
Abstract
Half metallic ferromagnetic (HMF) are key materials for quantum computing information storage devices and green energy. So, the structural, electronic, optical, and magnetic properties of SMoO2 (S = Na, K, Rb, Cs) are studied by quantum simulations based on density functional theory (DFT) within WIEN2K code. The Perdew − Burke − Ernzerhof (PBE + GGA) was used for the exchange correlation potential to study the said compounds. The volume optimization properties indicated that these compounds have minimum ground state energy in monoclinic crystal structure with space group 12 C2/m. Moreover, electronic band gap properties and density of states (DOS) graphs depicted the metallic nature for spin up a channel and semiconductor nature for spin down channel. This confirms the half-metallic ferromagnetism (HMF), and Heisenberg's classical model confirms the 100% spin polarization. Various frequency dependent optical parameters explain the optical properties. The double exchange process, crystal field energy ∆Ecry, direct Δx(d), and indirect exchange energies Δx(pd), is demonstrated by the quantum ferromagnetic behavior. The negative value of Δx(pd) and magnetic exchange constants confirm the ferromagnetism due to the quantum exchange mechanism of electrons. In addition to the above, it has been noted from the density of states that Mo d-states are responsible for the response of half metallic nature of these compounds. The magnetic parameter values of SMoO2 (S = Na, K, Rb, and Cs) show higher magnetic moment and major contribution coming from the Mo atom. Hence the studied results show that these compounds are valuable materials for spintronic applications.
Similar content being viewed by others
1 Introduction
Spintronics has become the emerging research area that explains the quantum ferromagnetic response by controlling the intrinsic properties of electrons (spin + charge) [1]. There are various types of magnetic semiconductors materials such as; Full Heusler alloys [2], Half Heusler alloys [3, 4], perovskites [5] diluted magnetic semiconductor materials DMS [6] and delafossites ABO2 materials. Commercially, these materials are used for the fabrication of spintronics devices. Especially, delafossite ABO2-based semiconductor magnetic materials have fascinating applications in giant magneto-resistance (GMR), tunnel magneto-resistance (TMR), and anisotropic magneto-resistance (AMR) [6, 7]. Furthermore, expanding the need for memory devices with high speed, higher capacity and smaller size has sparked research that has produced memory devices with ever increasing capacity in ever smaller packages. The size of a memory chip can only be decreased so far due to quantum restrictions. In addition to the charge of the electron used in typical semiconductor devices, one can use the spin of the electron to improve the capacity of memory of the small size [7].
The half metallic ferromagnetic materials (HMFM) have the ability to sustain the charge and spin of the electron. HMFM have two channels; for one channel it shows the metallic and for the other channel it shows the semiconductor nature. Due to these unique properties of HMFM become the desirable materials that are suitable for wide variety of innovative application in spintronics technology. The combined effect of both channels is very important and plays avital role in understanding the physics phenomenon among the spin and electronic transitions within materials. Therefore, in spintronics technology, the combined effect of both channels is very important and plays avital role to understand the physics among the spin and electronic mechanisms. The physics behind the HMFM opens the door for spintronics applications in the future. The control of electron spin and low Curie temperature are the main challenges for the spintronics industry. In the past decades, a large number of theoretical works have been done on half metallic semiconductor materials [8]. For example, Mavlanjan et al., predicted the electronic band structure and magnetic response of SrMO2 (M = Ni, Co, Mn) by ab initio method [9]. Mouatassimeet al., performed the theoretical calculation to check the half metallic in Co2FeGe compounds [10]. Similarly, Sachin al., predicted the optical, thermoelectric properties in BaCdS2 [11]. Guobao Li et al., check the F-doping effect on NaXO2 (X = V, Cr, Co, and Ni) by DFT scheme [12]. Moreover, Benmakhlouf et al. studied the half-metallic behavior of XMnSe2 (X = Rb, Cs) by DFT calculation [13]. In addition, Benmakhlouf et al. predicted the half-metallic magnetic characteristics of KMnX2 (X = O, S, Se, Te) chalcogenides by DFT method [14]. The metal oxides have great attention of the researchers during the past several years, as the structural qualities have a major impact on the shown physical properties. Therefore, it is crucial to investigate the structural parameters to fully understand the physical properties of the materials for future applications. Several researchers have examined different metal oxide semiconductor like MgO–NiO [6].
To the best of our knowledge till date, theoretical and experimental literature is limited on these S1+Mo3+O2 compounds. Therefore, the present aim of this computational work is to investigate the structural, electronic, magnetic, and optical properties of S1+Mo3+O2 (S = Na, K, Rb, Cs) delafossite based compounds by DFT with Perdew–Burke–Ernzerhof generalized gradient approximation (PBE + GGA) within WIEN2K code.
