sink (impairing propagation).
Pharmacological treatment of ventricular fibrillation is currently limited [47]. Class I antiarrhythmic drugs are contraindicated due to increased risk for arrhythmia-associated sudden cardiac death as shown in the “Cardiac Arrhythmia Suppression Trial” (CAST) [92].
After the CAST study had abolished the use of class I antiarrhythmic drugs in ICM, several antiarrhythmic compounds underwent evaluation, as the alternative compound amiodarone, besides its antiarrhythmic potential, had been shown to have cumulative toxicity in many body organs.
The Class III antiarrhythmic compound, MS551 (nifekalant), was described to have antiarrhythmic properties shortly after the CAST results had been published [93]. This pyrimidine derivative leads to frequency-dependent AP-prolongation. It has voltage- and frequency-dependent inhibitory properties on HERG-encoded IKr with high affinity for the “open state” of IKr. As described above, this is known to possibly induce an aquired form of QT-interval prolongation with the risk for fatal polymorphic tachycardias including Torsade de Pointes. However, unlike other class III agents, it also has agonistic/facilitating effects on HERG current [94]. Nifekalant has a high potency for destabilization and early termination of spiral wave reentry and to prevent VT/ventricular fibrillation after acute myocardial infarction. It also improves electrical defibrillation efficacy in this setting [95]. However, nifekalant is only approved in Japan for the treatment of life threatening ventricular tachyarrhythmias.
Another class III antiarrhythmic compound, SSR149744C (celivarone), is, like amiodarone, a benzofuran derivative; however, unlike amiodarone, it is not iodinated and thus exerts less organ toxicity [96]. Celivarone has antiadrenergic and angiotensin II antagonistic effects and is a multi ion-channel blocker. Similar to amiodarone it is a weak sodium channel blocker with additional IKr, IKs and IK1 blocking properties, but exerts less effective IK,ACh-, IKv1.5- or ICa,L blockade [96]. In the ICARIOS-trial, celivarone showed a 46% (non-statistically significant) reduction in VT/-ventricular fibrillation-triggered ICD shock therapies; however, in the ALPHEE Study, celivarone was not effective for the prevention of ICD interventions or death [97].
To date, according to the European Society of Cardiology guidelines, only beta-blockers (in the non-acute phase of MI) and amiodarone are recommended in ischaemia-related ventricular fibrillation or VT associated with heart failure [47]. Therefore, it seems urgently necessary to search for other treatment options of ischaemic cardiomyopathy-associated arrhythmias.
A very recent and interesting approach comes from experimental data from a pig animal model which evaluated cardiac remodelling in an ischaemia/reperfusion setting with or without administration of the Class III antiarrhythmic compound dronedarone [98]. In a meticulous transcriptome profiling and combined proteome analysis of post-infarction remodelling the authors found the levels of 879 transcripts in the infarction border zone, seven transcripts in the myocardial infarction area, as well as 51 proteins in the unaffected left ventricle and 15 proteins in the border zone affected by dronedarone treatment. All findings were supported by disease/function charts and an integrated network established by combined “omics”. Table 1 shows transcriptomics with the most predominant changes in gene expression. Although dronedarone is not approved for ventricular arrhythmias, this approach highlights the importance of myocardial infarction border zone in ICM. Secondly, it may lead to similar evaluation approaches for future antiarrhythmic compounds that can improve our understanding of arrhythmogenesis on a subcellular transcriptome and proteome level.
Autonomic nervous system
Autonomic imbalance characterized by increased sympathetic activation and parasympathetic withdrawal, along with changes in density and spatial distribution of the intrinsic efferent innervation of the ventricles, may account for the timing of clinical presentation of arrhythmias after ventricular injury [99]. Sympathetic hyperinnervation in the ventricle occurs post-MI and has been linked to ventricular arrhythmias. Whereas nerve growth factor (NGF) stimulates axon growth, its precursor, proNGF, triggers axon degeneration and may be involved in regional denervation after myocardial injury. Additionally, denervated reperfused infarcts display β-adrenoreceptor supersensitivity [100]. Both sympathetic hyperinnervation and denervation of the ventricles can lead to heterogeneous β-adrenoreceptor activation, either through localized catecholamine release or localized β-adrenoreceptor supersensitivity. This non-uniform sympathetic activation increases the risk of focal triggers and creates gradients of repolarization, increasing the susceptibility for reentry. Attempts to therapeutically reduce sympathetic activation or sympathetic nerve sprouting (e.g. by cardiac sympathetic denervation or renal denervation) [101,102,103] or to increase cardiac parasympathetic tone (e.g. by baroreceptor stimulation) [104] reduce arrhythmias in animal models and selected cohorts of patients.
Non-ischaemic cardiomyopathy
Non-ischaemic cardiomyopathies include genuine dilated or congestive cardiomyopathy (DCM) and special cases such as arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D).
While in ICM usually focal areas with post ischaemic/MI fibrosis and scarring can be found that lead to reduced myocardial contractility, in DCM the impaired myocardial contractility is generally dispersed.
Pathophysiology of VT generation in DCM is as complex as its variety of genetic causes. Nowadays, an abundance of genes and alleles that contribute to phenotypic DCM have been identified [105]. Gene alterations of TTN (coding for Titin, 12–25% of DCM), LMNA (coding for Lamina/C, 4–8% of DCM), DES, VLC and FLNC (coding for cytoskeletal proteins, each around 1% of DCM) significantly contribute to the DCM phenotype [105]. While DSP, coding for desmoplakin, also contributes to DCM, other desmosome changes instead result in ARVC/M. Loss of RNA-binding protein 20 (RMB20), which is a RNA-binding protein of a spliceosome of TTN and calcium/calmodulin-dependent protein kinase II (CaMKII) delta, leads to a clinically aggressive form of DCM [106]. Despite similar left ventricular function, altered calcium handling increased arrhythmic burden (44% vs. 5%) when RBM20 mutation carriers were compared to TTN mutation carriers [106]. Therefore, ICa,L-blockers may possibly reduce arrhythmia burden in this disease entity. Besides playing a role in LQTS3 and BrS, SCN5A mutations can also lead to DCM or ARVC/D [107]. In some cases with a gain-of-function mutation in the sodium channel (i.e. in p.R222Q mutation carriers) class I antiarrhythmic agents have been reported to decrease arrhythmogenic burden and improve left ventricular function [107].
In ARVC/D, several desmosomal and non-desmosomal gene mutations have been identified to induce phenotypic disease. Desmosomal genes include plakophilin 2, desmoglein 2 and desmoplakin gene mutations contributing to half of all cases [108]. Non-desmosomal changes include amongst others gene mutations in the RyR2, phospholamban, Lamin A/C, Desmin, Titin and transforming growth factor 2 [108]. Pharmacological treatment options include beta-blockers in all ARVC/D phenotype patients, as well as sotalol, amiodarone and mexiletine. In selected patients epicardial ablation may be considered.
Besides antiarrhythmic drugs, experimental data of mouse models have shown reduced structural and electrical remodelling leading to arrhythmia reduction by blockade of the renin-angiotensin-system. The direct renin inhibitor aliskiren was shown to have antiarrhythmic potential by reestablishing normal ventricular conduction velocities due to restoration of connexin 43 expression in a DCM mouse model [109]. Also, the angiotensin II receptor antagonist candesartan was able to partially reverse pro-arrhythmic down-regulation of Kv4.2 (Ito channel protein), KChIP2 (auxiliary subunit of Kv4.2) and Kv1.5 (IKur channel protein) in another DCM mouse model [110].
To date, however, most treatment approaches in DCM are symptomatic or prophylactic and not driven by pathophysiologic understanding of the disease or underlying genetic pathology. Therefore, the main goal for the future is to gain further knowledge about underlying disease pathology at the organ, cellular and subcellular levels in order to translate this knowledge into mechanism-based therapeutic approaches.
Atrial arrhythmias
Atrial arrhythmias, particularly AF, are the most common cardiac rhythm disorders and are associated with increased rate of stroke, HF and death. Great efforts have been undertaken to understand the underlying arrhythmic mechanisms and improve treatment options. Although there are now many sophisticated therapeutic options available, antiarrhythmic therapy remains unsatisfactory [111]. In contrast, pacemakers are the accepted standard therapy for patients with bradycardia. Similarly, patients with accessory-pathway or AV-nodal reentry syndromes, such as Wolff–Parkinson–White syndrome, are usually successfully treated with ablation therapy [112]. A thorough understanding of the molecular mechanisms underlying the latter has had little impact on therapeutic options and these arrhythmias will therefore not be discussed in further detail.
Atrial tachycardia (AT)
ATs are subdivided based on underlying mechanisms into focal tachycardias and macroreentrant tachycardias, also designated as atrial flutter [113]. Focal ATs commonly arise in the absence of preexisting structural heart disease and can occur at any age with no gender preference [114]. The autonomic nervous system likely contributes to the initiation of focal AT, which can be triggered by changes in posture, belching and swallowing. Pathomechanisms of focal AT include abnormal automaticity (19%), triggered activity (25%) and microreentry (56%) [115].
Focal ATs due to triggered activity are likely to be mediated by DADs. Focal ATs may also be caused by microreentry, which refers to a small reentrant loop which is below the resolution of current mapping systems [113]. Adenosine can be used to distinguish reentry from other mechanisms underlying AT. Adenosine binding to A1-receptors leads to dissociation of Gi-proteins and to activation of a repolarizing potassium current (IK,ACh), due to direct interaction with the Gi-protein βγ-subunit. The resulting stabilization of the RMP terminates focal AT caused by afterdepolarizations, transiently suppresses AT due to abnormal automaticity, but does not affect AT based on reentry mechanisms [116].
The current knowledge surrounding underlying AT mechanisms is not yet sufficient to guide antiarrhythmic drug choice, but may help to understand why certain treatment options may be effective in some patients while ineffective in others. There is hope that the future development of selective drugs may provide new therapeutic options for mechanism-based therapy of AT [117].
Atrial fibrillation
Remodelling; progression of AF
The “atrial fibrillation begets atrial fibrillation” paradigm [118] states that the longer AF persists, the higher the inducibility and stability of AF, i.e. the higher the likelihood that AF will perpetuate. These basic research findings recapitulate the clinical observation that AF is a highly progressive disease [111, 119]. The progressive nature of AF is caused by atrial remodelling processes on the cellular and tissue level, and basic research has been instrumental in unravelling these processes [9]. Atrial remodelling comprises electrical, contractile, structural and calcium-handling remodelling. Electrical remodelling is characterized by a dramatic reduction in atrial ERP caused by shortening of atrial myocyte APD. The latter is due to altered regulation and expression of ion channels, e.g. reduced ICa,L and transient outward potassium current (Ito) and increased inward rectifier potassium currents and TASK-1 (K2P 3.1) current [119,120,121,122]. Contractile remodelling is caused by multiple mechanisms including impaired calcium handling and dysregulation of the sarcomeres and myofilaments [119]. Prominent features of structural remodelling are atrial dilatation, atrial myocyte hypertrophy and fibrosis, which create a substrate for AF [119]. Calcium-handling remodelling includes altered expression and/or regulation (e.g. phosphorylation) of L-type calcium channels, NCX, RyR2, SERCA2a or CaMKII [123,124,125]. Calcium-handling abnormalities (1) contribute to electrical, contractile, structural and intracellular signalling remodelling, (2) may provide both a trigger (e.g. by spontaneous SR calcium release triggering DADs) and a substrate for AF (by contributing to electrical and structural remodelling) and (3) may be both a cause and consequence of AF [125].
