
Abstract
Purpose
Colorectal cancer is one of the major leading causes of death worldwide, and available treatments for advanced colorectal cancer are not successful. Therefore, early detection of colorectal cancer is essential to improve patient survival, and biomarkers are potential tools to achieve this goal. Considering the key role of lncRNAs in cancers, the aim of this study is to identify lncRNAs involved in colorectal cancer as new potential prognosis biomarkers for CRC.
Methods
In this observational study, gene expression data obtained from the TCGA database were analyzed, Identification of differentially expressed mRNAs, miRNAs, and lncRNAs was performed, and ceRNA network was drawn. Also, survival analysis of patients was performed in order to identify potential biomarkers related to the diagnosis and prognosis of colon cancer. After confirming the results using the GSE39582 dataset, the expression of target lncRNAs in colorectal tumor tissues was also investigated to confirm the bioinformatic data.
Results
Analysis of the TCGA data showed that the expression of three lncRNAs—SNHG7, ASMTL-AS1, and LINC02604—that had the highest interaction with other miRNAs and mRNAs identified based on the ceRNA network was increased in colorectal cancer. Also, based on the ceRNA network, three microRNAs, hsa-let-7d-5p, hsa-mir-92a-3p, and hsa-mir-423-5p, and eight mRNAs, including CPA4, MSI2, RRM2, IGF2BP1, ONECUT2, HMGA1, SOX4, and SRM, were associated with all three mentioned lncRNAs, the expression of microRNAs was decreased and the expression of mRNAs was increased. By enrichment analysis, it was found that the target lncRNAs are involved in the processes of cell proliferation, apoptosis, and metastasis, indicating their importance in the development and malignancy of colorectal cancer. Furthermore, Kaplan–Meier analysis showed a significant increase in mortality in patients with higher expression levels of these lncRNAs. Analysis of the GSE39582 dataset, and real-time RT-PCR analysis, confirmed our bioinformatic results. Also, ROC analysis showed that SNHG7 was a relatively good promising biomarker (AUC = 0.73, p value = 0.02), while ASMTL-AS1 (AUC = 0.92, p value < 0.0001) and LINC02604 (AUC = 1.00, p value < 0.0001) emerged as excellent diagnostic biomarkers in colorectal cancer.
Conclusion
It seems that increased expression of lncRNAs ASMTL-AS1 and LINC02604 can serve as molecular biomarkers for CRC, possibly through the sponge hsa-let-7d-5p, hsa-mir-92a-3p, and hsa-mir-423 5p, which increases target mRNAs, which are effective in the carcinogenesis process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer is one of the leading causes of death worldwide [1]. Despite extensive efforts in developing diagnostic and prognostic methods, a significant proportion of patients are diagnosed in advanced stages, resulting in unsatisfactory treatment outcomes [2].
Although various environmental and genetic risk factors have been identified in relation to colorectal cancer [3], the exact molecular mechanisms involved in its development remain unclear. Therefore, it is crucial to gain a comprehensive understanding of the molecular mechanisms underlying colorectal cancer and discover biomarkers for early diagnosis.
Recent studies have uncovered significant findings regarding the essential role of long non-coding RNAs (lncRNAs) in various aspects of cell biology, including cell proliferation, apoptosis, metastasis, and treatment resistance. These molecules have the potential to contribute to the occurrence and progression of different types of tumors, including colorectal cancer [4,5,6,7]. LncRNAs are RNA molecules that exceed 200 nucleotides in length and lack the ability to encode proteins [7, 8]. However, they actively participate in the gene expression network through diverse mechanisms, such as miRNA sequestration, histone and chromatin modification, and protein translation [9, 10].
Emerging evidence suggests that the aberrant expression of lncRNAs may play a crucial role in the initiation and progression of colorectal cancer. As a result, these lncRNAs have been proposed as biomarkers and potential therapeutic targets for the diagnosis and management of the disease [11, 12]. Therefore, the identification of these lncRNAs holds promise for aiding in the early diagnosis and treatment of colorectal cancer.
In recent years, the development of high-throughput technologies, such as microarray and next-generation sequencing, has enabled the identification of numerous key genes associated with colorectal cancer that may contribute to its initiation and development [13].
Considering the pivotal role of lncRNAs as vital biomarkers in cancer, the present study was conducted with the aim of identifying lncRNAs involved in colorectal cancer. For this purpose, gene expression data obtained from TCGA were analyzed and ceRNA network was drawn. Also, survival analysis of patients was performed in order to identify potential biomarkers related to the diagnosis and prognosis of colon cancer. Next, the results of TCGA analysis were analyzed in the GSE39582 dataset from the GEO database, and finally, in order to confirm the results of bioinformatics analysis, the expression of target lncRNAs in tumor and healthy colorectal tissues was also investigated, and for the first time, two new biomarkers were introduced for the diagnosis of colorectal disease.
Materials and methods
In this section, we used the STREGA reporting guidelines [14].