2 Method of calculations
The spin-polarized electronic band structure, optical, thermal, and magnetic response of S1+Mo3+O2 (S = Na, K, Rb, Cs) compounds have been studied by quantum simulations based on density functional theory (DFT) within WIEN2K code. The DFT calculation is based on electron full potential linearized augmented plane wave (FP–LAPW). The ground states energy Eg (eV) of a unit crystal structure, lattice constants a = b & c (Å), and Bulk modulus (B) are computed by the Birch–Murnaghan equation [2]. To compute the spin-polarized electronic band structure, optical, thermal and magnetic properties, self-converging field (SCF) converge for the exchange correlation energy of these materials by employing the Perdew–Burke–Ernzerhof generalized gradient approximation (GGA-PBE) approximation [15]. The space region in (FP–LAPW) has been divided into spherical harmonics and plane wave regions. The angular momentum expansion Lmax = 10 and Gaussian factors of the Fourier expansion Gmax = 16 are adjusted as all the electrons remains in the muffin-tin region. The wave vector in reciprocal lattice and muffin-tin radius is chosen as RMT × Kmax = 8. For convergence of exchange correlation energy of the materials, the order of k-mesh is 12 × 12 × 12. The self-consisting field (SCF) calculation is repeated until the energy converging value of the unit crystal is less than 10−5Ry. In addition to the above, the characterization has been done by various techniques. The optical behavior is checked by dielectric constants which explain by Kramers–Kronig model [16].
3 Result and discussion
3.1 Structural properties
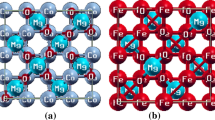
The crystal structures give information about lattice constants and the sitting arrangement of individual atoms in the unit crystal of the materials. So, unit crystal structures of full Heusler based S1+Mo3+O2 (S = Na, K, Rb and Cs) compounds are shown in Fig. 1. These studied compounds are stable in monoclinic crystal structure with space group 12 (C 2/m). It can be noted from Fig. 1, which shows the crystal structures of SMoO2 (S = Na, K, Rb, and Cs), the oxygen (O) atoms are denoted by red balls, Mo atoms are denoted by grey balls, and Na, K, Rb, and Cs atoms are denoted by various colors. We have optimized the monoclinic crystal structures of SMoO2 (Na, K, Rb, and Cs) with the aid of the GGAPBEsol approximation. Table 1 represents the theoretically optimized parameters. Lattice constants values are increasing as the atomic number of elements increases; it may be due to an increased number of electronic shells which minimize the effect of nucleus. The sitting atomic position of (Na, K, Rb and Cs) is (0, 0, 0) and the sitting atomic position of molybdenum (Mo) is (0.5, 0.5, 0.5) in the unit crystal cell. In the case of the oxygen atom (O2), the sitting atomic position arrangement for one oxygen atom is (0.01, 0.75, and 0.50) and for the second oxygen atom is (0.98, 0.24, and 0.50) respectively.

The unit crystal structure of SMoO2 (S = Na, K, Rb and Cs) compounds formed by using quantum based DFT method within WIEN2K software
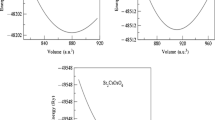
The stability of the materials is very important for the fabrication of the application of the device. Therefore, the crystal structure stability of these SMoO2 (S = Na, K, Rb, Cs) compounds are checked by the volume optimization method. The unit crystal structural stability can be ensured by the energy formation and volume optimization process. The volume optimization is done by employing the Birch–Murnaghan equation to obtain the minimum ground state energy of these compounds [2]. The optimization plots graph between energy versus volume are shown in Fig. 2 which shows minimum ground states energy of unit crystal structure of each compound are negative. The calculated values of lattice constants a, b, and c (Å), Bulk modulus B (GPa) and energy bandgap Eg (eV) values are presented in Table 1. All the negative values of the ground states energy of these compounds have been ordered as CsMoO2 > RbMoO2 > KMoO2 > NaMoO2, which ensures that unit crystal structures of these materials are energetically sustainable.

The unit crystal structure of SMoO2 (S = Na, K, Rb and Cs) compounds formed by using quantum based DFT method within WIEN2K software
3.2 Electronic properties
In the electronic properties, the spin-polarized band structure and density of states of the materials are demonstrated. So, the spin-polarized band structure of SMoO2 (S = Na, K, Rb and Cs) materials are depicted in Fig. 3a–d, respectively. It is noticed from spin-polarized band structure figures that these studied compounds have a semiconductor nature for down spin channels and show the metallic nature for up spin channels. As a result, the combination of down spin channels and up spin channels of these compounds makes them half metallic ferromagnetic materials. In addition to the above, the bottom of conduction band minima (CBM) and top of valance band maxima (VBM) lie at the same highly symmetric points (Η, Γ) near the Fermi level (EF) in the first Brillouin Zone. Which confirm the direct band gap semiconductor behavior of these materials for down spin channels [2]. The energy band gap Eg (eV) values in spin down channel are 1.26, 2.17, 2.21, and 2.31 for NaMoO2, KMoO2, RbMoO2, and CsMoO2, respectively.