There are notable differences in the time course and reversibility of electrical and structural remodelling with important clinical ramifications. Electrical remodelling is an early event starting within hours or days after initiation of AF and is reversible upon restoration of sinus rhythm [118]. Fibrosis (structural remodelling), on the other hand, is a late event [12] and is considered poorly reversible. In patients, the degree of atrial fibrosis correlates with the occurrence of postoperative AF as well as with the persistence and recurrence of AF [126, 127]. Thus, a high degree of atrial fibrosis may be regarded as a marker of terminally-remodelled atria highly susceptible to AF. These findings about the time course and progression of atrial remodelling may explain the clinical observation that cardioversion and catheter ablation for rhythm control are more successful in younger patients with shorter history of AF and less atrial structural remodelling [111]. They stress the clinical paradigm that, in order to be treated successfully, AF has to be diagnosed early, i.e. before the development of excessive structural remodelling, which may represent a “point of no return” to sinus rhythm.
Recent evidence from cellular electrophysiology indicates that calcium-handling remodelling exhibits important differences between paroxysmal and chronic AF [123, 124]. These findings have important clinical implications: firstly, they indicate that paroxysmal and chronic AF may be different entities and that (calcium-handling) remodelling does not necessarily represent a continuum from paroxysmal to chronic AF, where a certain alteration simply progresses and aggravates over time. Secondly, it has been suggested that optimal treatment for paroxysmal and chronic AF could differ and, ideally, should be tailored to the individual remodelling processes in a given patient.
The “complex of structural, architectural, contractile or electrophysiological changes affecting the atria with the potential to produce clinically-relevant manifestations” has been summarized as “atrial cardiomyopathy” [128].
Mechanism-based stratification of AF subtypes: impact of comorbidities
Patient stratification and antiarrhythmic therapy planning relies primarily on symptoms and the duration of AF (i.e. paroxysmal, persistent, long-standing persistent, or permanent) [111]. This classification is limited by a lack of regard for arrhythmia burden, the predominant underlying mechanism of the arrhythmia, or the severity of the atrial substrate. Specifically, the impact of basic scientific findings on AF categorization and therapeutic decision-making has been low in the past, despite significant advances in the field [129]. From a cellular electrophysiologist’s perspective, among the most striking differences between AF patients are changes in APD of atrial myocytes. In patients with persistent or permanent AF but without pronounced LV dysfunction, the “classical” mechanism of atrial arrhythmogenesis is observed, characterized by reentry-promoting shortening of atrial APD [119, 120, 123]. In these patients, inhibition of repolarizing K+ channels that are expressed in human atrium by class III antiarrhythmic drugs is expected to be particularly effective in suppressing AF. The coexistence of AF with severe LV dysfunction poses a clinically significant therapeutic challenge attributed to a distinct atrial substrate that was recently studied in detail. In affected patients with paroxysmal AF, atrial APs are prolonged [130]. The hypothesis that manipulations to activate potassium currents could exert antiarrhythmic effects in these cases remains to be tested in translational approaches. Of note, HF-associated APD prolongation and APD shortening in chronic forms of AF offset each other, resulting in apparently normal APD in patients with long-standing persistent AF and severe LV dysfunction.
The development and application of methodologies evaluating the extent of atrial remodelling in patients such as electro-anatomic mapping or late gadolinium enhancement magnetic resonance imaging (LGE-MRI), as well as mapping techniques applied during ongoing arrhythmias (e.g. mapping of focal or re-entrant activity) may help to improve arrhythmia characterization and the initiation of individualized treatment of AF patients. In translational research strategies evaluating antiarrhythmic concepts to be applied in patients with paroxysmal AF or following rhythm control by ablation or cardioversion, the use of cells obtained from subjects with persistent or even permanent AF exhibiting different cellular electrophysiological mechanisms constitutes a limitation that requires careful consideration. Cells obtained from specific patient populations targeted by antiarrhythmic interventions (e.g. paroxysmal AF) should be preferred when studying antiarrhythmic concepts.
Vagal AF
AF patients with structurally normal hearts tend to show a vagal pattern of AF onset (nocturnal AF, AF during rest or after exercise), while patients with structural heart disease tend to show a sympathetic pattern (AF during daytime or during exercise) [131]. Other studies in “lone” AF patients suggest that AF onset might be associated with a combined symatho-vagal activation rather than with an increase in vagal or sympathetic drive alone [119]. Nocturnal arrhythmias might be triggered by autonomic activation due to sleep disordered breathing like obstructive sleep apnea. During obstructive respiratory events a profound peri-apneic vagal activation [132] followed by combined sympathetic activation at the end of the apnea during the arousal reaction may trigger AF [133]. Additionally, endurance athletes are more likely to develop AF than non-athletes. The type, intensity and amount of sport appears to influence the risk of developing AF and different mechanisms involving increased vagal tone at rest, increased cardiomyocyte-sensitivity to cholinergic stimulation [134], structural atrial remodelling, sinus bradycardia and genetic predisposition may contribute to AF in athletes [135]. Treatment of sleep apnea or detraining in athletes may reduce AF burden in these patients.
Classical and novel antiarrhythmic drug paradigms
In contrast to the inherited channelopathies, where insights into mechanisms of arrhythmia have greatly advanced antiarrhythmic drug therapy, the ICD remains the cornerstone of antiarrhythmic therapy in patients at high risk for ventricular arrhythmias due to acquired conditions [47]. Indeed, with the exception of beta-blockers, antiarrhythmic drugs have not shown a consistent efficacy in the primary management of arrhythmias in this setting, perhaps because currently available antiarrhythmic drugs have mainly been identified based on chance observations during clinical studies, without a precise understanding of the molecular mechanisms underlying initiation and maintenance of arrhythmias [117].
The discovery of sodium-channel block as the major mechanism underlying antiarrhythmic effects of quinidine led to the development of class I antiarrhythmic agents such as flecainide and propafenone. Improved understanding of the fundamental biophysical determinants of state-dependent block of sodium channels subsequently led to the subdivision of agents into classes IA, IB and IC by Vaughan Williams [136]. Ranolazine is a multi-channel blocker, primarily inhibiting IKr and the late component of the cardiac sodium current, which was originally developed as an antianginal drug. Several studies have supported the therapeutic potential of ranolazine for ventricular arrhythmias. However, the large, randomized, double-blind, placebo-controlled Ranolazine Implantable Cardioverter-Defibrillator (RAID) trial in which high-risk ICD patients with ischaemic or nonischaemic cardiomyopathy were randomly assigned to ranolazine or placebo only found a non-significant 16% reduction in the primary composite outcome of VT/ventricular fibrillation or death. Furthermore, although more selective blockers of the late sodium current, such as eleclazine, showed strong antiarrhythmic effects in animal studies, all subsequent clinical trials (ClinicalTrials.gov numbers: NCT02104583, NCT02291237, NCT02300558) have recently been discontinued.
Besides beta-blockers, amiodarone is most often used due to contraindications for other antiarrhythmic drugs in the presence of advanced structural heart disease, which is common in this population. The antiarrhythmic effects of amiodarone were also a coincidental discovery during amiodarone use in the therapy of angina. Further investigations revealed that potassium-channel inhibition by amiodarone reduces the likelihood of reentry by prolonging APD and ERP. In addition, amiodarone blocks sodium and calcium channels and inhibits the effects of alpha- and beta-adrenoceptors, thereby possessing properties of all four Vaughan Williams classes of antiarrhythmic agents. Therefore, combined blockade of multiple ion channels has recently been suggested to underlie the relatively high anti-AF efficacy of amiodarone [137]. However, amiodarone’s extracardiac toxicity has motivated the search for alternatives [138]. Dronedarone is the first amiodarone-analog and exhibits reduced toxicity and lipophilicity. Initial clinical studies with dronedarone did not show significant extracardiovascular toxicity and the ATHENA trial demonstrated a reduction in stroke associated with dronedarone use in AF patients [139]. However, dronedarone is less effective in maintaining sinus rhythm than amiodarone and it is contraindicated in patients with HF [111].
A number of alternative antiarrhythmic strategies for patients with ventricular arrhythmias have been proposed based on preclinical studies. Among these, inhibition of CaMKII, which appears to play a nodal role in both atrial and ventricular arrhythmias, has received significant interest. CaMKII activity is increased in multiple cardiovascular diseases including AF and HF [123] and targets a large number of ion channels and calcium-handling proteins, including L-type calcium channels, RyR2 and phospholamban. Recent studies have highlighted the antiarrhythmic potential of several new CaMKII inhibitors, particularly for conditions in which CaMKII activity is increased. Some of these compounds are currently undergoing clinical testing for rheumatoid arthritis [140], but their antiarrhythmic potential has not yet been evaluated in clinical studies. Alternatively, since RyR2 dysfunction plays a major role in maladaptive cardiac remodelling, e.g. in the setting of HF [141], RyR2 stabilizing drugs (e.g. K201/JTV-519, S107, carvedilol-analogues), may have a more general therapeutic use, beyond their role in RyR2-associated channelopathies like CPVT.
Atrial-specific antiarrhythmic drugs
Despite the rapid development of radiofrequency ablation strategies for the treatment of AF, their effectiveness and safety remain inadequate. Furthermore, these methods can only be applied in a restricted number of patients and therefore pharmacological approaches remain clinically relevant, especially due to the large and growing size of the AF population [111].
The ongoing search for new agents against AF has led to the development of atrial-selective antiarrhythmic approaches [142]. Vernakalant and ranolazine, for example, are the first approaches to selectively target atrial sodium-channels by taking advantage of the biophysical differences between atrial and ventricular channels [143]. In atrial tissue, a higher fraction of sodium channels is in the inactivated state because of the more depolarized resting membrane potential. Since vernakalant and ranolazine bind with higher affinity to activated/inactivated sodium channels than to channels in the closed state, this may contribute to an atrial specific effect on peak sodium current of these drugs [143, 144]. In addition, ranolazine predominantly inhibits late sodium current, which is increased in patients with AF and has been suggested to contribute to AF pathophysiology [145, 146].
Another approach to developing atrial specific compounds aims to target potassium channels which are predominantly expressed in the atrium, such as IKur, IKACh, two-pore K+-channels or Ca2+-dependent K+-channels [147]. Interestingly, some of the already available antiarrhythmic agents, such as flecainide, amiodarone, quinidine, chloroquine or verapamil, inhibit some of these atrial-selective K+-channels, which may contribute to their antiarrhythmic effect in AF [147].