Data source
To identify mRNAs, miRNAs, and lncRNAs associated with the development and pathogenesis of colorectal cancer, the TCGA data was utilized. The raw transcriptomic data of colorectal cancer (TCGA-COAD) was downloaded in HTseq-Counts format using the TCGAbiolinks package. Genes with zero or minimal expression were filtered out based on the criterion of less than 10 counts per million (CPM) in 70% of the samples, employing the edgeR package. Subsequently, the data underwent normalization using the TMM method (trimmed mean of M values) and the limma package. The resulting expression matrix was used for all subsequent analyses, comprising 480 tumor samples and 41 normal samples for mRNAs and lncRNAs, as well as 457 tumor samples and 8 normal samples for miRNAs. Additionally, the most recent clinical information was downloaded for all samples and incorporated into the analysis process.
Also, the raw data from the GSE39582 dataset was obtained from the GEO database, consisting of 19 normal and 542 colorectal cancer samples (www.ncbi.nlm.nih.gov/geo). The data underwent preprocessing steps, including background correction, data normalization using the RMA (robust multichip average) method, and transformation to logarithmic mode base 2 using the limma package.
Identification of differentially expressed mRNAs, miRNAs, and lncRNAs
The normalized expression matrix was utilized to identify mRNAs, miRNAs, and lncRNAs that displayed significant expression changes in colorectal cancer samples compared to normal samples. The linear model method was employed to assess the expression differences between the groups, and a criterion of |logFC|> 1 and FDR < 0.01 was applied to select genes. Gene lists and information for mRNAs and lncRNAs were extracted using the Bio Mart tool. Furthermore, the expression changes were visualized using the Enhanced Volcano package through a volcano plot.
Construction of the ceRNA network
The genes displaying significant expression differences were used to construct the ceRNA network. To investigate the interaction between miRNAs and mRNAs, the mirwalk database (http://mirwalk.umm.uni-heidelberg.de) and the mirTarBase database (http://miRTarBase.cuhk.edu.cn/) were employed. Only miRNA-mRNA pairs confirmed by both databases were selected.
Furthermore, the DIANA-LncBase v3 database (www.microrna.gr/LncBase) was utilized to explore miRNA-lncRNA interactions. The selection criteria included validation type = direct, miRNA Confidence level = high, and species = human. Subsequently, miRNA-mRNA and miRNA-lncRNA pairs were chosen based on their expression differences in colorectal cancer, meeting the criteria of |logFC|> 1 and FDR < 0.01 from the previous steps.
Finally, the data were visualized in Cytoscape software to represent the ceRNA network. The criterion of degree > 12 was used to identify the important lncRNAs within the ceRNA network.
Preprocessing of clinical and prognosis data
Colorectal clinical data from TCGA-COAD were used to investigate the relationship between gene expression and patient prognosis. Preprocessing of clinical data was performed by removing normal samples, samples from patients with survival of 1 day or missing data (NA), and those without tumor-related mortality status.
Next, the expression levels of all genes were extracted for the samples meeting the specified clinical criteria. Z-scores were calculated to standardize the expression of each gene across all samples. The relationship between gene expression and patient prognosis was examined using the univariate Cox regression test.
Furthermore, Kaplan–Meier analysis was employed to confirm the obtained results. The median expression of candidate genes in cancer samples served as the cutoff point. A log Rank p value of less than 0.05 was considered statistically significant.
Quantitative RT-PCR
Thirty-two colorectal tumor tissues and also adjacent normal tissue samples, based on the diagnosis of the attending physician and confirmation of the pathology results, from the Iranian race, were obtained from the Cancer Research Center, Cancer Institute (Tehran University of Medical Sciences, Tehran, Iran). RNA extraction was performed using TRIzol reagent (Invitrogen). Subsequently, cDNA synthesis was carried out using the cDNA Synthesis Kit (YTA), and RT-qPCR was performed using the SYBR green master-mix kit (YTA) and Rotor-gene 6000.
The GAPDH gene was used as the reference gene to normalize the expression level. Each reaction was repeated three times to ensure accuracy. The results were analyzed using the 2−∆Ct method, a commonly employed quantitative analysis approach for gene expression studies.
Using Beacon software, specific primers were designed for all isoforms of LncRNAs SNHG7, LINC02604, ASMTL-AS1, mRNAs CPA4, SRM, SOX4, and GAPDH gene based on the sequence of genes in NCBI. The primer sequences used were as follows:
SNHG7, forward: 5′TCTCCTCCCGGCCAGTTC3′, reverse: 5′GCACCCGGAGGCCAGCAG3′; LINC02604, forward: 5′GCTAGACCATTTTTGTGCC3′, reverse: 5′CTGAAGGGACAATGCAAAC3′; ASMTL-AS1, forward: 5′GTTTACAGACGCATTTCAGCC3′, reverse: 5′GCTATGGAGTGGCAGTTCTC3′; GAPDH, forward: 5′GCCAAAAGGGTCATCATCTCTCTGC3′, reverse: 5′GGTCACGAGTCCTTCCACGATAC3′; CPA4, forward: 5′GACAACCCTTGCTCCGAAGT3′, reverse: 5′AGTAGCTGTGCAGGTCGATG3′; SRM, forward: 5′CTTTGTGCTGCCCGAGTTTG3′, reverse: 5′GTAACACTTGGTTGGTGGGC3′; SOX4, forward: 5′GACTTCGAGTTTGCTCCCCT3′, reverse: 5′TAACTCGCCTTCTTGCTGGG3′
Statistical analysis
All preprocessing and data analysis were performed using the R programming language (version 4.0.2). GraphPad Prism software (version 8) was utilized for graphing and visualization purposes. Expression differences were calculated using the linear model method, and the significance level between the groups was assessed through multiple hypothesis testing. A threshold of FDR < 0.01 was applied for all analyses.