a Spin polarized band structure of NaMoO2 show the half metallic nature. b Spin polarized band structure of KMoO2 show the half metallic nature. c Spin polarized band structure of RbMoO2 show the half metallic nature. d Spin polarized band structure of CsMoO2 show the half metallic nature
The spin-polarized density of states (DOS) is related to the electronic properties of the materials which give information about (s, p, d & f) atomic orbital electronic states contribution of each element in the compounds. The density of states also explains the spin polarization and natures of the materials near the Fermi level (EF) either are semiconductors, metal or insulators. The spin polarization (P) is computed by this relation [18].
Haft metallic ferromagnetic (HFM) materials fulfil the demand to sustain the 100% spin polarization near the Fermi level (EF). It is observed from Figs. 4, 5, 6, 7, the maximum states occur in up spin (\(\uparrow\)) channel and zero states in the down spin (\(\downarrow\)) channel represents the 100% spin polarization near the Fermi level (EF). This ensure that these materials may be potential candidates for the fabrication of spintronics devices applications [19]. The (HMF) arises due to the hybridization process that occurs between the s-states of Na, K, Rb and Cs, p-states of oxygen O2 and d-states of molybdenum Mo atoms. During the hybridization, electrons exchange their spin of electrons in orbitals, which is the main properties of spintronics materials [20, 21].

a Total density of states (DOS) of NaMoO2 and individual atomic orbital electronic states of b molybdenum (Mo), c sodium (Na), and d oxygen (O), respectively

a Total density of states (DOS) of KMoO2 and individual atomic orbital electronic states of b molybdenum (Mo), c potassium (K), and d oxygen (O), respectively

a Total density of states (DOS) of RbMoO2 and individual atomic orbital electronic states of b molybdenum (Mo), c rubidium (Rb), and d oxygen (O), respectively

a Total density of states (DOS) of CsMoO2 and individual atomic orbital electronic states of b molybdenum (Mo), c cesium (Cs), and d oxygen (O), respectively
The hybridization process can be demonstrated by the double exchange process [22]. From Figs. 7, 8, 9, 10, the d-states of molybdenum split into degeneracy states like dxy, dyz, dxz, dz2 and dx2−y2 due to the interaction of anion of oxygen with the cation of molybdenum atom, which hybridized with s-states of Na, K, Rb and Cs and p-states of oxygen atom individually for to generate the ferromagnetism. In addition to the above, s-states of Na, K, Rb and Cs have less contribution in conduction as well as valance band to making the total DOS of SMoO2 near the EF. However, d-states and p-states of molybdenum and oxygen atom represent the higher contribution in conduction as well as valance band to making the total DOS of SMoO2 near the EF. Therefore, p–d hybridization is more dominant than s–d hybridization and plays a vital role to stabilize the half metallic ferromagnetism in these compounds.

Graph of a real part of dielectric function ɛ1(ω), b imaginary part of dielectric function ɛ2(ω), c refractive index n(ω), d extinction coefficient K(ω), e reflectivity R(ω), f absorption coefficient α(ω), g optical conductivity σ(ω) and h energy loss function L(ω)

a Total density of states (DOS) of RbMoO2 and individual atomic orbital electronic states of b molybdenum (Mo), c rubidium (Rb), and d oxygen (O), respectively

a Total density of states (DOS) of CsMoO2 and individual atomic orbital electronic states of b molybdenum (Mo), c cesium (Cs), and d oxygen (O), respectively.
3.3 Magnetic properties
The crystal field theory explains the splitting mechanism due to which magnetic properties arise in these studied SMoO2 compounds [23, 24]. When oxygen legend atoms interacted with molybdenum atoms in the octahedral environment as shown in Fig. 1. The d-states of molybdenum splitting in three-fold dxy, dyz, dxz lower energy t2g and two-fold dz2, dx2−y2 higher energy eg states. The crystal field splitting process explain the electrons distribution of Mo+3 in splitting d-states. These splitting d-states hybridized with s-states of Na, K, Rb and Cs, and p-states of oxygen atom due to this hybridization mechanism magnetic moment is arises at the atomic sites and interstitial sites in the unit crystal structure. The total magnetic moments of SMoO2 and partial magnetic moments of contributed elements and interstitial magnetic moments are shown in Table 2.
In addition to the above, quantum ferromagnetic origination and exchange coupling mechanism in the conduction band and valance band of SMoO2 compounds explained by studying the magnetic parameters like valance band edges splitting energy ∆Ev(eV) and conduction band edges splitting energy ∆Ec (eV), crystal field energy (∆Ecry), indirect or direct splitting energy and magnetic exchange constants; that are very helpful to explain the magnetic properties in detail. These magnetic parameters are computed by mean-field theory relation [25, 26]. Theoretically reported values of this magnetic parameter are presented in Table 3.