The unique effectiveness and low arrhythmogenic potential of amiodarone has been attributed, amongst other factors, to its broad spectrum of ion-channel blocking effects [117, 147]. Therefore, identification of specific combinations of ion-channel modulating activities could optimize antiarrhythmic efficacy and atrial selectivity. Accordingly, the combination of ranolazine and amiodarone, dronedarone or dofetilide has higher antiarrhythmic efficacy, compared with either drug alone [148, 149] and AF-selectivity of sodium channel blockers can be improved by adding potassium channel blockade [150]. In the HARMONY trial the combination of ranolazine and dronedarone showed synergistic effects in reducing AF burden in paroxysmal AF patients [151]. However, at present, currently available evidence is insufficient to recommend antiarrhythmic drug combinations and further research is necessary to define the required channel blocking profile and to validate these approaches in clinical studies.
Basic research during recent years on the mechanisms underlying AF pathology also led to the identification of new potential antiarrhythmic approaches, including the normalization of atrial calcium-handling abnormalities, atrial metabolism or autonomic-tone manipulation. These are currently not implemented in therapeutic strategies and we refer the interested reader to recent reviews [12, 117].
Opportunities, challenges and future perspectives
Personalized diagnostic and therapeutic approaches
An increased understanding of patho-mechanisms underlying specific disease subtypes may open the avenue for more specific, personalized therapies. In monogenic channelopathies such as LQTS, for example, this has already led to genotype-specific approaches, e.g. late sodium channel blocker therapy in LQT3 [27]. However, even mutation-specific, personalized therapies may develop as (1) different mutations may convey pronounced differences in arrhythmic risk [32] and (2) pronounced mutation-specific differences are observed in response to a given antiarrhythmic drug. The SQTS gain-of-function mutations HERG-N588K and KCNQ1-V307L, for example, alter the extent of IKr/IKs-blocking effects of various beta-blocking agents [41] and HERG-N588K may similarly diminish IKr-blocking (and hence therapeutic) effects of a variety of class I and III agents such as sotalol, quinidine and amiodarone [152]. Here, the development of patient-specific hiPSC-CM may help to test personalized anti-arrhythmic approaches [153].
Not only in monogenic arrhythmia disorders but also in more common arrhythmias such as AF, a more detailed classification of the disease into “mechanistic” subtypes (see chapter Mechanism-based stratification of AF subtypes) may promote patient-oriented rather than generalized therapeutic strategies. miRNAs may help to characterize the substrate or the electrical phenotype in individual AF patients and predict the outcome of interventional therapy [154]. Reduced expression of anti-fibrotic miR29, for example, is associated with increased atrial fibrosis and vulnerability to AF in a canine ventricular tachypacing-induced HF model [155]. Similarly, miR29 expression was reduced in patients with cAF and plasma levels of miR29b could be used to predict the outcome in pAF-patients [156].
Taken together, substratification of AF phenotypes may allow the combination of different mechanism-specific drugs (direct anti-arrhythmic atrial-selective channel-modifying drugs as well as modifiers of remodelling processes, inflammation etc.) and interventional therapies guided by the disease subtypes [130].
Application of human induced pluripotent stem cell-derived cardiomyocytes in experimental and clinical electrophysiology
Application of hIPSC-CM may theoretically provide the unique opportunity to investigate cardiac electrophysiology of any individual in a non-invasive way and is expected to help close the gap between experimental and clinical electrophysiology. Ideally, hIPSC-CM could predict arrhythmogenic risk and efficacy of drugs, contributing to personalized medicine. From a broader perspective, studies on cellular electrophysiology would no longer be restricted to tissue harvested during open-heart surgery, allowing intense work on undiseased ventricular tissue. hIPSC-CM should enable repeated investigations, making long and complex functional studies even at different laboratories possible. Classic transgene approaches applied in hIPSC-CM could be used to study heart electrophysiology on a human background. Since hIPSC-CM can be kept in culture over weeks or even months; they could be used to study effects of “long-term” exposure to drugs or increased mechanical load. From this perspective, one could assume that hIPSC-CM could dominate cellular cardiac electrophysiology in the near future; however, there are several critical issues to be addressed.
A central question is how closely hIPSC-CM resemble native human cardiomyocytes, in particular: (1) what type of cardiomyocytes do hIPSC-CM present? Ventricular, atrial, nodal or just a mixture of them [157]? (2) What is the repolarization reserve of hIPSC-CM compared to classic approaches based on animal and undiseased human ventricular tissue [158]? (3) Is the relatively depolarized RMP an intrinsic peculiarity of hIPSC-CM? (4) Are there other reasons for automaticity in hIPSC-CM than low RMP [159]? (5) How can APD be normalized when measured at different spontaneous beating rates? Can hIPSC-CM be used as biological pacemakers [160]? (6) Do hIPSC-CM recapitulate basic findings on cAMP/PKA-mediated contribution to heart pathophysiology? (7) Can hIPSC-CM be used as a model to study human atrial electrophysiology [161]?
To answer these questions, a wide range of cellular electrophysiological techniques is needed. Since it is likely that many of these questions relate to differences in the differentiation and culture of hIPSC-CM, close collaboration between cellular cardiac electrophysiologists and experts in stem cell biology will be mandatory.
Gene- and cell-based therapy
Gene therapy offers greater selectivity than small molecule-based or interventional treatment. The gene of interest is packaged into viral or non-viral carriers and delivered to the target area via direct injection or using catheter-based techniques, providing the advantage of site-restricted action in contrast to systemic application of drugs. Gene therapy for heart rhythm disorders is currently being evaluated in preclinical stages. To date, no antiarrhythmic gene therapy drug is commercially available or has been investigated in clinical trials. Antiarrhythmic effects against AF and ventricular tachycardia, and restoration or suppression of pacemaker activity were successfully achieved in promising pre-clinical gene therapeutic approaches [162]. Cell-based approaches for the treatment of heart rhythm disorders have almost exclusively focused on the generation of biological pacemaker activity [163]. However, in terms of clinical translation most gene and cell therapy strategies are still in early stages of a complex developmental process that involves extensive research and caution prior to widespread human application.
Limitations of the translation of current approaches
One major challenge of efficient translation is the presence of multiple pathophysiological mechanisms in a single disease entity or even an individual patient. Computational cardiology technologies, comprehensive genetics, or the use of biomarkers may help to overcome this challenge. In addition, the effect of underlying mechanisms on clinical arrhythmia manifestations may vary with time, reducing the potential for the establishment of more generalized treatment recommendations in some cases. Furthermore, experiments using cells derived from animal models and hIPSC-CM do not fully reconstitute phenotypes observed in arrhythmia patients with arrhythmia highlighting the increased need for research employing human cells. In terms of personnel, it is important to recognize that clinical heart rhythm specialists and translational scientists are rarely located in the same unit. In fact, in most cases they do not share the same academic institution or hospital, preventing multidisciplinary exchange, interaction and collaboration. Other organizational factors pose limitations to translation as well. In clinical electrophysiology departments, the combined workload produced by clinical and scientific work, in addition to administrative duties, writing of grant applications and academic obligations, create an unfavorable environment for focused application of basic findings into optimized patient care. Finally, limited options to obtain substantial funding, particularly for translational electrophysiologists in training, require optimization to promote this emerging field in the long term.
Future perspectives
The ability of an antiarrhythmic intervention to prevent cardiac arrhythmia depends on its capacity to suppress the underlying disease mechanisms. Despite great advances in the mechanistic understanding of inherited channelopathies, translation of mechanism-based novel therapies into clinical use remains challenging due to problems in designing and funding clinical studies with sufficient power in these rare diseases, particularly for “second use” applications of already approved drugs. Additional electrophysiological targets currently in the pipeline include previously unrecognized ion channels, regulatory signalling pathways, structural alterations such as fibrosis that may be targeted by “upstream therapy” and epigenetic modulation of cardiac electrophysiology. The impact of different comorbidities on proarrhythmic mechanisms is increasingly recognized as well. Identification of remodelling associated with specific concomitant diseases is expected to enable personalized approaches, for example in patients with AF. It would thus be of great importance to improve funding for translational studies implementing basic science into clinical approaches.
A call to establish and support cellular electrophysiology programs in clinical electrophysiology
Multiple efforts are underway to standardize training and education of heart rhythm specialists in Germany and Europe. At the National level in Germany, a curriculum required to achieve heart rhythm specialist qualification (“Zusatzqualifikation Spezielle Rhythmologie”) conveys detailed clinical content on heart rhythm disorders [164]. By contrast, cellular and, particularly, translational electrophysiology is not addressed in detail. Similarly, within the European Core Curriculum for the Heart Rhythm Specialist proposed by the European Heart Rhythm Association [165], training focuses virtually entirely on clinical knowledge and practical skills, and basic scientific knowledge on arrhythmias is included only to a small extent. However, basic science plays a crucial role in clinical medicine and the health care system by producing novel technologies, drugs, biomarkers and mechanistic knowledge as basis for patient-oriented therapy that may ultimately serve to save health care budgets by delivering the right therapy to a specific patient. In accordance, the Diploma of Advanced Studies in Cardiac Arrhythmia Management (DAS-CAM), organized by the European Society of Cardiology and European Heart Academy in collaboration with Maastricht University, explicitly includes sessions on arrhythmia mechanisms and translational electrophysiology.
Despite the recognition and appreciation of translational electrophysiology by European and United States societies for heart rhythm disorders [166, 167], a relevant gap is evident between current patient therapy on one side and the implementation of basic research knowledge into clinical decision making on the other. The German Cardiac Society Working Group on Cellular Electrophysiology assumes the responsibility for the promotion of translational electrophysiology within the field of heart rhythm disorders. Therefore, this group of authors proposes the establishment of cellular electrophysiology programs in institutions providing medical care to arrhythmia patients, to optimize patient care through multi-disciplinary diagnostic and therapeutic approaches. In addition, the Working Group intends to provide a platform for the coordination of nationwide research efforts in the field of translational electrophysiology. This platform is designed to optimize collaborative scientific projects and third-party funded research programs. Furthermore, a “Pathophysiology and Translational Electrophysiology Curriculum” is called for to provide a platform for standardized education of heart rhythm specialists in this hitherto underrepresented field. Ultimately, this educational curriculum may be advanced into a formal “Sachkundekurs: Cellular and Translational Electrophysiology” as part of the “Zusatzqualifikation Spezielle Rhythmologie”. Finally, improved funding for research and education in the field of translational electrophysiology, especially within dedicated translational units, is required to successfully achieve the proposed goals. Anticipated advantages and added value of such infrastructure and programs are summarized in Table 2. A coordinated effort of researchers, translational scientists and clinicians is required to achieve “true” translation and to obtain maximum benefit for optimized therapy of patients affected by heart rhythm disorders.