The relationship between the expression of candidate genes and patient prognosis was investigated using the log Rank test. A log Rank p value below 0.05 was considered statistically significant. In addition, Cytoscape software (version 4) was employed to display the ceRNA network and visualize the association of genes with the identified lncRNAs.
Results
Differentially expressed genes in colorectal cancer
To identify key genes involved in the progression and development of colorectal cancer, the TCGA data was evaluated. Differential expression analysis was conducted to compare lncRNAs, mRNAs, and miRNAs between cancer samples and normal samples.
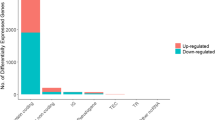
The analysis revealed that among the examined genes, 403 lncRNAs, 3088 mRNAs, and 161 miRNAs exhibited increased expression in cancer samples compared to normal samples. Conversely, 280 lncRNAs, 3893 mRNAs, and 123 miRNAs showed decreased expression (Fig. 1) and may play important roles in the pathogenesis of colorectal cancer.

Volcano plots illustrating the expression levels of lncRNAs (A), mRNAs (B), and miRNAs (C). In these plots, red dots represent differentially expressed genes that fulfill both criteria of |logFC|> 1 and FDR < 0.01. Blue dots represent genes that do not meet the |logFC|> 1 criterion. Green dots represent genes that do not meet the FDR < 0.01 criterion, and gray dots represent genes that do not meet both the |logFC|> 1 and FDR < 0.01 criteria
Association of identified lncRNAs and mRNAs with the survival rate of patients
Our findings revealed that 1269 mRNAs were associated with poor prognosis (HR > 1, log Rank < 0.05), while 124 mRNAs were associated with good prognosis (HR < 1, log Rank < 0.05) (Fig. 2).

The volcano plot of the association of mRNAs with the survival rate of CRC patients
Additionally, the results revealed that out of the differentially expressed mRNAs, 209 upregulated mRNAs were associated with poor prognosis of patients, whereas 22 downregulated mRNAs were associated with good prognosis of patients (Fig. 3).

Intersection of differentially expressed mRNAs and survival-related mRNAs. A The intersected upregulated mRNAs associated with survival; B the intersected downregulated mRNAs associated with survival
Next, the association between lncRNAs and the survival rate of colorectal cancer (CRC) patients was investigated. The results revealed that 142 lncRNAs were associated with poor prognosis (HR > 1, log Rank < 0.05), while 17 lncRNAs were associated with good prognosis (HR < 1, log Rank < 0.05) (Fig. 4).

Volcano plot depicting the association of lncRNAs with the survival rate of colorectal cancer patients
Subsequently, an analysis was conducted to determine the overlap between differentially expressed lncRNAs and the identified survival-related lncRNAs. The findings revealed that 38 upregulated lncRNAs were associated with poor prognosis in patients, while 3 downregulated lncRNAs were associated with good prognosis (Fig. 5). These results suggest that the identified lncRNAs and mRNAs may play a significant role in the development of colorectal cancer.

Intersection of differentially expressed lncRNAs and survival-related lncRNAs. A The intersected upregulated lncRNAs associated with survival; B the intersected downregulated lncRNAs associated with survival
Identification of hub lncRNAs in the ceRNA network and their association with survival
A ceRNA network was constructed using 161 upregulated and 123 downregulated miRNAs, along with differentially expressed mRNAs and lncRNAs associated with patient mortality. This network included 224 miRNA-mRNA pairs and 272 miRNA-lncRNA pairs, which were merged using Cytoscape. The results highlighted three lncRNAs, namely SNHG7, ASMTL-AS1, and LINC02604, which exhibited the most interactions with other identified miRNAs and mRNAs (Fig. 6).

Illustration of the ceRNA network between all differentially expressed genes. In this network, blue diamonds represent lncRNAs, green ellipses represent mRNAs, and yellow triangles represent miRNAs
To validate our results obtained from TCGA, we assessed the expression levels of SNHG7, ASMTL-AS1, and LINC02604 in the GSE39582 dataset. The findings revealed a significant and substantial increase in the expression levels of SNHG7, ASMTL-AS1, and LINC02604 genes in cancer samples compared to normal samples (Fig. 7).

