
Abstract
Background
Sensenbrenner syndrome, also known as cranioectodermal dysplasia (CED), is a genetically heterogeneous ciliopathy, characterized by dysmorphic features including dolichocephaly (with inconstant sagittal craniosynostosis), chronic kidney disease (CKD), hepatic fibrosis, retinitis pigmentosa, and brain abnormalities, with a partial clinical overlap with other ciliopathies.
Patients and methods
A retrospective review of four children with Sensenbrenner syndrome treated at the Femme Mère Enfant University Hospital of Lyon from 2005 to 2020 was conducted.
Results
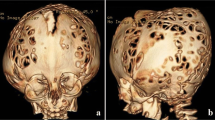
Variants in WDR35 or WDR19 were found in all children. Two of them underwent surgery for a scaphocephaly in the first months of life. All patients developed CKD leading to end-stage renal disease during the first/second decades.
Discussion
The diagnosis of scaphocephaly may precede the diagnosis of the underlying Sensenbrenner syndrome, thus highlighting the importance of a systematic multidisciplinary assessment and follow-up for craniosynostoses, in order to identify syndromic forms requiring specific management. In Sensenbrenner syndrome, patients’ management should be coordinated by multidisciplinary teams of reference centers for rare diseases, with expertise in the management of craniofacial malformations as well as rare skeletal and renal disorders. Indeed, a prompt etiological diagnosis will result in an early diagnosis of multisystemic complications, notably renal involvement, thus improving global prognosis.

Similar content being viewed by others

References
Amar MJ, Sutphen R, Kousseff BG (1997) Expanded phenotype of cranioectodermal dysplasia (Sensenbrenner syndrome). Am J Med Genet 70:349–352
Antony D, Nampoory N, Bacchelli C, Melhem M, Wu K, James CT, Beales PL, Hubank M, Thomas D, Mashankar A, Behbehani K, Schmidts M, Alsmadi O (2017) Exome sequencing for the differential diagnosis of ciliary chondrodysplasias: example of a WDR35 mutation case and review of the literature. Eur J Med Genet 60(12):658–666. https://doi.org/10.1016/j.ejmg.2017.08.019
Arts H, Knoers N (2013) Cranioectodermal dysplasia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews®. University of Washington, Seattle, Seattle 1993-2020 Initial Posting:September 12, 2013; Last Revision:April 12, 2018
Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, Leh SM, Midtbø M, Filhol E, Bole-Feysot C, Nitschké P, Gilissen C, Haugen OH, Sanders JS, Stolte-Dijkstra I, Mans DA, Steenbergen EJ, Hamel BC, Matignon M, Pfundt R, Jeanpierre C, Boman H, Rødahl E, Veltman JA, Knappskog PM, Knoers NV, Roepman R, Arts HH (2011) Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet 89:634–643
Caparrós-Martín JA, De Luca A, Cartault F, Aglan M, Temtamy S, Otaify GA, Mehrez M, Valencia M, Vázquez L, Alessandri JL, Nevado J, Rueda-Arenas I, Heath KE, Digilio MC, Dallapiccola B, Goodship JA, Mill P, Lapunzina P, Ruiz-Perez VL (2015) Specific variants in WDR35 cause a distinctive form of Ellis-van Creveld syndrome by disrupting the recruitment of the EvC complex and SMO into the cilium. Hum Mol Genet 24(14):4126–4137. https://doi.org/10.1093/hmg/ddv152
Córdova-Fletes C, Becerra-Solano LE, Rangel-Sosa MM, Rivas-Estilla AM, Alberto Galán-Huerta K, Ortiz-López R, Rojas-Martínez A, Juárez-Vázquez CI, García-Ortiz JE (2018) Uncommon runs of homozygosity disclose homozygous missense mutations in two ciliopathy-related genes (SPAG17 and WDR35) in a patient with multiple brain and skeletal anomalies. Eur J Med Genet 61(3):161–167. https://doi.org/10.1016/j.ejmg.2017.11.011
Di Rocco F, Gleizal A, Szathmari A, Beuriat PA, Paulus C, Mottolese C (2019) Sagittal suture craniosynostosis or craniosynostoses? The heterogeneity of the most common premature fusion of the cranial sutures. Neurochirurgie. 65(5):232–238. https://doi.org/10.1016/j.neuchi.2019.09.011
Fenglan L, Yu-Hong T (2018) Nephronophthisis: a review of genotype-phenotype correlation. Nephrology (Carlton) 23(10):904–911. https://doi.org/10.1111/nep.13393
Genitori L, Lang D, Philip N, Cavalheiro S, Lena G, Choux M (1992) Cranioectodermal dysplasia with sagittal craniosynostosis (Sensenbrenner’s syndrome): case report and review of the literature. Br J Neurosurg 6:601–606
Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, van Lier B, Steehouwer M, van Reeuwijk J, Kant SG, Roepman R, Knoers NVAM, Veltman JA, Brunner HG (2010) Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet 87:418–423
Hildebrandt F, Zhou W (2007) Nephronophthisis-associated Ciliopathies. J Am Soc Nephrol 18(6):1855–1871
Lin AE, Traum AZ, Sahai I, Keppler-Noreuil K, Kukolich MK, Adam MP, Westra SJ, Arts HH (2013) Sensenbrenner syndrome (cranioectodermal dysplasia): clinical and molecular analyses of 39 patients including two new patients. Am J Med Genet A 161A:2762–2776
Muttusamy T, Ma A, Sinnerbrink I, Quinton AE, Peek MJ, Joung S (2017) Prenatal sonographic features of cranioectodermal dysplasia. Prenat Diagn 37(6):628–630. https://doi.org/10.1002/pd.5037
Obikane K, Nakashima T, Watarai Y, Morita K, Cho K, Tonoki H, Nagata M, Sasaki S (2006) Renal failure due to tubulointerstitial nephropathy in an infant with cranioectodermal dysplasia. Pediatr Nephrol 21:574–576
Sensenbrenner JA, Dorst JP, Owens RP (1975) New syndrome of skeletal, dental and hair anomalies. Birth Defects Orig Artic Ser 11:372–379
Smith C, Lamont RE, Wade A, Bernier FP, Parboosingh JS, Innes AM (2016) A relatively mild skeletal ciliopathy phenotype consistent with cranioectodermal dysplasia is associated with a homozygous nonsynonymous mutation in WDR35. Am J Med Genet A 170(3):760–765. https://doi.org/10.1002/ajmg.a.37514
Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, Hennekam RC, Torre G, Garshasbi M, Tzschach A, Szczepanska M, Krawczynski M, Zachwieja J, Zwolinska D, Beales PL, Ropers HH, Latos-Bielenska A, Kuss AW (2010) Cranioectodermal dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet 86:949–956
Yoshikawa T, Kamei K, Nagata H, Saida K, Sato M, Ogura M, Ito S, Miyazaki O, Urushihara M, Kondo S, Sugawara N, Ishizuka K, Hamasaki Y, Shishido S, Morisada N, Iijima K, Nagata M, Yoshioka T, Ogata K, Ishikura K (2017) Diversity of renal phenotypes in patients with WDR19 mutations: two case reports. Nephrology (Carlton) 22(7):566–571. https://doi.org/10.1111/nep.12996
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Quinaux, T., Custodi, V., Putoux, A. et al. Sensenbrenner syndrome: a further challenge in evaluating sagittal synostosis and a need for a multidisciplinary approach. Childs Nerv Syst 37, 1695–1701 (2021). https://doi.org/10.1007/s00381-021-05075-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-021-05075-1