References
Hill JA, Ardehali R, Clarke KT, Del Zoppo GJ, Eckhardt LL, Griendling KK, Libby P, Roden DM, Sadek HA, Seidman CE, Vaughan DE, American Heart Association Council on Basic Cardiovascular S, Council on Clinical C, Council on E, Prevention, Council on Functional G, Translational B, Stroke C (2017) Fundamental cardiovascular research: returns on societal investment: a scientific statement from the American Heart Association. Circ Res 121(3):e2–e8. https://doi.org/10.1161/RES.0000000000000155
Atienza F, Martins RP, Jalife J (2012) Translational research in atrial fibrillation: a quest for mechanistically based diagnosis and therapy. Circ Arrhythm Electrophysiol 5(6):1207–1215. https://doi.org/10.1161/CIRCEP.111.970335
Busch S, Forkmann M, Kuck KH, Lewalter T, Ince H, Straube F, Wieneke H, Julian Chun KR, Eckardt L, Schmitt C, Hochadel M, Senges J, Brachmann J (2018) Acute and long-term outcome of focal atrial tachycardia ablation in the real world: results of the german ablation registry. Clin Res Cardiol 107(5):430–436. https://doi.org/10.1007/s00392-018-1204-8
Desteghe L, Hendriks JML, McEvoy RD, Chai-Coetzer CL, Dendale P, Sanders P, Heidbuchel H, Linz D (2018) The why, when and how to test for obstructive sleep apnea in patients with atrial fibrillation. Clin Res Cardiol 107(8):617–631. https://doi.org/10.1007/s00392-018-1248-9
Fink T, Schluter M, Kuck KH (2018) From early beginnings to elaborate tools: contribution of German electrophysiology to the interventional treatment of cardiac arrhythmias: the German Cardiac Society welcomes ESC in Munich 2018. Clin Res Cardiol. https://doi.org/10.1007/s00392-018-1319-y
Murray MI, Arnold A, Younis M, Varghese S, Zeiher AM (2018) Cryoballoon versus radiofrequency ablation for paroxysmal atrial fibrillation: a meta-analysis of randomized controlled trials. Clin Res Cardiol 107(8):658–669. https://doi.org/10.1007/s00392-018-1232-4
van der Graaf AW, Bhagirath P, de Hooge J, de Groot NM, Gotte MJ (2016) A priori model independent inverse potential mapping: the impact of electrode positioning. Clin Res Cardiol 105(1):79–88. https://doi.org/10.1007/s00392-015-0891-7
Walsh KA, Galvin J, Keaney J, Keelan E, Szeplaki G (2018) First experience with zero-fluoroscopic ablation for supraventricular tachycardias using a novel impedance and magnetic-field-based mapping system. Clin Res Cardiol 107(7):578–585. https://doi.org/10.1007/s00392-018-1220-8
Heijman J, Algalarrondo V, Voigt N, Melka J, Wehrens XH, Dobrev D, Nattel S (2016) The value of basic research insights into atrial fibrillation mechanisms as a guide to therapeutic innovation: a critical analysis. Cardiovasc Res 109(4):467–479. https://doi.org/10.1093/cvr/cvv275
Haissaguerre M, Vigmond E, Stuyvers B, Hocini M, Bernus O (2016) Ventricular arrhythmias and the His-Purkinje system. Nat Rev Cardiol 13(3):155–166. https://doi.org/10.1038/nrcardio.2015.193
Dukes JW, Dewland TA, Vittinghoff E, Mandyam MC, Heckbert SR, Siscovick DS, Stein PK, Psaty BM, Sotoodehnia N, Gottdiener JS, Marcus GM (2015) Ventricular ectopy as a predictor of heart failure and death. J Am Coll Cardiol 66(2):101–109. https://doi.org/10.1016/j.jacc.2015.04.062
Heijman J, Voigt N, Nattel S, Dobrev D (2014) Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res 114(9):1483–1499. https://doi.org/10.1161/CIRCRESAHA.114.302226
Antzelevitch C, Nesterenko V, Shryock JC, Rajamani S, Song Y, Belardinelli L (2014) The role of late INa in development of cardiac arrhythmias. Handb Exp Pharmacol 221:137–168. https://doi.org/10.1007/978-3-642-41588-3_7
Eisner DA, Caldwell JL, Kistamas K, Trafford AW (2017) Calcium and excitation–contraction coupling in the heart. Circ Res 121(2):181–195. https://doi.org/10.1161/CIRCRESAHA.117.310230
Allessie MA, Bonke FI, Schopman FJ (1977) Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ Res 41(1):9–18
Berenfeld O, Zaitsev AV, Mironov SF, Pertsov AM, Jalife J (2002) Frequency-dependent breakdown of wave propagation into fibrillatory conduction across the pectinate muscle network in the isolated sheep right atrium. Circ Res 90(11):1173–1180
Ciaccio EJ, Coromilas J, Wit AL, Peters NS, Garan H (2018) Source-sink mismatch causing functional conduction block in re-entrant ventricular tachycardia. JACC Clin Electrophysiol 4(1):1–16. https://doi.org/10.1016/j.jacep.2017.08.019
Joyner RW, Sugiura H, Tan RC (1991) Unidirectional block between isolated rabbit ventricular cells coupled by a variable resistance. Biophys J 60(5):1038–1045. https://doi.org/10.1016/S0006-3495(91)82141-5
Haissaguerre M, Hocini M, Denis A, Shah AJ, Komatsu Y, Yamashita S, Daly M, Amraoui S, Zellerhoff S, Picat MQ, Quotb A, Jesel L, Lim H, Ploux S, Bordachar P, Attuel G, Meillet V, Ritter P, Derval N, Sacher F, Bernus O, Cochet H, Jais P, Dubois R (2014) Driver domains in persistent atrial fibrillation. Circulation 130(7):530–538. https://doi.org/10.1161/CIRCULATIONAHA.113.005421
Narayan SM, Krummen DE, Shivkumar K, Clopton P, Rappel WJ, Miller JM (2012) Treatment of atrial fibrillation by the ablation of localized sources: CONFIRM (conventional ablation for atrial fibrillation with or without focal impulse and rotor modulation) trial. J Am Coll Cardiol 60(7):628–636. https://doi.org/10.1016/j.jacc.2012.05.022
Gourdie RG, Dimmeler S, Kohl P (2016) Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat Rev Drug Discov 15(9):620–638. https://doi.org/10.1038/nrd.2016.89
Greener ID, Sasano T, Wan X, Igarashi T, Strom M, Rosenbaum DS, Donahue JK (2012) Connexin43 gene transfer reduces ventricular tachycardia susceptibility after myocardial infarction. J Am Coll Cardiol 60(12):1103–1110. https://doi.org/10.1016/j.jacc.2012.04.042
Verrier RL, Klingenheben T, Malik M, El-Sherif N, Exner DV, Hohnloser SH, Ikeda T, Martinez JP, Narayan SM, Nieminen T, Rosenbaum DS (2011) Microvolt T-wave alternans physiological basis, methods of measurement, and clinical utility-consensus guideline by International Society for Holter and Noninvasive Electrocardiology. J Am Coll Cardiol 58(13):1309–1324. https://doi.org/10.1016/j.jacc.2011.06.029
Kockskamper J, Blatter LA (2002) Subcellular Ca2+ alternans represents a novel mechanism for the generation of arrhythmogenic Ca2+ waves in cat atrial myocytes. J Physiol 545(Pt 1):65–79
Cutler MJ, Wan X, Plummer BN, Liu H, Deschenes I, Laurita KR, Hajjar RJ, Rosenbaum DS (2012) Targeted sarcoplasmic reticulum Ca2+ ATPase 2a gene delivery to restore electrical stability in the failing heart. Circulation 126(17):2095–2104. https://doi.org/10.1161/CIRCULATIONAHA.111.071480
Therasse D, Sacher F, Babuty D, Mabo P, Mansourati J, Kyndt F, Redon R, Schott JJ, Barc J, Probst V, Gourraud JB (2017) Value of the sodium-channel blocker challenge in Brugada syndrome. Int J Cardiol 245:178–180. https://doi.org/10.1016/j.ijcard.2017.05.099
Fabritz L, Damke D, Emmerich M, Kaufmann SG, Theis K, Blana A, Fortmuller L, Laakmann S, Hermann S, Aleynichenko E, Steinfurt J, Volkery D, Riemann B, Kirchhefer U, Franz MR, Breithardt G, Carmeliet E, Schafers M, Maier SK, Carmeliet P, Kirchhof P (2010) Autonomic modulation and antiarrhythmic therapy in a model of long QT syndrome type 3. Cardiovasc Res 87(1):60–72. https://doi.org/10.1093/cvr/cvq029
Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC (2009) Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 15(4):380–383. https://doi.org/10.1038/nm.1942
Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C (2013) HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 10(12):1932–1963. https://doi.org/10.1016/j.hrthm.2013.05.014
Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT (1995) A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80(5):795–803
Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT (1995) SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80(5):805–811
Wilde AAM, Amin A (2017) Channelopathies, genetic testing and risk stratification. Int J Cardiol 237:53–55. https://doi.org/10.1016/j.ijcard.2017.03.063
Chua HC, Servatius H, Asatryan B, Schaller A, Rieubland C, Noti F, Seiler J, Roten L, Baldinger SH, Tanner H, Fuhrer J, Haeberlin A, Lam A, Pless SA, Medeiros-Domingo A (2018) Unexplained cardiac arrest: a tale of conflicting interpretations of KCNQ1 genetic test results. Clin Res Cardiol 107(8):670–678. https://doi.org/10.1007/s00392-018-1233-3
Lang CN, Koren G, Odening KE (2016) Transgenic rabbit models to investigate the cardiac ion channel disease long QT syndrome. Prog Biophys Mol Biol 121(2):142–156. https://doi.org/10.1016/j.pbiomolbio.2016.05.004
Liu GX, Choi BR, Ziv O, Li W, de Lange E, Qu Z, Koren G (2012) Differential conditions for early after-depolarizations and triggered activity in cardiomyocytes derived from transgenic LQT1 and LQT2 rabbits. J Physiol 590(5):1171–1180. https://doi.org/10.1113/jphysiol.2011.218164
Shimizu W, Tanabe Y, Aiba T, Inagaki M, Kurita T, Suyama K, Nagaya N, Taguchi A, Aihara N, Sunagawa K, Nakamura K, Ohe T, Towbin JA, Priori SG, Kamakura S (2002) Differential effects of beta-blockade on dispersion of repolarization in the absence and presence of sympathetic stimulation between the LQT1 and LQT2 forms of congenital long QT syndrome. J Am Coll Cardiol 39(12):1984–1991
Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR, Mathur R, Hajjiri M, Odening KE, Steinberg E, Folco EJ, Pringa E, Centracchio J, Macharzina RR, Donahay T, Schofield L, Rana N, Kirk M, Mitchell GF, Poppas A, Zehender M, Koren G (2008) Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest 118(6):2246–2259. https://doi.org/10.1172/JCI33578