Comparison of the expression levels of SNHG7, ASMTL-AS1, and LINC02604 in the GSE39582 dataset
Furthermore, hsa-let-7d-5p, hsa-mir-92a-3p, and hsa-mir-423-5p were found to be associated with each of the three mentioned lncRNAs, as well as with a total of eight mRNAs, including CPA4, MSI2, RRM2, IGF2BP1, ONECUT2, HMGA1, SOX4, and SRM (Fig. 8). Interestingly, our analysis revealed that the expression levels of the three miRNAs were decreased in tumor samples compared to normal samples, while the expression levels of the three lncRNAs and eight mRNAs were increased. Furthermore, the enrichment analysis of the genes in the ceRNA network revealed that a majority of these genes are involved in pathways related to cell proliferation, apoptosis, and metastasis (Fig. 9).

Association of target lncRNAs with hsa-let-7d-5p, hsa-mir-92a-3p, and hsa-mir-423-5p with the corresponding mRNAs

Enrichment analysis of all differentially expressed genes
To validate the obtained results, the relationship between the expression of SNHG7, ASMTL-AS1, and LINC02604 with the mortality rate of patients was investigated using Kaplan–Meier analysis. As depicted in Fig. 10, an increase in the expression levels of SNHG7, ASMTL-AS1, and LINC02604 in cancer samples was significantly associated with a higher mortality rate among patients.

Association of expression levels of SNHG7, ASMTL-AS1, and LINC02604 with patients’ survival
These findings suggest that SNHG7, ASMTL-AS1, and LINC02604 may play a crucial role in the disease’s pathogenesis and the survival rate of patients.
qRT-PCR
Next, the expression levels of lncRNAs SNHG7, ASMTL-AS1, and LINC02604 were examined using qRT-PCR in 32 tumor and 32 adjacent normal colorectal tissues. The findings revealed an upregulation in the expression levels of all three lncRNAs in tumor tissues when compared to the adjacent healthy tissues (Fig. 11).

Comparison of the expression level of SNHG7, ASMTL-AS1, and LINC02604 lncRNAs in tumor tissues compared to adjusted normal tissues
Furthermore, ROC analysis revealed that SNHG7 (AUC = 0.73, p value = 0.02) showed promise as a relatively good biomarker, while ASMTL-AS1 (AUC = 0.92, p value < 0.0001) and LINC02604 (AUC = 1.00, p value < 0.0001) emerged as excellent diagnostic biomarkers in colorectal cancer (Fig. 12).

ROC curves of SNHG7, ASMTL-AS1, and LINC02604 lncRNAs
Finally, in the sub-networks related to target lncRNAs (Fig. 8), the expression levels of SOX4, SRM, and CPA4 (with the highest level of expression change based on bioinformatics analysis) mRNAs, in 32 tumor and 32 normal colorectal tissues, were investigated using qRT-PCR. Data analysis showed that the expression of SRM and CPA4 mRNAs in tumor tissue increased compared to the adjacent normal tissue, while the expression of SOX4 mRNA did not change (Fig. 13).

Comparison of the expression level of SRM, CPA4, and SOX4 mRNAs in tumor tissues compared to adjusted normal tissues
Also, ROC analysis revealed that SRM (AUC = 0.89, p value < 0.0001) acts as an excellent diagnostic biomarker in colorectal cancer (Fig. 14).

ROC curves of SRM, CPA4, and SOX4 mRNAs
Discussion
A number of studies are currently looking for biomarkers to help achieve the goal of early detection of colorectal cancer, which is crucial to improving patient survival [15]. Long non-coding RNAs (lncRNAs) can significantly affect the development of tumors by controlling gene expression in a variety of cellular processes, including epigenetic modifications, mRNA stability, translation, alternative splicing, transcription, and miRNA regulation [16,17,18].
According to our results, the expression of three lncRNAs—SNHG7, ASMTL-AS1, and LINC02604—which have the highest number of interactions with other identified miRNAs and mRNAs, is increased in colorectal cancer and confirmed by analyzing the GSE39582 dataset from GEO and real-time RT-PCR analysis on colorectal tumor tissues compared to adjacent normal tissues.
Three lncRNAs were found to be involved in the processes of cell proliferation, apoptosis, and metastasis by enrichment analysis, suggesting their significance in the growth and malignancy of colorectal cancer. Additionally, Kaplan–Meier analysis indicated a significant increase in mortality in patients with higher expression levels of these lncRNAs.
In addition, based on bioinformatics data, a total of eight mRNAs, including CPA4, MSI2, RRM2, IGF2BP1, ONECUT2, HMGA1, SOX4, and SRM, were related to each of the three lncRNAs, as well as hsa-let-7d-5p, hsa-mir-92a-3p, and hsa-mir-423-5p. Considering the decrease in the expression of these miRNAs and the increase in the expression of target mRNAs, it seems that the mRNA/miRNA/LncRNA axes can be taken into consideration.