From Table 3, it has been observed that the value of crystal field energy ΔEcrys are low and direct exchange energy Δx(d) higher which confirm the ferromagnetism. In addition to the above, p-orbital electronic states and d-orbital electronic states hybridizations mechanism between p-states of O and d-states of Mo atom give the value of indirect exchange energy Δx(pd) of these compounds. The negative values of Δx(pd) depicted the down spin channel at valence band edge, which is responsible for double exchange phenomena and minimized the energy of these compounds [27,28,29]. Moreover, the magnetic exchange constant (Nοα) and (Nοβ) play a vital role to understand the exchange splitting mechanism in the conduction and valence band of the materials. The Hamiltonian based mean-field theory demonstrated the magnetic exchange constant (Nοα) and (Nοβ). Here, Nοα represents the s–d coupling process between the s-states of Na, K, Rb, Cs, and d-states of Mo atoms. Whereas Nοβ represents the p–d coupling process between the p-states of O atom and d-states of Mo atom [30]. These magnetic exchange constants calculated by the following relations, Nοα = \(\frac{\mathrm{\Delta Ec}}{x\langle \mathrm{S}\rangle }\) and Nοβ = \(\frac{\mathrm{\Delta Ev}}{x\langle \mathrm{S}\rangle }\), respectively. where x is the concentration of Mo atom and 〈S〉 half of the spin magnetic moment of Mo atom. The s–d (Nοβ) and p–d (Nοα) depicts the numerical value of conduction band and valence band edge splitting alternatively. From Table 3, it has been noticed that these half metallic semiconductor materials have opposite signs of numerical values for Nοα and Nοβ, which gives information about the opposite spin interaction between the valence and conduction states and exchange mechanism. In addition to the above, the anti-ferromagnetic mechanism is noticed among opposite parallel magnetic spins of p-states of O and d-states Mo atom, which is demonstrated by the hybridization between (p–d) states. Ferromagnetic interaction depicted among the parallel magnetic moment of Na, K, Rb, Cs, and O atoms, respectively. This is explained by the hybridization mechanism in (s–d) states of these compounds [31].
3.4 Optical properties
When electromagnetic radiations interact with materials, then the response of material towards these radiations is described in terms of optical properties. Optical properties include different parameters such as complex dielectric function, refractive index, reflectivity, energy loss, optical conductivity, extinction coefficient and absorption coefficient. Complex dielectric function, ɛ = ɛ1(ω) + iɛ2(ω) is one of the important terms to describe the optical properties of materials [31]. The imaginary part of the dielectric function is telling about the absorptive nature of the material and the real transition between unoccupied and occupied electric states totally depends on it. Both inter-band and intra-band transitions also contributes in imaginary part ɛ2(ω). The complex dielectric function ɛ(ω) = ɛ1(ω) + iɛ2(ω), here \({\varepsilon }_{1}\left(\omega \right)=1+{(\mathrm{\hslash }{\omega }_{p}/{E}_{g})}^{2}\,\mathrm{ and }\,{\varepsilon }_{2}\left(\omega \right)=\frac{{e}^{2}\mathrm{\hslash }}{\pi {m}^{2}{\omega }^{2}}\sum_{\upsilon \mathcal{C}}\int {|n,{n}^{\mathrm{^{\prime}}}\left(k,q\right)|}^{2}\partial \left[{\omega }_{n,{n}^{\mathrm{^{\prime}}}}\left(k\right)-\omega \right]{d}^{3}k\) [18, 32,33,34] is used to describe the optical nature of physical system. As seen from Fig. 8a, b of ɛ2(ω), the maximum absorption for all the compounds occurred in the lower energy region. However, NaMoO2 has the highest absorption as compared to other compounds which means that the maximum transition of electrons from the valence to conduction band happens for this compound in the lower energy region shows the semiconductor nature of material. After this, as the energy increases along the x-axis absorption property start decreasing. Moreover, the static dielectric constant value is approximately 7.2.
Graph of the real part of dielectric function ɛ1(ω) in Fig. 8a, b represents maximum dispersion in the lower energy region for all compounds after that it starts decreasing and at some point, becomes parallel to the x-axis depicting zero dispersion. Refractive index n(ω) discusses the materials transparency and opaque nature [35, 36]. Refractive index and ɛ1(ω) are interrelated and are showing the same trend. Figure 8c, d shows that the maximum refractive index for NaMoO2 is approximately 8.41 in the IR region making this material less transparent or opaque. As the refractive index moves towards decreasing, materials begin to gain transparency. The variations in refractive index n(ω) and the real part ε(ω) of dielectric function relate to each other by this relation ɛ1(ω) = n2-k2 [8]. The following equation is used to determine the refractive index \(n\left(\omega \right)={\left(\frac{{\left[{\varepsilon }_{1}^{2}\left(\omega \right)+{\varepsilon }_{2}^{2}\left(\omega \right)\right]}^\frac{1}{2}+{\varepsilon }_{1}\left(\omega \right)}{2}\right)}^\frac{1}{2}\).