Head CE, Balasubramaniam R, Thomas G, Goddard CA, Lei M, Colledge WH, Grace AA, Huang CL (2005) Paced electrogram fractionation analysis of arrhythmogenic tendency in ΔKPQ Scn5a mice. J Cardiovasc Electrophysiol 16(12):1329–1340. https://doi.org/10.1111/j.1540-8167.2005.00200.x
Portero V, Casini S, Hoekstra M, Verkerk AO, Mengarelli I, Belardinelli L, Rajamani S, Wilde AAM, Bezzina CR, Veldkamp MW, Remme CA (2017) Anti-arrhythmic potential of the late sodium current inhibitor GS-458967 in murine Scn5a-1798insD+/− and human SCN5A-1795insD+/− iPSC-derived cardiomyocytes. Cardiovasc Res 113(7):829–838. https://doi.org/10.1093/cvr/cvx077
Tian XL, Yong SL, Wan X, Wu L, Chung MK, Tchou PJ, Rosenbaum DS, Van Wagoner DR, Kirsch GE, Wang Q (2004) Mechanisms by which SCN5A mutation N1325S causes cardiac arrhythmias and sudden death in vivo. Cardiovasc Res 61(2):256–267
Bodi I, Franke G, Pantulu ND, Wu K, Perez-Feliz S, Bode C, Zehender M, zur Hausen A, Brunner M, Odening KE (2013) Differential effects of the beta-adrenoceptor blockers carvedilol and metoprolol on SQT1- and SQT2-mutant channels. J Cardiovasc Electrophysiol 24(10):1163–1171. https://doi.org/10.1111/jce.12178
Chorin E, Hu D, Antzelevitch C, Hochstadt A, Belardinelli L, Zeltser D, Barajas-Martinez H, Rozovski U, Rosso R, Adler A, Benhorin J, Viskin S (2016) Ranolazine for congenital long-QT syndrome type III: experimental and long-term clinical data. Circ Arrhythm Electrophysiol. https://doi.org/10.1161/CIRCEP.116.004370
El-Bizri N, Xie C, Liu L, Limberis J, Krause M, Hirakawa R, Nguyen S, Tabuena DR, Belardinelli L, Kahlig KM (2018) Eleclazine exhibits enhanced selectivity for long QT syndrome type 3-associated late Na+ current. Heart Rhythm 15(2):277–286. https://doi.org/10.1016/j.hrthm.2017.09.028
Mazzanti A, Maragna R, Faragli A, Monteforte N, Bloise R, Memmi M, Novelli V, Baiardi P, Bagnardi V, Etheridge SP, Napolitano C, Priori SG (2016) Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3. J Am Coll Cardiol 67(9):1053–1058. https://doi.org/10.1016/j.jacc.2015.12.033
Moss AJ, Windle JR, Hall WJ, Zareba W, Robinson JL, McNitt S, Severski P, Rosero S, Daubert JP, Qi M, Cieciorka M, Manalan AS (2005) Safety and efficacy of flecainide in subjects with Long QT-3 syndrome (ΔKPQ mutation): a randomized, double-blind, placebo-controlled clinical trial. Ann Noninvasive Electrocardiol 10(4 Suppl):59–66. https://doi.org/10.1111/j.1542-474X.2005.00077.x
Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL (2008) Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol 19(12):1289–1293. https://doi.org/10.1111/j.1540-8167.2008.01246.x
Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez-Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ, Group ESCSD (2015) 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 36(41):2793–2867. https://doi.org/10.1093/eurheartj/ehv316
Odening KE, Choi BR, Liu GX, Hartmann K, Ziv O, Chaves L, Schofield L, Centracchio J, Zehender M, Peng X, Brunner M, Koren G (2012) Estradiol promotes sudden cardiac death in transgenic long QT type 2 rabbits while progesterone is protective. Heart Rhythm 9(5):823–832. https://doi.org/10.1016/j.hrthm.2012.01.009
Odening KE, Jung BA, Lang CN, Cabrera Lozoya R, Ziupa D, Menza M, Relan J, Franke G, Perez Feliz S, Koren G, Zehender M, Bode C, Brunner M, Sermesant M, Foll D (2013) Spatial correlation of action potential duration and diastolic dysfunction in transgenic and drug-induced LQT2 rabbits. Heart Rhythm 10(10):1533–1541. https://doi.org/10.1016/j.hrthm.2013.07.038
Lang CN, Menza M, Jochem S, Franke G, Perez Feliz S, Brunner M, Koren G, Zehender M, Bugger H, Jung BA, Foell D, Bode C, Odening KE (2016) Electro-mechanical dysfunction in long QT syndrome: role for arrhythmogenic risk prediction and modulation by sex and sex hormones. Prog Biophys Mol Biol 120(1–3):255–269. https://doi.org/10.1016/j.pbiomolbio.2015.12.010
Steinfurt J, Brunner M, Franke G, Perez-Feliz S, Bodi I, Pantulu ND, Lang CN, Ziupa D, Zehender M, zur Hausen A, Jolivet G, Bode C, Odening KE (2016) In vivo EP studies in transgenic short QT syndrome type 1 rabbits reveal shortened atrial and ventricular effective refractory periods and higher atrial and ventricular tachyarrhythmia inducibility. Heart Rhythm 13(Supplement):PO06–87
Brugada P, Brugada J (1992) Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 20(6):1391–1396
Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O’Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q (1998) Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392(6673):293–296. https://doi.org/10.1038/32675
Antzelevitch C, Yan GX, Ackerman MJ, Borggrefe M, Corrado D, Guo J, Gussak I, Hasdemir C, Horie M, Huikuri H, Ma C, Morita H, Nam GB, Sacher F, Shimizu W, Viskin S, Wilde AA (2016) J-wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Heart Rhythm 13(10):e295–324. https://doi.org/10.1016/j.hrthm.2016.05.024
Crotti L, Marcou CA, Tester DJ, Castelletti S, Giudicessi JR, Torchio M, Medeiros-Domingo A, Simone S, Will ML, Dagradi F, Schwartz PJ, Ackerman MJ (2012) Spectrum and prevalence of mutations involving BrS1-through BrS12-susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: implications for genetic testing. J Am Coll Cardiol 60(15):1410–1418. https://doi.org/10.1016/j.jacc.2012.04.037
Fukuyama M, Ohno S, Makiyama T, Horie M (2016) Novel SCN10A variants associated with Brugada syndrome. Europace 18(6):905–911. https://doi.org/10.1093/europace/euv078
Hu D, Barajas-Martinez H, Pfeiffer R, Dezi F, Pfeiffer J, Buch T, Betzenhauser MJ, Belardinelli L, Kahlig KM, Rajamani S, DeAntonio HJ, Myerburg RJ, Ito H, Deshmukh P, Marieb M, Nam GB, Bhatia A, Hasdemir C, Haissaguerre M, Veltmann C, Schimpf R, Borggrefe M, Viskin S, Antzelevitch C (2014) Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol 64(1):66–79. https://doi.org/10.1016/j.jacc.2014.04.032
Behr ER, Savio-Galimberti E, Barc J, Holst AG, Petropoulou E, Prins BP, Jabbari J, Torchio M, Berthet M, Mizusawa Y, Yang T, Nannenberg EA, Dagradi F, Weeke P, Bastiaenan R, Ackerman MJ, Haunso S, Leenhardt A, Kaab S, Probst V, Redon R, Sharma S, Wilde A, Tfelt-Hansen J, Schwartz P, Roden DM, Bezzina CR, Olesen M, Darbar D, Guicheney P, Crotti L, Consortium UK, Jamshidi Y (2015) Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res 106(3):520–529. https://doi.org/10.1093/cvr/cvv042
Nademanee K, Raju H, de Noronha SV, Papadakis M, Robinson L, Rothery S, Makita N, Kowase S, Boonmee N, Vitayakritsirikul V, Ratanarapee S, Sharma S, van der Wal AC, Christiansen M, Tan HL, Wilde AA, Nogami A, Sheppard MN, Veerakul G, Behr ER (2015) Fibrosis, Connexin-43, and conduction abnormalities in the Brugada syndrome. J Am Coll Cardiol 66(18):1976–1986. https://doi.org/10.1016/j.jacc.2015.08.862
Nademanee K, Veerakul G, Chandanamattha P, Chaothawee L, Ariyachaipanich A, Jirasirirojanakorn K, Likittanasombat K, Bhuripanyo K, Ngarmukos T (2011) Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 123(12):1270–1279. https://doi.org/10.1161/CIRCULATIONAHA.110.972612
Antzelevitch C (2012) Genetic, molecular and cellular mechanisms underlying the J wave syndromes. Circ J 76(5):1054–1065
Schweizer PA, Becker R, Katus HA, Thomas D (2010) Successful acute and long-term management of electrical storm in Brugada syndrome using orciprenaline and quinine/quinidine. Clin Res Cardiol 99(7):467–470. https://doi.org/10.1007/s00392-010-0145-7
Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, Brugada J, Brugada R, Antzelevitch C (1999) Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res 85(9):803–809
Lieve KV, van der Werf C, Wilde AA (2016) Catecholaminergic polymorphic ventricular tachycardia. Circ J 80(6):1285–1291. https://doi.org/10.1253/circj.CJ-16-0326
Landstrom AP, Dobrev D, Wehrens XHT (2017) Calcium signaling and cardiac arrhythmias. Circ Res 120(12):1969–1993. https://doi.org/10.1161/CIRCRESAHA.117.310083
Glukhov AV, Kalyanasundaram A, Lou Q, Hage LT, Hansen BJ, Belevych AE, Mohler PJ, Knollmann BC, Periasamy M, Gyorke S, Fedorov VV (2015) Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex1. Eur Heart J 36(11):686–697. https://doi.org/10.1093/eurheartj/eht452
Salvage SC, Chandrasekharan KH, Jeevaratnam K, Dulhunty AF, Thompson AJ, Jackson AP, Huang CL (2018) Multiple targets for flecainide action: implications for cardiac arrhythmogenesis. Br J Pharmacol 175(8):1260–1278. https://doi.org/10.1111/bph.13807
Sedej S, Heinzel FR, Walther S, Dybkova N, Wakula P, Groborz J, Gronau P, Maier LS, Vos MA, Lai FA, Napolitano C, Priori SG, Kockskamper J, Pieske B (2010) Na+-dependent SR Ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human CPVT mutation. Cardiovasc Res 87(1):50–59. https://doi.org/10.1093/cvr/cvq007
Zhou Q, Xiao J, Jiang D, Wang R, Vembaiyan K, Wang A, Smith CD, Xie C, Chen W, Zhang J, Tian X, Jones PP, Zhong X, Guo A, Chen H, Zhang L, Zhu W, Yang D, Li X, Chen J, Gillis AM, Duff HJ, Cheng H, Feldman AM, Song LS, Fill M, Back TG, Chen SR (2011) Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat Med 17(8):1003–1009. https://doi.org/10.1038/nm.2406
Jung CB, Moretti A, Mederos y Schnitzler M, Iop L, Storch U, Bellin M, Dorn T, Ruppenthal S, Pfeiffer S, Goedel A, Dirschinger RJ, Seyfarth M, Lam JT, Sinnecker D, Gudermann T, Lipp P, Laugwitz KL (2012) Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med 4(3):180–191. https://doi.org/10.1002/emmm.201100194