Recent studies have shown that SNHG7 acts as an oncogene, and its expression significantly increased in a wide range of carcinomas, including pancreatic cancer, thyroid cancer, bladder cancer, breast cancer, cervical cancer, gastric cancer, hepatocellular carcinoma, hypopharyngeal cancer, melanoma, and neuroblastoma [19]. It has been reported that SNHG7 acts as a ceRNA by sponging miR-34a, which controls the expression of the GALNT7 target gene and promotes the progression of colorectal cancer (CRC) via the PI3K/Akt/mTOR pathway [15]. Additionally, through the miR-216b sponge, SNHG7 can upregulate GALNT1 expression. This can result in oncogenic effects through the SNHG7/miR-216b/GALNT1 axis, and it has been proposed that targeting this axis could be a promising therapeutic approach for CRC [20].
The antisense acetyl serotonin O-methyltransferase 1 (ASMTL-AS1) long non-coding RNA was found recently at the Xp22.33 and Yp11.2 locus, and it plays a dual role in cancer. In breast cancer, ASMTL-AS1 expression is significantly downregulated, which triggers the Wnt/β-catenin signaling pathway via the miR-1228-3p/SOX17 axis [21]. However, in hepatocellular carcinoma, ASMTL-AS1 upregulation activates the YAP signaling pathway through the ASMTL-AS1/miR-1343-3p/LAMC1 axis, which results in a recurrence of the cancer or its metastasis [22]. Nevertheless, little is known about the expression and function of ASMTL-AS1 lncRNA in colorectal cancer; our study gives the first evidence of its oncogenic role in colorectal cancer.
In addition, there is not much information about the lncRNA LINC02604. According to Shi et al., in glioblastoma tumors, high expression of LINC02604 increases the probability of cell proliferation, invasion, and migration, which is associated with poor prognosis and decreased patient survival [23]. Also, in a bioinformatics study, Jing et al. identified LINC02604 as a potential biomarker to identify colon cancer patients who benefited from antitumor immunotherapy [24]. The results of both studies are in line with the results of the present study.
According to the ceRNA network identified in this study and the evaluation of target mRNAs of these lncRNAs, it can be suggested that target lncRNAs can exert these oncogenic effects through hsa-mir-423-5p/SRM or hsa-let-7d-5p/CPA4 axes. Also, other hsa-let-7d-5p/MSI2-RRM2-IGF2BP1-ONECUT2-HMGA1 axis should be investigated.
MiR-423-5p has been found to be downregulated in several tumors, including ovarian cancer, osteosarcoma, cervical cancer, and colon cancer [25]. In colon cancer, miR-423-5p as a tumor suppressor has been reported to induce cell apoptosis through caspase activation, and interestingly, it has been introduced as a biomarker for early detection of colon cancer [26]. Also, Let-7d, a member of the let-7 family, acts as a tumor suppressor in various cancers, including colorectal cancer [27], and hsa-let-7d-5p expression is significantly downregulated in CRC tumoral tissues compared to adjacent normal tissues [28, 29]. As a member of the miR-17–92 family, miR-92a-3p, as an oncogene or tumor suppressor gene [30], plays an important role in regulating cell viability, apoptosis, and metastasis of tumor cells [31, 32], and its dysregulation is associated with tumor progression and prognosis [33].
Carboxypeptidase A4 (CPA4) is a zinc-dependent metallocarboxypeptidase that is overexpressed in various cancer tissues, including colorectal cancer (CRC), which is in line with the results of the present study. Increased expression of CPA4 promotes the growth of CRC cells, while its knockdown results in decreased proliferation, arrest in the G1/S phase transition, and induction of apoptosis. Accordingly, CPA4 has been considered a prognostic factor or therapeutic target for CRC [34].
The level of several polyamines increases in various cancers such as skin, breast, colon, lung, and prostate cancer [35], which play an important role in cell proliferation, tumor invasion, and metastasis by modulating gene expression and signaling pathways [36, 37]. Spermidine synthase (SRM) is an enzyme involved in the biosynthesis of various polyamines, including putrescine, spermidine, and spermine [38]. In this regard, it has been reported that SRM gene expression is upregulated in colorectal cancer through the function of lncRNA ELFN1-AS1 and miR-423 sponge [39] which can be a confirmation of our results.
Also, the increase in the expression of MSI2, RRM2, IGF2BP1, ONECUT2, and HMGA1 mRNAs has been reported in different studies. The RRM2 gene has been introduced as an oncogene in several cancers, including CRC, so its increased expression is associated with advanced tumor grade, poor prognosis, and reduced patient survival, and its inhibition through the induction of cell apoptosis can be a potential therapeutic strategy for CRC [40,41,42,43].
Sex-determining region high mobility group box 4 (SOX4), as a transcription factor, regulates multiple signaling pathways, including PI3K, Wnt, and TGFβ, and promotes tumorigenesis by increasing cell survival, migration, invasion, and metastasis. Therefore, it is not surprising that the expression of SOX4 is increased in various malignancies, including breast, prostate, stomach, and colon cancer, and that it acts as an oncogene [44,45,46] which was not confirmed in the present study. It seems that the role of miR-92a-3p in colorectal cancer should be clearly defined.