The relationship between the imaginary component ɛ2(ω) and the extinction coefficient K(ω), which tells us how much light has been dimmed by the investigated materials, is 2nk = 2. Extinction coefficient is represented by K, which is a product of the absorption rate and wavelength. The k-value denotes the amplitude of the damping oscillation of the electric field of light by this relation \(k\left(\omega \right)=\frac{\alpha \lambda }{4\pi }\).
Extinction coefficient K(ω) described the efficiency of material to absorb electromagnetic radiations and it is related to the imaginary part of dielectric function ɛ2(ω) [37]. The maximum absorption efficiency is found for the KMoO2 material which is approximately 5.35. Reflectivity R(ω) discussed that part of incident light which reflected from the surface of the material. Because it shows the surface composition of the material, reflectivity R(ω) is essential for the development of photonic surfaces in devices. The way light reflects off a material’s surface reveals the uniformity of its surface, the placements of its atoms, and its atomic levels. Figure 8 displays the computed results for light reflection in terms of materials, where the reflectivity R(ω) reaches a maximum value, and the absorption drops to a lowest value. Most of the light is likewise reflected in the region of 1 eV since this is where the value ɛ1(ω) of becomes negative, demonstrating the semiconductor nature of the materials under research. This behavior results in total reflection of the light incident on the substance’s surface according to this relation \(R\left(\omega \right)=\frac{{[n\left(\omega \right)-1]}^{2}+{k}^{2}(\omega )}{{[n\left(\omega \right)+1]}^{2}+{k}^{2}(\omega )}\).
Graph of Fig. 8e, f shows which materials surface is more reflected. NaMoO2 has maximum reflection than other materials. Reflectivity and absorption are inversely proportional, the region in which reflectivity is maximum has minimum absorption. So, it can be concluded that NaMoO2 is a good reflector material. Absorption coefficient α(ω) explains material ability to absorb incident radiation. The energy required for any inter-band transitions is supplied by the material's absorption rate or α(ω). As the absorption doesn't start until a substance absorbs energy over a threshold, it is always zero for bandgap materials at 0 eV. The absorption coefficient α(ω), which calculates the radiation’s attenuation when it travels through a medium, reveals details about that substance's ability to absorb the light. The substance starts to absorb light when the energy of the particles is equal to or greater than the band gap of the materials under examination. As a result, absorption is always associated with k(ω) and ɛ1(ω). It is linked with extinction coefficient and imaginary part of dielectric function ɛ2(ω) because both these parameters are also absorption related. The maximum absorption coefficient value observed for KMoO2 same as the extinction coefficient because the more efficient material is more capable to absorb radiations. This clarifies the strong UV absorption. The absorption coefficient explains the energy that exists above and below the substance's absorption and transmission. The absorption coefficient [35] is provided by the following equation \(\alpha \left(\omega \right)=\frac{4\pi }{\lambda }{\left(\frac{{\left[{\varepsilon }_{1}^{2}\left(\omega \right)+{\varepsilon }_{2}^{2}\left(\omega \right)\right]}^\frac{1}{2}+{\varepsilon }_{1}\left(\omega \right)}{2}\right)}^\frac{1}{2}.\)
In the presence of an electric field, the conduction of electrons from the valence to the conduction band is known as optical conductivity. It is also noted that the optical conductivity is a result of intense electromagnetic radiation interacting with the materials, which causes the material's connection to be broken, creating an electronic current. It is evident that light energy can be converted into electrical energy, and this outcome was consistent with the Maxwell formulas for electromagnetism in optics. When an intense photon collides with a material's surface, three things happen: propagation, absorption, and reflection. Various peaks appear as a result of the inter band transition and are related to the fictitious component of the dielectric constant via a relation. Various peaks appear because of the inter band transition and are related, hypothetically, to the fraction of the dielectric constant. It should also be noted that the absorption peaks change towards higher energy levels as the voltage increases.
Through this formula σ (ω) = ω/4πε2(ω), the optical conductivity can be determined [38]. This phenomenon converts light energy into electrical energy. From Fig. 8g, it is noticed that KMoO2 material has the highest value of optical conductivity at about 0.4 eV. Figure 8h shows the energy loss function verses energy. The energy loss occurs due to different processes such as heating, scattering of electrons, dispersion and losses occur due to interaction between photoelectrons and other electrons of material known as plasmon losses [39, 40]. The maximum energy loss is observed for RbMoO2. This process regulates how rapidly electrons flow through a substance. The electron energy loss function L(ω) plays a significant role in revealing the energy loss of incoming photons as they pass through the material. The greatest peak value for optical energy loss is determined for the energy range of 0–10 eV. The energy loss factor L(ω), however, ranges from 0 to 9 eV. All of the compounds under investigation show comparable drift in L(ω), which makes them the best candidates for creating future devices with a limited energy range. To calculate energy loss as given relation \(L\left(\omega \right)=\mathrm{Im}\left(\frac{-1}{\varepsilon (\omega )}\right)\) [41].