Penttinen K, Swan H, Vanninen S, Paavola J, Lahtinen AM, Kontula K, Aalto-Setala K (2015) Antiarrhythmic effects of dantrolene in patients with catecholaminergic polymorphic ventricular tachycardia and replication of the responses using iPSC models. PLoS One 10(5):e0125366. https://doi.org/10.1371/journal.pone.0125366
Hofman N, Tan HL, Alders M, Kolder I, de Haij S, Mannens MM, Lombardi MP, Dit Deprez RH, van Langen I, Wilde AA (2013) Yield of molecular and clinical testing for arrhythmia syndromes: report of 15 years’ experience. Circulation 128(14):1513–1521. https://doi.org/10.1161/CIRCULATIONAHA.112.000091
Schott JJ, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, Hulsbeek M, Wilde AA, Escande D, Mannens MM, Le Marec H (1999) Cardiac conduction defects associate with mutations in SCN5A. Nat Genet 23(1):20–21. https://doi.org/10.1038/12618
Kruse M, Schulze-Bahr E, Corfield V, Beckmann A, Stallmeyer B, Kurtbay G, Ohmert I, Schulze-Bahr E, Brink P, Pongs O (2009) Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J Clin Invest 119(9):2737–2744. https://doi.org/10.1172/JCI38292
Brodt C, Siegfried JD, Hofmeyer M, Martel J, Rampersaud E, Li D, Morales A, Hershberger RE (2013) Temporal relationship of conduction system disease and ventricular dysfunction in LMNA cardiomyopathy. J Card Fail 19(4):233–239. https://doi.org/10.1016/j.cardfail.2013.03.001
Gollob MH, Seger JJ, Gollob TN, Tapscott T, Gonzales O, Bachinski L, Roberts R (2001) Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation 104(25):3030–3033
Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG (1998) Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281(5373):108–111
Basson CT, Huang T, Lin RC, Bachinsky DR, Weremowicz S, Vaglio A, Bruzzone R, Quadrelli R, Lerone M, Romeo G, Silengo M, Pereira A, Krieger J, Mesquita SF, Kamisago M, Morton CC, Pierpont ME, Muller CW, Seidman JG, Seidman CE (1999) Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations. Proc Natl Acad Sci U S A 96(6):2919–2924
Choudhury M, Boyett MR, Morris GM (2015) Biology of the sinus node and its disease. Arrhythm Electrophysiol Rev 4(1):28–34. https://doi.org/10.15420/aer.2015.4.1.28
Duhme N, Schweizer PA, Thomas D, Becker R, Schroter J, Barends TR, Schlichting I, Draguhn A, Bruehl C, Katus HA, Koenen M (2013) Altered HCN4 channel C-linker interaction is associated with familial tachycardia-bradycardia syndrome and atrial fibrillation. Eur Heart J 34(35):2768–2775. https://doi.org/10.1093/eurheartj/ehs391
Schweizer PA, Duhme N, Thomas D, Becker R, Zehelein J, Draguhn A, Bruehl C, Katus HA, Koenen M (2010) cAMP sensitivity of HCN pacemaker channels determines basal heart rate but is not critical for autonomic rate control. Circ Arrhythm Electrophysiol 3(5):542–552. https://doi.org/10.1161/CIRCEP.110.949768
Schweizer PA, Schroter J, Greiner S, Haas J, Yampolsky P, Mereles D, Buss SJ, Seyler C, Bruehl C, Draguhn A, Koenen M, Meder B, Katus HA, Thomas D (2014) The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J Am Coll Cardiol 64(8):757–767. https://doi.org/10.1016/j.jacc.2014.06.1155
Baruscotti M, Bucchi A, Milanesi R, Paina M, Barbuti A, Gnecchi-Ruscone T, Bianco E, Vitali-Serdoz L, Cappato R, DiFrancesco D (2017) A gain-of-function mutation in the cardiac pacemaker HCN4 channel increasing cAMP sensitivity is associated with familial Inappropriate Sinus Tachycardia. Eur Heart J 38(4):280–288. https://doi.org/10.1093/eurheartj/ehv582
Heusch G, Kleinbongard P (2016) Ivabradine: cardioprotection by and beyond heart rate reduction. Drugs 76(7):733–740. https://doi.org/10.1007/s40265-016-0567-2
Turker I, Ai T, Itoh H, Horie M (2017) Drug-induced fatal arrhythmias: acquired long QT and Brugada syndromes. Pharmacol Ther 176:48–59. https://doi.org/10.1016/j.pharmthera.2017.05.001
Heijman J, Voigt N, Carlsson LG, Dobrev D (2014) Cardiac safety assays. Curr Opin Pharmacol 15:16–21. https://doi.org/10.1016/j.coph.2013.11.004
Odening KE, Hyder O, Chaves L, Schofield L, Brunner M, Kirk M, Zehender M, Peng X, Koren G (2008) Pharmacogenomics of anesthetic drugs in transgenic LQT1 and LQT2 rabbits reveal genotype-specific differential effects on cardiac repolarization. Am J Physiol Heart Circ Physiol 295(6):H2264–H2272. https://doi.org/10.1152/ajpheart.00680.2008
Colatsky T, Fermini B, Gintant G, Pierson JB, Sager P, Sekino Y, Strauss DG, Stockbridge N (2016) The comprehensive in vitro proarrhythmia assay (CiPA) initiative—update on progress. J Pharmacol Toxicol Methods 81:15–20. https://doi.org/10.1016/j.vascn.2016.06.002
Wakili R, Voigt N, Kaab S, Dobrev D, Nattel S (2011) Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest 121(8):2955–2968. https://doi.org/10.1172/JCI46315
Hausenloy DJ, Yellon DM (2013) Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 123(1):92–100. https://doi.org/10.1172/JCI62874
Walters AM, Porter GA Jr, Brookes PS (2012) Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ Res 111(9):1222–1236. https://doi.org/10.1161/CIRCRESAHA.112.265660
Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, Arensberg D, Baker A, Friedman L, Greene HL et al (1991) Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med 324(12):781–788. https://doi.org/10.1056/NEJM199103213241201
Kamiya J, Ishii M, Katakami T (1992) Antiarrhythmic effects of MS-551, a new class III antiarrhythmic agent, on canine models of ventricular arrhythmia. JpnJ Pharmacol 58(2):107–115
Hosaka Y, Iwata M, Kamiya N, Yamada M, Kinoshita K, Fukunishi Y, Tsujimae K, Hibino H, Aizawa Y, Inanobe A, Nakamura H, Kurachi Y (2007) Mutational analysis of block and facilitation of HERG current by a class III anti-arrhythmic agent, nifekalant. Channels (Austin) 1(3):198–208
Yamazaki M, Honjo H, Nakagawa H, Ishiguro YS, Okuno Y, Amino M, Sakuma I, Kamiya K, Kodama I (2007) Mechanisms of destabilization and early termination of spiral wave reentry in the ventricle by a class III antiarrhythmic agent, nifekalant. Am J Physiol Heart CircPhysiol 292(1):H539–H548. https://doi.org/10.1152/ajpheart.00640.2006
Gautier P, Guillemare E, Djandjighian L, Marion A, Planchenault J, Bernhart C, Herbert JM, Nisato D (2004) In vivo and in vitro characterization of the novel antiarrhythmic agent SSR149744C: electrophysiological, anti-adrenergic, and anti-angiotensin II effects. J Cardiovasc Pharmacol 44(2):244–257
Kowey PR, Crijns HJ, Aliot EM, Capucci A, Kulakowski P, Radzik D, Roy D, Connolly SJ, Hohnloser SH (2011) Efficacy and safety of celivarone, with amiodarone as calibrator, in patients with an implantable cardioverter-defibrillator for prevention of implantable cardioverter-defibrillator interventions or death: the ALPHEE study. Circulation 124(24):2649–2660. https://doi.org/10.1161/CIRCULATIONAHA.111.072561
Chilukoti RK, Lendeckel J, Darm K, Bukowska A, Goette A, Suhling M, Utpatel K, Peters B, Homuth G, Volker U, Wolke C, Scharf C, Lendeckel U (2018) Integration of “omics” techniques: Dronedarone affects cardiac remodeling in the infarction border zone. Exp Biol Med (Maywood) 243(11):895–910. https://doi.org/10.1177/1535370218788517
Ripplinger CM, Noujaim SF, Linz D (2016) The nervous heart. Prog Biophys Mol Biol 120(1–3):199–209. https://doi.org/10.1016/j.pbiomolbio.2015.12.015
Gardner RT, Wang L, Lang BT, Cregg JM, Dunbar CL, Woodward WR, Silver J, Ripplinger CM, Habecker BA (2015) Targeting protein tyrosine phosphatase sigma after myocardial infarction restores cardiac sympathetic innervation and prevents arrhythmias. Nat Commun 6:6235. https://doi.org/10.1038/ncomms7235
Linz D, van Hunnik A, Hohl M, Mahfoud F, Wolf M, Neuberger HR, Casadei B, Reilly SN, Verheule S, Bohm M, Schotten U (2015) Catheter-based renal denervation reduces atrial nerve sprouting and complexity of atrial fibrillation in goats. Circ Arrhythm Electrophysiol 8(2):466–474. https://doi.org/10.1161/CIRCEP.114.002453
Linz D, Wirth K, Ukena C, Mahfoud F, Poss J, Linz B, Bohm M, Neuberger HR (2013) Renal denervation suppresses ventricular arrhythmias during acute ventricular ischemia in pigs. Heart Rhythm 10(10):1525–1530. https://doi.org/10.1016/j.hrthm.2013.07.015
Ukena C, Mahfoud F, Ewen S, Bollmann A, Hindricks G, Hoffmann BA, Linz D, Musat D, Pavlicek V, Scholz E, Thomas D, Willems S, Bohm M, Steinberg JS (2016) Renal denervation for treatment of ventricular arrhythmias: data from an International Multicenter Registry. Clin Res Cardiol 105(10):873–879. https://doi.org/10.1007/s00392-016-1012-y
Linz D, Ukena C, Mahfoud F, Neuberger HR, Bohm M (2014) Atrial autonomic innervation: a target for interventional antiarrhythmic therapy? J Am Coll Cardiol 63(3):215–224. https://doi.org/10.1016/j.jacc.2013.09.020
McNally EM, Mestroni L (2017) Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res 121(7):731–748. https://doi.org/10.1161/CIRCRESAHA.116.309396
van den Hoogenhof MMG, Beqqali A, Amin AS, van dM I, Aufiero S, Khan MAF, Schumacher CA, Jansweijer JA, van Spaendonck-Zwarts KY, Remme CA, Backs J, Verkerk AO, Baartscheer A, Pinto YM, Creemers EE (2018) RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.117.031947
Zaklyazminskaya E, Dzemeshkevich S (2016) The role of mutations in the SCN5A gene in cardiomyopathies. Biochim Biophys Acta 1863(7 Pt B):1799–1805. https://doi.org/10.1016/j.bbamcr.2016.02.014
Akdis D, Brunckhorst C, Duru F, Saguner AM (2016) Arrhythmogenic cardiomyopathy: electrical and structural phenotypes. Arrhythm Electrophysiol Rev 5(2):90–101. https://doi.org/10.15420/AER.2016.4.3