The Musashi-2 (MSI-2) gene is overexpressed in various tumors, including colorectal cancer, and plays an important role as an oncogene in carcinogenesis and tumor progression [47]. Increased cytoplasmic expression of MSI-2 in CRC patients is associated with an unfavorable prognosis and has been proposed as a potential biomarker for CRC prognosis [48]. Increasing expression of transcription factor high mobility group protein A1 (HMGA1) can cause the development of cancer by regulating the transcription of targets of several biological pathways, which is associated with poor clinical results, distant metastasis, and advanced tumor stage in many cancers. HMGA1 expression has been reported to be increased in colorectal cancer and can serve as a diagnostic indicator for CRC [49].
Increased expression of the transcription factor one-cut domain 2 (ONECUT2) has been observed in various cancers, including colorectal cancer, which is associated with tumor growth, metastasis, chemoresistance, and poor prognosis and is considered a potential therapeutic target in CRC [50]. Finally, in limited adult tissues, increased expression of the insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) gene has been reported in cancers, including gastrointestinal cancers, lung cancer, melanoma, and colorectal cancer [51].
Conclusion
The data of this research showed that the increased expression of target lncRNA, especially ASMTL-AS1 and LINC02604, can act as a biomarker in the identification of colorectal tumors. Based on bioinformatic data, it seems that SNHG7, ASMTL-AS1, and LINC02604 can act as molecular sponges, through the reduction of hsa-let-7d-5p and hsa-mir-423-5p, which leads to the increase of target mRNAs, specially RSM and CPA4 mRNAs which are related to the development and progression of colorectal cancer through the regulation of cell proliferation, apoptosis, invasion, metastasis, and response to treatment. Investigating the functions and interactions of these genes in a broader way can provide new insight into the underlying mechanisms of colorectal cancer and potentially develop therapeutic strategies.
Data availability
The datasets analyzed during the current study are available in Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/projects/geo/) and The Cancer Genome Atlas (TCGA) (https://tcga-data.nci.nih.gov/).
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA cancer J Clin 68(6):394–424
Mo S, Zhang L, Dai W, Han L, Wang R, Xiang W, Wang Z, Li Q, Yu J, Yuan J, Cai S (2020) Antisense lncRNA LDLRAD4-AS1 promotes metastasis by decreasing the expression of LDLRAD4 and predicts a poor prognosis in colorectal cancer. Cell Death Dis 11(2):1–16
Sawicki T, Ruszkowska M, Danielewicz A, Niedźwiedzka E, Arłukowicz T, Przybyłowicz KE (2021) A review of colorectal cancer in terms of epidemiology, risk factors, development, symptoms and diagnosis. Cancers 13(9):2025
Poursheikhani A, Abbaszadegan MR, Nokhandani N, Kerachian MA (2020) Integration analysis of long non-coding RNA (lncRNA) role in tumorigenesis of colon adenocarcinoma. BMC Med Genomics 13(1):1–16
Xu W, Zhou G, Wang H, Liu Y, Chen B, Chen W, Lin C, Wu S, Gong A, Xu M (2020) Circulating lncRNA SNHG11 as a novel biomarker for early diagnosis and prognosis of colorectal cancer. Int J Cancer 146(10):2901–2912
Zhao K, Ye Z, Li Y, Li C, Yang X, Chen Q, Xing C (2020) LncRNA FTX contributes to the progression of colorectal cancer through regulating miR-192-5p/EIF5A2 axis. Onco Targets Ther 13:2677–2688
Su M, Xiao Y, Ma J, Cao D, Zhou Y, Wang H, Liao Q, Wang W (2018) Long non-coding RNAs in esophageal cancer: molecular mechanisms, functions, and potential applications. J Hematol Oncol 11(1):1–14
Iyer MK, Niknafs YS, Malik R et al (2015) The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 47(3):199–208
Ponjavic J, Ponting CP, Lunter G (2007) Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res 17(5):556–565
Geisler S, Coller J (2013) RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol 14(11):699–712
Jiang C, Li X, Zhao H, Liu H (2016) Long non-coding RNAs: potential new biomarkers for predicting tumor invasion and metastasis. Mol Cancer 15(1):1–15