4 Conclusions
In summary, half metallic ferromagnetic (HMF) are key materials for optoelectronics and information storage applications like spintronics devices. Therefore, physical properties such as, structural, electronic, optical, and magnetic properties of SMoO2 (S = Na, K, Rb, Cs) studied by density functional theory (DFT) method that implemented within WIEN2K code. The Perdew−Burke−Ernzerhof (PBE + GGA) was used for the exchange correlation potential to study the physical properties of these compounds. The volume optimization graph determines the minimum ground states energy. The electronic band structure and density of states (DOS) graphs reveal that these studied compounds have metallic properties for spin up channel and the semiconductor properties for spin down channel, that confirms the HMF of these studied compounds. Moreover, Heisenberg's classical model confirms the 100% spin polarization. frequency dependent parameters like, dielectric function, absorption, optical conductivity, refractive index, and energy loss function explain the optical properties in infra-red, visible, and ultraviolet spectrum. The double exchange process, crystal field energy ∆Ecry, direct Δx(d), and indirect exchange energies Δx(pd), is demonstrated by the quantum ferromagnetic behavior. The negative value of Δx(pd) and magnetic exchange constants confirm the ferromagnetism due to the quantum exchange mechanism of electrons. Hence the studied results show that these compounds are valuable materials for spintronic applications. Moreover, the oxides resurfaced in the spotlight many years after the halcyon days of high temperature superconductivity. These materials not only offer intriguing physics, but also made oxide electronics and spintronics possible in new ways. Due to having the benefit of a significantly stronger spin orbit connection. It is attempted to cover the impressive advances made, notably in the last five years, in this cutting-edge review of the interesting materials. There are also several concerns that remain unresolved, along with recommendations for future research topics and prospective uses.
Data availability
Not applicable.
References
W. Xu, S. Ali, X.W. YuanjunJin, X. Hu, Intrinsic ferromagnetic semiconductors in two-dimensional alkali-based chromium chalcogenides. ACS Appl. Electr. Mater. 2(12), 3853–3858 (2020)
S. Ali, H. Ullah, A.A. AlObaid, T.I. Al-Muhimeed, Crystal field splitting, half metallic ferromagnetism, structural, mechanical and magneto-electronic properties of spinels type structure compounds MgX2O4 (X= Fe and Co) for spintronic applications. Eur. Phys. J. Plus 136(7), 1–15 (2021)
A. Benmakhlouf, Y. Bourourou, A. Bouhemadou, A. Bentabet, F. Khemloul, S. Maabed, M. Bouchenafa, I. Galanakis, Structural, electronic and magnetic properties of the manganese telluride layers AMnTe2 (A= K, Rb, Cs) from first-principles calculations. J. Magn. Magn. Mater. 465, 430–436 (2018)
B. Tariq, G. Murtaza, H. Ali, R.M. Samia Razzaq, A. Khalil, M.I. Hussain, K. Ismail, G. Nazir, N.H. Alotaibi, First principles study of the structural, half-metallic ferromagnetism, magnetic, and transport properties of KXO2 (X= Pr, Nd, and Pm) hexagonal oxides. Solid State Commun. 370, 1129 (2023)
M.K. Butt, Y. Muhammad, I. Javed, S.A. Abeer, M. Adil, I. Munawar, A. Laref, Structural, electronic, half–metallic ferromagnetic and optical properties of cubic MAlO3 (M= Ce, Pr) perovskites: a DFT study. J. Phys. Chem. Solids 154, 1184 (2021)
K. Ismail, A. Hind, G. Murtaza, I.A.-M. Tahani, A.A.O. Abeer, Q. Mahmood, Study of half metallic ferromagnetism, transport and mechanical properties of X0.9375 Ti0.0625Te (X= Ca, Sr, and Ba) alloys: for spintronics application. Phys. Scr. 96(9), 095802 (2021)
H. Wang, L. Chengliang, C. Jun, L. Yong, S.L. Yuan, C. Sang-Wook, D. Shuai, L. Jun-Ming, Giant anisotropic magnetoresistance and nonvolatile memory in canted antiferromagnet Sr2IrO4. Nat. Commun. 10(1), 1–7 (2019)