Yamada C, Kuwahara K, Yamazaki M, Nakagawa Y, Nishikimi T, Kinoshita H, Kuwabara Y, Minami T, Yamada Y, Shibata J, Nakao K, Cho K, Arai Y, Honjo H, Kamiya K, Nakao K, Kimura T (2016) The renin-angiotensin system promotes arrhythmogenic substrates and lethal arrhythmias in mice with non-ischaemic cardiomyopathy. Cardiovasc Res 109(1):162–173. https://doi.org/10.1093/cvr/cvv248
Odagiri F, Inoue H, Sugihara M, Suzuki T, Murayama T, Shioya T, Konishi M, Nakazato Y, Daida H, Sakurai T, Morimoto S, Kurebayashi N (2014) Effects of candesartan on electrical remodeling in the hearts of inherited dilated cardiomyopathy model mice. PLoS One 9(7):e101838. https://doi.org/10.1371/journal.pone.0101838
Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, Castella M, Diener HC, Heidbuchel H, Hendriks J, Hindricks G, Manolis AS, Oldgren J, Popescu BA, Schotten U, Van Putte B, Vardas P, Agewall S, Camm J, Baron Esquivias G, Budts W, Carerj S, Casselman F, Coca A, De Caterina R, Deftereos S, Dobrev D, Ferro JM, Filippatos G, Fitzsimons D, Gorenek B, Guenoun M, Hohnloser SH, Kolh P, Lip GY, Manolis A, McMurray J, Ponikowski P, Rosenhek R, Ruschitzka F, Savelieva I, Sharma S, Suwalski P, Tamargo JL, Taylor CJ, Van Gelder IC, Voors AA, Windecker S, Zamorano JL, Zeppenfeld K (2016) 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Europace 18(11):1609–1678. https://doi.org/10.1093/europace/euw295
Page RL, Joglar JA, Caldwell MA, Calkins H, Conti JB, Deal BJ, Estes NAM 3rd, Field ME, Goldberger ZD, Hammill SC, Indik JH, Lindsay BD, Olshansky B, Russo AM, Shen WK, Tracy CM, Al-Khatib SM (2016) 2015 ACC/AHA/HRS guideline for the management of adult patients with supraventricular tachycardia: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 67(13):e27–e115. https://doi.org/10.1016/j.jacc.2015.08.856
Katritsis DG, Boriani G, Cosio FG, Hindricks G, Jais P, Josephson ME, Keegan R, Kim YH, Knight BP, Kuck KH, Lane DA, Lip GY, Malmborg H, Oral H, Pappone C, Themistoclakis S, Wood KA, Blomstrom-Lundqvist C, Gorenek B, Dagres N, Dan GA, Vos MA, Kudaiberdieva G, Crijns H, Roberts-Thomson K, Lin YJ, Vanegas D, Caorsi WR, Cronin E, Rickard J (2017) European Heart Rhythm Association (EHRA) consensus document on the management of supraventricular arrhythmias, endorsed by Heart Rhythm Society (HRS), Asia-Pacific Heart Rhythm Society (APHRS), and Sociedad Latinoamericana de Estimulacion Cardiaca y Electrofisiologia (SOLAECE). Europace 19(3):465–511. https://doi.org/10.1093/europace/euw301
Medi C, Kalman JM, Haqqani H, Vohra JK, Morton JB, Sparks PB, Kistler PM (2009) Tachycardia-mediated cardiomyopathy secondary to focal atrial tachycardia: long-term outcome after catheter ablation. J Am Coll Cardiol 53(19):1791–1797. https://doi.org/10.1016/j.jacc.2009.02.014
Chen SA, Chiang CE, Yang CJ, Cheng CC, Wu TJ, Wang SP, Chiang BN, Chang MS (1994) Sustained atrial tachycardia in adult patients. Electrophysiological characteristics, pharmacological response, possible mechanisms, and effects of radiofrequency ablation. Circulation 90(3):1262–1278
Leonelli F, Bagliani G, Boriani G, Padeletti L (2017) Arrhythmias originating in the Atria. Card Electrophysiol Clin 9(3):383–409. https://doi.org/10.1016/j.ccep.2017.05.002
Heijman J, Voigt N, Dobrev D (2013) New directions in antiarrhythmic drug therapy for atrial fibrillation. Future Cardiol 9(1):71–88. https://doi.org/10.2217/fca.12.78
Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA (1995) Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 92(7):1954–1968
Schotten U, Verheule S, Kirchhof P, Goette A (2011) Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev 91(1):265–325. https://doi.org/10.1152/physrev.00031.2009
Schmidt C, Wiedmann F, Voigt N, Zhou XB, Heijman J, Lang S, Albert V, Kallenberger S, Ruhparwar A, Szabo G, Kallenbach K, Karck M, Borggrefe M, Biliczki P, Ehrlich JR, Baczko I, Lugenbiel P, Schweizer PA, Donner BC, Katus HA, Dobrev D, Thomas D (2015) Upregulation of K2P3.1 K+ current causes action potential shortening in patients with chronic atrial fibrillation. Circulation 132(2):82–92. https://doi.org/10.1161/CIRCULATIONAHA.114.012657
Dobrev D, Friedrich A, Voigt N, Jost N, Wettwer E, Christ T, Knaut M, Ravens U (2005) The G protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation 112(24):3697–3706. https://doi.org/10.1161/CIRCULATIONAHA.105.575332
Makary S, Voigt N, Maguy A, Wakili R, Nishida K, Harada M, Dobrev D, Nattel S (2011) Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine-regulated potassium channels in atrial remodeling. Circ Res 109(9):1031–1043. https://doi.org/10.1161/CIRCRESAHA.111.253120
Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH, Dobrev D (2012) Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+–Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125(17):2059–2070. https://doi.org/10.1161/CIRCULATIONAHA.111.067306
Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XHT, Nattel S, Dobrev D (2014) Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 129(2):145–156. https://doi.org/10.1161/CIRCULATIONAHA.113.006641
Greiser M, Lederer WJ, Schotten U (2011) Alterations of atrial Ca2+ handling as cause and consequence of atrial fibrillation. Cardiovasc Res 89(4):722–733. https://doi.org/10.1093/cvr/cvq389
Goette A, Juenemann G, Peters B, Klein HU, Roessner A, Huth C, Rocken C (2002) Determinants and consequences of atrial fibrosis in patients undergoing open heart surgery. Cardiovasc Res 54(2):390–396
Xu J, Cui G, Esmailian F, Plunkett M, Marelli D, Ardehali A, Odim J, Laks H, Sen L (2004) Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation 109(3):363–368. https://doi.org/10.1161/01.CIR.0000109495.02213.52
Goette A, Kalman JM, Aguinaga L, Akar J, Cabrera JA, Chen SA, Chugh SS, Corradi D, D’Avila A, Dobrev D, Fenelon G, Gonzalez M, Hatem SN, Helm R, Hindricks G, Ho SY, Hoit B, Jalife J, Kim YH, Lip GY, Ma CS, Marcus GM, Murray K, Nogami A, Sanders P, Uribe W, Van Wagoner DR, Nattel S (2017) EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: definition, characterization, and clinical implication. Heart Rhythm 14(1):e3–e40. https://doi.org/10.1016/j.hrthm.2016.05.028
Fabritz L, Guasch E, Antoniades C, Bardinet I, Benninger G, Betts TR, Brand E, Breithardt G, Bucklar-Suchankova G, Camm AJ, Cartlidge D, Casadei B, Chua WW, Crijns HJ, Deeks J, Hatem S, Hidden-Lucet F, Kaab S, Maniadakis N, Martin S, Mont L, Reinecke H, Sinner MF, Schotten U, Southwood T, Stoll M, Vardas P, Wakili R, West A, Ziegler A, Kirchhof P (2016) Expert consensus document: defining the major health modifiers causing atrial fibrillation: a roadmap to underpin personalized prevention and treatment. Nat Rev Cardiol 13(4):230–237. https://doi.org/10.1038/nrcardio.2015.194
Schmidt C, Wiedmann F, Zhou XB, Heijman J, Voigt N, Ratte A, Lang S, Kallenberger SM, Campana C, Weymann A, De Simone R, Szabo G, Ruhparwar A, Kallenbach K, Karck M, Ehrlich JR, Baczko I, Borggrefe M, Ravens U, Dobrev D, Katus HA, Thomas D (2017) Inverse remodelling of K2P3.1 K+ channel expression and action potential duration in left ventricular dysfunction and atrial fibrillation: implications for patient-specific antiarrhythmic drug therapy. Eur Heart J 38(22):1764–1774. https://doi.org/10.1093/eurheartj/ehw559
Coumel P (1996) Autonomic influences in atrial tachyarrhythmias. J Cardiovasc Electrophysiol 7(10):999–1007
Linz D, Schotten U, Neuberger HR, Bohm M, Wirth K (2011) Negative tracheal pressure during obstructive respiratory events promotes atrial fibrillation by vagal activation. Heart Rhythm 8(9):1436–1443. https://doi.org/10.1016/j.hrthm.2011.03.053
Linz D, McEvoy RD, Cowie MR, Somers VK, Nattel S, Levy P, Kalman JM, Sanders P (2018) Associations of obstructive sleep apnea with atrial fibrillation and continuous positive airway pressure treatment: a review. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2018.0095
Guasch E, Benito B, Qi X, Cifelli C, Naud P, Shi Y, Mighiu A, Tardif JC, Tadevosyan A, Chen Y, Gillis MA, Iwasaki YK, Dobrev D, Mont L, Heximer S, Nattel S (2013) Atrial fibrillation promotion by endurance exercise: demonstration and mechanistic exploration in an animal model. J Am Coll Cardiol 62(1):68–77. https://doi.org/10.1016/j.jacc.2013.01.091
Elliott AD, Maatman B, Emery MS, Sanders P (2017) The role of exercise in atrial fibrillation prevention and promotion: finding optimal ranges for health. Heart Rhythm 14(11):1713–1720. https://doi.org/10.1016/j.hrthm.2017.07.001
Vaughan Williams EM (1984) A classification of antiarrhythmic actions reassessed after a decade of new drugs. J Clin Pharmacol 24(4):129–147
Burashnikov A, Di Diego JM, Sicouri S, Ferreiro M, Carlsson L, Antzelevitch C (2008) Atrial-selective effects of chronic amiodarone in the management of atrial fibrillation. Heart Rhythm 5(12):1735–1742. https://doi.org/10.1016/j.hrthm.2008.09.015
Dobrev D, Nattel S (2010) New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet 375(9721):1212–1223. https://doi.org/10.1016/S0140-6736(10)60096-7
Hohnloser SH, Crijns HJ, van Eickels M, Gaudin C, Page RL, Torp-Pedersen C, Connolly SJ, Investigators A (2009) Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 360(7):668–678. https://doi.org/10.1056/NEJMoa0803778
Neef S, Mann C, Zwenger A, Dybkova N, Maier LS (2017) Reduction of SR Ca2+ leak and arrhythmogenic cellular correlates by SMP-114, a novel CaMKII inhibitor with oral bioavailability. Basic Res Cardiol 112(4):45. https://doi.org/10.1007/s00395-017-0637-y
Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skardal K, Wisloff U, Wieland T, Ai X, Pogwizd SM, Dobrev D, Wehrens XH (2012) Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ Res 110(11):1474–1483. https://doi.org/10.1161/CIRCRESAHA.112.268094