Poursheikhani A, Abbaszadegan MR, Kerachian MA (2021) Mechanisms of long non-coding RNA function in colorectal cancer tumorigenesis. Asia Pac J Clin Oncol 17(1):7–23
Ghasemi T, Khalaj-Kondori M, Hosseinpour Feizi MA, Asadi P (2021) Aberrant expression of lncRNAs SNHG6, TRPM2-AS1, MIR4435-2HG, and hypomethylation of TRPM2-AS1 promoter in colorectal cancer. Cell Biol Int 45(12):2464–2478
Little J, Higgins JP, Ioannidis JP, Moher D, Gagnon F, von Elm E, Khoury MJ, Cohen B, Davey-Smith G, Grimshaw J, Scheet P, Gwinn M, Williamson RE, Zou GY, Hutchings K, Johnson CY, Tait V, Wiens M, Golding J, van Duijn C, McLaughlin J, Paterson A, Wells G, Fortier I, Freedman M, Zecevic M, King R, Infante-Rivard C, Stewart A, Birkett N (2009) Strengthening the reporting of genetic association studies (STREGA)—an extension of the STROBE statement. Genetic Epidemiology: The Official Publication of the International Genetic Epidemiology Society 33(7):581–598
Li Y, Zeng C, Hu J, Pan Y, Shan Y, Liu B, Jia L (2018) Long non-coding RNA-SNHG7 acts as a target of miR-34a to increase GALNT7 level and regulate PI3K/Akt/mTOR pathway in colorectal cancer progression. J Hematol Oncol 11(1):1–7
Yang H, Jiang Z, Wang S, Zhao Y, Song X, Xiao Y, Yang S (2019) Long non-coding small nucleolar RNA host genes in digestive cancers. Cancer Med 8(18):7693–7704
Zhang L, Meng X, Zhu XW, Yang DC, Chen R, Jiang Y, Xu T (2019) Long non-coding RNAs in Oral squamous cell carcinoma: biologic function, mechanisms and clinical implications. Mol Cancer 18:1–19
Zhang Y, Tian Q, Huang S, Wang Q, Wu H, Dong Q, Chen X (2022) Prognostic effect of lncRNA SNHG7 on cancer outcome: a meta and bioinformatic analysis. BMC Cancer 22(1):1–13
Bian Z, Ji W, Xu B, Huang W, Jiao J, Shao J, Zhang X (2020) The role of long noncoding RNA SNHG7 in human cancers. Mol Clin Oncol 13(5):1–1
Shan Y, Ma J, Pan Y, Hu J, Liu B, Jia L (2018) LncRNA SNHG7 sponges miR-216b to promote proliferation and liver metastasis of colorectal cancer through upregulating GALNT1. Cell Death Dis 9(7):722
Sun J, Li X, Yu E, Liu J, Sun L, He Q, Lu Q (2021) A novel tumor suppressor ASMTL-AS1 regulates the miR-1228-3p/SOX17/β-catenin axis in triple-negative breast cancer. Diagn Pathol 16(1):1–9
Ma D, Gao X, Liu Z, Lu X, Ju H, Zhang N (2020) Exosome-transferred long non-coding RNA ASMTL-AS1 contributes to malignant phenotypes in residual hepatocellular carcinoma after insufficient radiofrequency ablation. Cell Prolif 53(9):e12795
Shi J, Wang YJ, Sun CR, Qin B, Zhang Y, Chen G (2017) Long noncoding RNA lncHERG promotes cell proliferation, migration and invasion in glioblastoma. Oncotarget 8(64):108031
Xing XL, Zhang T, Yao ZY, Xing C, Wang C, Liu YW, Huang M (2021) Immune-related gene expression analysis revealed three lncRNAs as prognostic factors for colon cancer. Front Genet 12:690053
Ke R, Lv L, Zhang S, Zhang F, Jiang Y (2020) Functional mechanism and clinical implications of MicroRNA-423 in human cancers. Cancer Med 9(23):9036–9051
Jia W, Yu T, An Q, Cao X, Pan H (2018) MicroRNA-423-5p inhibits colon cancer growth by promoting caspase-dependent apoptosis. Exp Ther Med 16(2):1225–1231
Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME (2010) The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer 17(1):F19-36
Niculae AM, Dobre M, Herlea V, Manuc TE, Trandafir B, Milanesi E, Hinescu ME (2022) Let-7 microRNAs are possibly associated with perineural invasion in colorectal cancer by targeting IGF axis. Life 12(10):1638
Pliakou E, Lampropoulou DI, Dovrolis N, Chrysikos D, Filippou D, Papadimitriou C, Vezakis A, Aravantinos G, Gazouli M (2022) Circulating miRNA expression profiles and machine learning models in association with response to irinotecan-based treatment in metastatic colorectal cancer. Int J Mol Sci 24(1):46
Zhu S, Zhang L, Zhao Z, Fu W, Fu K, Liu G, Jia W (2018) MicroRNA-92a-3p inhibits the cell proliferation, migration and invasion of Wilms tumor by targeting NOTCH1. Oncol Rep 40(2):571–578
Ma H, Pan JS, Jin LX, Wu J, Ren YD, Chen P, Xiao C, Han J (2016) MicroRNA-17~ 92 inhibits colorectal cancer progression by targeting angiogenesis. Cancer Lett 376(2):293–302
Zhou P, Ma L, Zhou J, Jiang M, Rao E, Zhao Y, Guo F (2016) miR-17-92 plays an oncogenic role and conveys chemo-resistance to cisplatin in human prostate cancer cells. Int J Oncol 48(4):1737–1748