G. Pourebrahim, F. Ahmadian, Prediction of Half-metallicity in the New Heusler Cs YX 2 (Y= Sc, Ti, V, Cr, and Mn; X= O and N) Compounds. J. Supercond. Novel Magn. 30(10), 2811–2822 (2017)
R. Mavlanjan, K.-C. Zhou, Y.-Z. Nie, G.-H. Guo, Electronic structure and magnetism of layered compounds SrBO2 (B= Ni Co, Mn): a theoretical investigation. Solid State Commun. 266, 6–10 (2017)
M. Mouatassime, Y. Selmani, S. Idrissi, L. Bahmad, F. Goumrhar, H. Labrim, A. Benyoussef, Magnetic properties and half metallic behavior of the Full-Heusler Co2FeGe alloy: DFT and Monte Carlo studies. J. Solid State Chem. 304, 122534 (2021)
S. Kumar, K. Naveen, Y. Kamna, K. Arvind, R.P. Singh, DFT investigations on optoelectronic spectra and thermoelectric properties of barium cadmium disulphide (BaCdS2). Optik 207, 163797 (2020)
G. Li, W. Zhu, First-principles investigation on the crystal, electronic structures and diffusion barriers of F-doped NaMO2 (M= V, Cr, Co and Ni) for rechargeable Na-ion batteries. J. Solid State Chem. 302, 122440 (2021)
A. Benmakhlouf, A. Bentabet, A. Bouhemadou, A. Benghia, Prediction of half-metallic properties for the AMnSe2 (A= Rb, Cs) compounds from first-principle calculations. J. Magn. Magn. Mater. 399, 179–184 (2016)
A. Benmakhlouf, A. Bentabet, A. Bouhemadou, S. Maabed, A. Benghia, R. Khenata, S. Bin-Omran, Structural, half-metallic magnetism and elastic properties of the KMnQ2 (Q= O, S, Se, Te) chalcogenides from first-principles calculations. J. Magn. Magn. Mater. 408, 199–205 (2016)
M.B. Saddique, R. Muhammad, A. Adnan, M.R. Shahid, A. Farooq, M. Asif, Ground state opto-electronic and thermoelectric response of cubic XSnO3 (X= Ba, Sr) compounds. Curr. Appl. Phys. 17(8), 1079–1086 (2017)
F. Wooten, Optical properties of solids, Academic Press, New York and London, 1972, 260×23.5× 16cm, 4,660. J. Phys. Soc. Jpn. 28(9), 803–804 (1973)
N. Katayama, H. Takeda, T. Yamaguchi, Y. Yamada, K. Iida, M. Takigawa, Y. Ohta, H. Sawa, Robust atomic orbital in the cluster magnet LiMoO 2. Phys. Rev. B 102(8), 081106 (2020)
N. Yaqoob, B. Sabir, G. Murtaza, M. Rana, K. Arif, M. Nawaz, A. Laref, Structural, electronic, magnetic, optical and thermoelectric response of half-metallic AMnTe2 (A= Li, Na, K): an ab-initio calculations. Phys. B 574, 3656 (2019)
H.S. Saini, A.K. Pundir, V. Mehta, M. Poonam, K.K. Manish, Investigation of half-metallic ferromagnetism in Ti-doped BeS DMS compound: A promising spintronic material. AIP Conf. Proc. 2006(1), 030019 (2018)
G.Y. Gao, K.L. Yao, E. Şaşıoğlu, L.M. Sandratskii, Z.L. Liu, J.L. Jiang, Half-metallic ferromagnetism in zinc-blende CaC, SrC, and BaC from first principles. Phys. Rev. B 75(17), 174442 (2007)
Q. Mahmood, S.M. Alay-e-Abbas, M. Yaseen, A. Mahmood, M. Rashid, N.A. Noor, Theoretical investigation of half-metallic ferromagnetism in Mg 0.75 Ti 0.25 Y (Y= S, Se, Te) alloys by using DFT-mBJ studies. J. Supercond. Nov. Magn. 29(5), 1387–1397 (2016)
H. Saini, S. Poonam, A.K. Pundir, M. Singh, J. Thakur, M.K. Kashyap, Prediction of half metallicity in Ti-doped BeSe: A spintronics material. AIP Conf. Proc. 2115(1), 030498 (2019)
L.V. Pourovskii, J. Boust, G. Rafik Ballou, G. Eslava, D. Givord, Higher-order crystal field and rare-earth magnetism in rare-earth–Co 5 intermetallics. Phys. Rev. B 101(21), 214433 (2020)
W. Bronger, A. Kyas, P. Müller, The antiferromagnetic structures of KFeS2, RbFeS2, KFeSe2, and RbFeSe2 and the correlation between magnetic moments and crystal field calculations. J. Solid State Chem. 70(2), 262–270 (1987)
S. Sanvito, P. Ordejón, N.A. Hill, First-principles study of the origin and nature of ferromagnetism in Ga 1–x Mn x As. Phys. Rev. B 63(16), 165206 (2001)
B. Doumi, M. Allel, T. Lahouari, B. Nadir, E. Mohammed, D. Fethallah, S. Adlane, T. Abdelkader, I.-B. Mustapha, First-principle investigation of half-metallic ferromagnetism in octahedrally bonded Cr-doped rock-salt SrS, SrSe, and SrTe. Eur. Phys. J. B 88(4), 1–9 (2015)