Ravens U, Poulet C, Wettwer E, Knaut M (2013) Atrial selectivity of antiarrhythmic drugs. J Physiol 591(17):4087–4097. https://doi.org/10.1113/jphysiol.2013.256115
Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C (2007) Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 116(13):1449–1457. https://doi.org/10.1161/CIRCULATIONAHA.107.704890
Wettwer E, Christ T, Endig S, Rozmaritsa N, Matschke K, Lynch JJ, Pourrier M, Gibson JK, Fedida D, Knaut M, Ravens U (2013) The new antiarrhythmic drug vernakalant: ex vivo study of human atrial tissue from sinus rhythm and chronic atrial fibrillation. Cardiovasc Res 98(1):145–154. https://doi.org/10.1093/cvr/cvt006
Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, Schmitto JD, Seipelt R, Schondube FA, Hasenfuss G, Belardinelli L, Maier LS (2010) Altered Na+ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol 55(21):2330–2342. https://doi.org/10.1016/j.jacc.2009.12.055
Burashnikov A, Antzelevitch C (2010) New developments in atrial antiarrhythmic drug therapy. Nat Rev Cardiol 7(3):139–148. https://doi.org/10.1038/nrcardio.2009.245
Voigt N, Dobrev D (2016) Atrial-selective potassium channel blockers. Card Electrophysiol Clin 8(2):411–421. https://doi.org/10.1016/j.ccep.2016.02.005
Burashnikov A, Sicouri S, Di Diego JM, Belardinelli L, Antzelevitch C (2010) Synergistic effect of the combination of ranolazine and dronedarone to suppress atrial fibrillation. J Am Coll Cardiol 56(15):1216–1224. https://doi.org/10.1016/j.jacc.2010.08.600
Hartmann N, Mason FE, Braun I, Pabel S, Voigt N, Schotola H, Fischer TH, Dobrev D, Danner BC, Renner A, Gummert J, Belardinelli L, Frey N, Maier LS, Hasenfuss G, Sossalla S (2016) The combined effects of ranolazine and dronedarone on human atrial and ventricular electrophysiology. J Mol Cell Cardiol 94:95–106. https://doi.org/10.1016/j.yjmcc.2016.03.012
Aguilar M, Xiong F, Qi XY, Comtois P, Nattel S (2015) Potassium channel blockade enhances atrial fibrillation-selective antiarrhythmic effects of optimized state-dependent sodium channel blockade. Circulation 132(23):2203–2211. https://doi.org/10.1161/CIRCULATIONAHA.115.018016
Reiffel JA, Camm AJ, Belardinelli L, Zeng D, Karwatowska-Prokopczuk E, Olmsted A, Zareba W, Rosero S, Kowey P, Investigators H (2015) The HARMONY Trial: combined ranolazine and dronedarone in the management of paroxysmal atrial fibrillation: mechanistic and therapeutic synergism. Circ Arrhythm Electrophysiol 8(5):1048–1056. https://doi.org/10.1161/CIRCEP.115.002856
McPate MJ, Duncan RS, Hancox JC, Witchel HJ (2008) Pharmacology of the short QT syndrome N588K-hERG K+ channel mutation: differential impact on selected class I and class III antiarrhythmic drugs. Br J Pharmacol 155(6):957–966. https://doi.org/10.1038/bjp.2008.325
Friedrichs S, Malan D, Sasse P (2013) Modeling long QT syndromes using induced pluripotent stem cells: current progress and future challenges. Trends Cardiovasc Med 23(4):91–98. https://doi.org/10.1016/j.tcm.2012.09.006
Clauss S, Sinner MF, Kaab S, Wakili R (2015) The role of MicroRNAs in antiarrhythmic therapy for atrial fibrillation. Arrhythm Electrophysiol Rev 4(3):146–155. https://doi.org/10.15420/aer.2015.4.3.146
Dawson K, Wakili R, Ordog B, Clauss S, Chen Y, Iwasaki Y, Voigt N, Qi XY, Sinner MF, Dobrev D, Kaab S, Nattel S (2013) MicroRNA29: a mechanistic contributor and potential biomarker in atrial fibrillation. Circulation 127(14):1466–1475. https://doi.org/10.1161/CIRCULATIONAHA.112.001207
Clauss S, Klier I, Schmidt V, Schuessler F, Siebermair J, Sinner M, Fichtner S, Estner H, Kääb S, Wakili R (2014) MicroRNAs as potential biomarkers of atrial fibrillation ablation therapy. Heart Rhythm 11(5 Supplement):AB36–A06
Du DT, Hellen N, Kane C, Terracciano CM (2015) Action potential morphology of human induced pluripotent stem cell-derived cardiomyocytes does not predict cardiac chamber specificity and is dependent on cell density. Biophys J 108(1):1–4. https://doi.org/10.1016/j.bpj.2014.11.008
Christ T, Horvath A, Eschenhagen T (2015) LQT1-phenotypes in hiPSC: are we measuring the right thing? Proc Natl Acad Sci USA 112(16):E1968. https://doi.org/10.1073/pnas.1503347112
Goversen B, van der Heyden MAG, van Veen TAB, de Boer TP (2018) The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: special focus on IK1. Pharmacol Ther 183:127–136. https://doi.org/10.1016/j.pharmthera.2017.10.001
Chauveau S, Anyukhovsky EP, Ben-Ari M, Naor S, Jiang YP, Danilo P Jr, Rahim T, Burke S, Qiu X, Potapova IA, Doronin SV, Brink PR, Binah O, Cohen IS, Rosen MR (2017) Induced pluripotent stem cell-derived cardiomyocytes provide in vivo biological pacemaker function. Circ Arrhythm Electrophysiol 10(5):e004508. https://doi.org/10.1161/CIRCEP.116.004508
Devalla HD, Schwach V, Ford JW, Milnes JT, El-Haou S, Jackson C, Gkatzis K, Elliott DA, Chuva de Sousa Lopes SM, Mummery CL, Verkerk AO, Passier R (2015) Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol Med 7(4):394–410. https://doi.org/10.15252/emmm.201404757
Lugenbiel P, Schweizer PA, Katus HA, Thomas D (2016) Antiarrhythmic gene therapy—will biologics replace catheters, drugs and devices? Eur J Pharmacol 791:264–273. https://doi.org/10.1016/j.ejphar.2016.09.001
Rosen MR (2014) Gene therapy and biological pacing. N Engl J Med 371(12):1158–1159. https://doi.org/10.1056/NEJMcibr1408897
Breithardt G, Krämer L, Willems S (2012) Curriculum spezielle Rhythmologie. Kardiologe 6:219–225
Merino JL, Arribas F, Botto GL, Huikuri H, Kraemer LI, Linde C, Morgan JM, Schalij M, Simantirakis E, Wolpert C, Villard MC, Poirey J, Karaim-Fanchon S, Deront K, Accreditation Committee of the European Heart Rhythm A (2009) Core curriculum for the heart rhythm specialist. Europace 11(Suppl 3):iii1–i26. https://doi.org/10.1093/europace/eup215
Albert CM, Chen PS, Anderson ME, Cain ME, Fishman GI, Narayan SM, Olgin JE, Spooner PM, Stevenson WG, Van Wagoner DR, Packer DL, Heart Rhythm Society Research Task F (2011) Full report from the first annual Heart Rhythm Society Research Forum: a vision for our research future, “dream, discover, develop, deliver”. Heart Rhythm 8(12):e1–e12. https://doi.org/10.1016/j.hrthm.2011.10.024
Lau DH, Volders PG, Kohl P, Prinzen FW, Zaza A, Kaab S, Oto A, Schotten U, European Heart Rhythm A (2015) Opportunities and challenges of current electrophysiology research: a plea to establish ‘translational electrophysiology’ curricula. Europace 17(5):825–833. https://doi.org/10.1093/europace/euu301
Ley TJ, Rosenberg LE (2005) The physician-scientist career pipeline in 2005: build it, and they will come. JAMA 294(11):1343–1351. https://doi.org/10.1001/jama.294.11.1343
Acknowledgements
The authors work was supported in part by research grants from the German Heart Foundation/German Foundation of Heart Research (Josef Freitag Foundation to A.G., F/08/14 to D.T., F/03/15 to D.L.), from the Else Kröner-Fresenius-Stiftung (2014_A242 to D.T., 2014_A306 to D.L., 2016_A20 to N.V.), from the Joachim Siebeneicher Foundation (to D.T.), from the Deutsche Forschungsgemeinschaft (German Research Foundation; TH 1120/7-1 and TH 1120/8-1 to D.T., KFO 196 to D.L. et al., BR2107/4-1 and OD 86/6-1 to K.E.O., SCHW 1611/1-1 to P.A.S., VO 1568/3-1 and IRTG1816 RP12 and SFB1002 TPA13 to N.V.), from the Ministry of Science, Research and the Arts Baden-Wuerttemberg (Sonderlinie Medizin to D.T.; Wrangell Programme to K.E.O.), from the Josef-Freitag-Stiftung (to A.G.), and from the German Cardiac Society (DGK0914 to D.L.). D.T. and N.V. were supported by the German Center for Cardiovascular Research (DZHK). A.G. and J.K. were supported by European Union Seventh Framework Programme (EUTRAF-261057). J.H. was supported by the Netherlands Organization for Scientific Research (ZonMW Veni 91616057) and the Young Talent Program of the CardioVascular Onderzoek Nederland (CVON) and Netherlands Heart Foundation PREDICT project, D.L. was supported by a Beacon Research Fellowship from the University of Adelaide. P.S. received support from the Molecular Medicine Partnership Unit, Heidelberg (Senior Career Fellowship).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethical standards
The manuscript does not contain clinical studies or patient data.
Conflict of interest
D.T. reports receiving lecture fees/honoraria from Bayer Vital, Bristol-Myers Squibb, Daiichi Sankyo, Medtronic, Pfizer Pharma, Sanofi-Aventis, St. Jude Medical and ZOLL CMS, and research grant support from Daiichi Sankyo. A.G. reports speaker fees from Astra Zeneca, Berlin Chemie, Biotronik, Boehringer Ingelheim, Bayer Health Care, Bristol-Myers Squibb/Pfizer, Daiichi-Sankyo, Medtronic. M.H. reports speaker fees from Astra Zeneca, Berlin Chemie, Boehringer Ingelheim, Bayer Health Care, Bristol-Myers Squibb/Pfizer, Daiichi-Sankyo. D.L. reports serving on the advisory board of LivaNova and Medtronic, receiving lecture fees/honoraria from LivaNova, Medtronic, Pfizer and ResMed, and receiving research grant support from Sanofi, ResMed and Medtronic. J.H. reports speaker fees from Pfizer. N.V. reports receiving research suppor from Nissan Biochemical.
Rights and permissions
About this article

Cite this article
Thomas, D., Christ, T., Fabritz, L. et al. German Cardiac Society Working Group on Cellular Electrophysiology state-of-the-art paper: impact of molecular mechanisms on clinical arrhythmia management. Clin Res Cardiol 108, 577–599 (2019). https://doi.org/10.1007/s00392-018-1377-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00392-018-1377-1