Song H, Zhang Y, Liu N, Zhao S, Kong Y, Yuan L (2016) miR-92a-3p exerts various effects in glioma and glioma stem-like cells specifically targeting CDH1/β-catenin and Notch-1/Akt signaling pathways. Int J Mol Sci 17(11):1799
Pan H, Pan J, Ji L, Song S, Lv H, Yang Z, Guo Y (2019) Carboxypeptidase A4 promotes cell growth via activating STAT3 and ERK signaling pathways and predicts a poor prognosis in colorectal cancer. Int J Biol Macromol 138:125–134
Nowotarski SL, Woster PM, Casero RA (2013) Polyamines and cancer: implications for chemotherapy and chemoprevention. Expert Rev Mol Med 15:e3
Thomas T, Thomas TJ (2003) Polyamine metabolism and cancer. J Cell Mol Med 7(2):113–126
Gerner EW, Meyskens FL Jr (2004) Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer 4(10):781–792
Casero RA Jr, Marton LJ (2007) Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discovery 6(5):373–390
Chen J, Song Y, Li M, Zhang Y, Lin T, Sun J, Wang D, Liu Y, Guo J, Yu W (2021) Comprehensive analysis of ceRNA networks reveals prognostic lncRNAs related to immune infiltration in colorectal cancer. BMC Cancer 21(1):1–7
Liu X, Zhang H, Lai L, Wang X, Loera S, Xue L, He H, Zhang K, Hu S, Huang Y, Nelson RA (2013) Ribonucleotide reductase small subunit M2 serves as a prognostic biomarker and predicts poor survival of colorectal cancers. Clin Sci 124(9):567–579
Fang Z, Gong C, Liu H, Zhang X, Mei L, Song M, Qiu L, Luo S, Zhu Z, Zhang R, Gu H (2015) E2F1 promote the aggressiveness of human colorectal cancer by activating the ribonucleotide reductase small subunit M2. Biochem Biophys Res Commun 464(2):407–415
Grolmusz VK, Karászi K, Micsik T, Tóth EA, Mészáros K, Karvaly G, Barna G, Szabó PM, Baghy K, Matkó J, Kovalszky I (2016) Cell cycle dependent RRM2 may serve as proliferation marker and pharmaceutical target in adrenocortical cancer. Am J Cancer Res 6(9):2041
Putluri N, Maity S, Kommagani R, Creighton CJ, Putluri V, Chen F, Nanda S, Bhowmik SK, Terunuma A, Dorsey T, Nardone A (2014) Pathway-centric integrative analysis identifies RRM2 as a prognostic marker in breast cancer associated with poor survival and tamoxifen resistance. Neoplasia 16(5):390–402
Moreno CS (2020) SOX4: the unappreciated oncogene. InSeminars in cancer biology Dec 1 (Vol. 67, pp. 57–64). Academic Press.
Hanieh H, Ahmed EA, Vishnubalaji R, Alajez NM (2020) SOX4: epigenetic regulation and role in tumorigenesis. InSeminars in cancer biology Dec 1 (Vol. 67, pp. 91–104). Academic Press.
Wang B, Li Y, Tan F, Xiao Z (2016) Increased expression of SOX4 is associated with colorectal cancer progression. Tumor Biology 37:9131–9137
Sun J, Sheng W, Ma Y, Dong M (2021) Potential role of musashi-2 RNA-binding protein in cancer EMT. Onco Targets Ther 17:1969–1980
Zong Z, Zhou T, Rao L, Jiang Z, Li Y, Hou Z, Yang B, Han F, Chen S (2016) Musashi2 as a novel predictive biomarker for liver metastasis and poor prognosis in colorectal cancer. Cancer Med 5(4):623–630
Wang Y, Hu L, Zheng Y, Guo L (2019) HMGA1 in cancer: cancer classification by location. J Cell Mol Med 23(4):2293–2302
Yu J, Li D, Jiang H (2020) Emerging role of ONECUT2 in tumors. Oncol Lett 20(6):1–1
Chen HM, Lin CC, Chen WS, Jiang JK, Yang SH, Chang SC, Ho CL, Yang CC, Huang SC, Chao Y, Liao TT (2021) Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) is a prognostic biomarker and associated with chemotherapy responsiveness in colorectal cancer. Int J Mol Sci 22(13):6940
Author information
Authors and Affiliations
Contributions
Fariba Shakeri performed data collection and analysis and wrote the first draft of the manuscript. Parisa Mohamadynejad designed, planned, and supervised the project and revised its manuscript. Mehdi Moghanibashi advised and helped in interpreting the results and writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
https://ethics.research.ac.ir/IR.IAU.SHK.REC.1401.084
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shakeri, F., Mohamadynejad, P. & Moghanibashi, M. Identification of ASMTL-AS1 and LINC02604 lncRNAs as novel biomarkers for diagnosis of colorectal cancer. Int J Colorectal Dis 39, 112 (2024). https://doi.org/10.1007/s00384-024-04692-x
Accepted:
Published:
DOI: https://doi.org/10.1007/s00384-024-04692-x