Q. Mahmood, A. Thamraa, U.H. Bakhtiar, T. Yasmeen, A.K. Nessrin, F. Mahvish, A. Laref, Role of 5 d orbital of Re in ferromagnetism and thermoelectric characteristics of Cs 2 ReCl/Br 6 double-perovskites: a density functional theory study. Eur. Phys. J. Plus 135(9), 1–13 (2020)
Z. Fang, K. Terakura, J. Kanamori, Strong ferromagnetism and weak antiferromagnetism in double perovskites: Sr 2 Fe M O 6 (M= Mo, W, and Re). Phys. Rev. B 63(18), 180407 (2001)
Q. Mahmood, M. Hassan, M.A. Faridi, B. Sabir, G. Murtaza, A. Mahmood, The study of electronic, elastic, magnetic and optical response of Zn1-xTixY (Y= S, Se) through mBJ potential. Curr. Appl. Phys. 16(5), 549–561 (2016)
B. Sabir, G. Murtaza, Q. Mahmood, R. Ahmad, K.C. Bhamu, First principles investigations of electronics, magnetic, and thermoelectric properties of rare earth based PrYO3 (Y= Cr, V) perovskites. Curr. Appl. Phys. 17(11), 1539–1546 (2017)
J. Singh, Optical properties of condensed matter and applications, vol 6 (John Wiley & Sons, 2006)
A. Bouhemadou, F. Djabi, R. Khenata, First principles study of structural, elastic, electronic and optical properties of the cubic perovskite BaHfO3. Phys. Lett. A 372(24), 4527–4531 (2008)
D.R. Penn, Wave-number-dependent dielectric function of semiconductors. Phys. Rev. 128(5), 2093 (1962)
M. Yaseen, K.B. Mehwish, A. Amna, I. Javed, M.A. Maha, I. Munawar, M. Adil, A. Laref, Phase transition and thermoelectric properties of cubic KNbO3 under pressure: DFT approach. J. Mater. Res. Technol. 11, 2106–2113 (2021)
C. Jing, C. Xiang-Rong, Z. Wei, Z. Jun, First-principles investigations on elastic and thermodynamic properties of zinc-blende structure BeS. Chin. Phys. B 17(4), 1377 (2008)
R. Khenata, M. Sahnoun, H. Baltache, A.H. Michel Rérat, N.I. Rashek, B. Bouhafs, First-principle calculations of structural, electronic and optical properties of BaTiO3 and BaZrO3 under hydrostatic pressure. Solid State Commun. 136(2), 120–125 (2005)
M.A. Pena, J.L.G. Fierro, Chemical structures and performance of perovskite oxides. Chem. Rev. 101(7), 1981–2018 (2001)
A.H. Reshak, Z. Charifi, H. Baaziz, Ab-initio calculation of structural, electronic, and optical characterizations of the intermetallic trialuminides ScAl3 compound. J. Solid State Chem. 183(6), 1290–1296 (2010)
A. Zia, G. Murtaza, I. Khawar, R.M. Arif-Khalil, I.H. Muhammad, Ab-initio calculations of the structural, electronic and optical response of KXCl3 (X= be, Ca and Sr) for optoelectronic applications. Comput. Condens. Matter 33, e00737 (2022)
K. Ismail, G. Murtaza, T. Shaista, N. Ghazanfar, A.K. Nessrin, A. Hind, U.H. Bakhtiar, M. Manal, Theoretical study of electronic, magnetic, optical and thermoelectric properties of XMnO2 (X= Au, Ag, Cu) oxides by DFT. J. Solid State Chem. 314, 123432 (2022)
S. Razzaq, I. Khawar, G. Murtaza, H.R. Hafiz, Theoretical study of Half-Heusler CsXAs (X= Ca, Sr, and Ba) from first principle calculations. J. Supercond. Novel Magn. 35(11), 3291–3299 (2022)
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Contributions
Conceptualization, KI, AP, and GM; methodology, KI, AP, and GM; software, KI, AP, AZ, SR, and GM; validation, KI, AP, AZ, SR, GM, SMA, and JF; formal analysis, KI, AP, AZ, SR, and GM; investigation, KI, AP, AZ, and SR; resources, KI, AP, AZ, SR, and GM; data curation, KI, AP, AZ, and SR; writing—original draft preparation, KI, AP, AZ, and SR; writing—review and editing, KI, GM, SMA, and JF; visualization, KI, and AP; supervision, GM, SMA, and JF; project administration, GM, SMA, and JF; funding acquisition, SMA, and JF.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ismail, K., Parveen, A., Zia, A. et al. Ab initio calculations of structural, electronic, optical, and magnetic properties of delafossite SMoO2 (S = Na, K, Rb, Cs) for spintronics. Appl. Phys. A 129, 676 (2023). https://doi.org/10.1007/s00339-023-06944-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-023-06944-z